Biology of Human Cutaneous Melanoma

Abstract
:1. Introduction
2. Clinical-Pathological Facts
2.1. Prognostic Factors
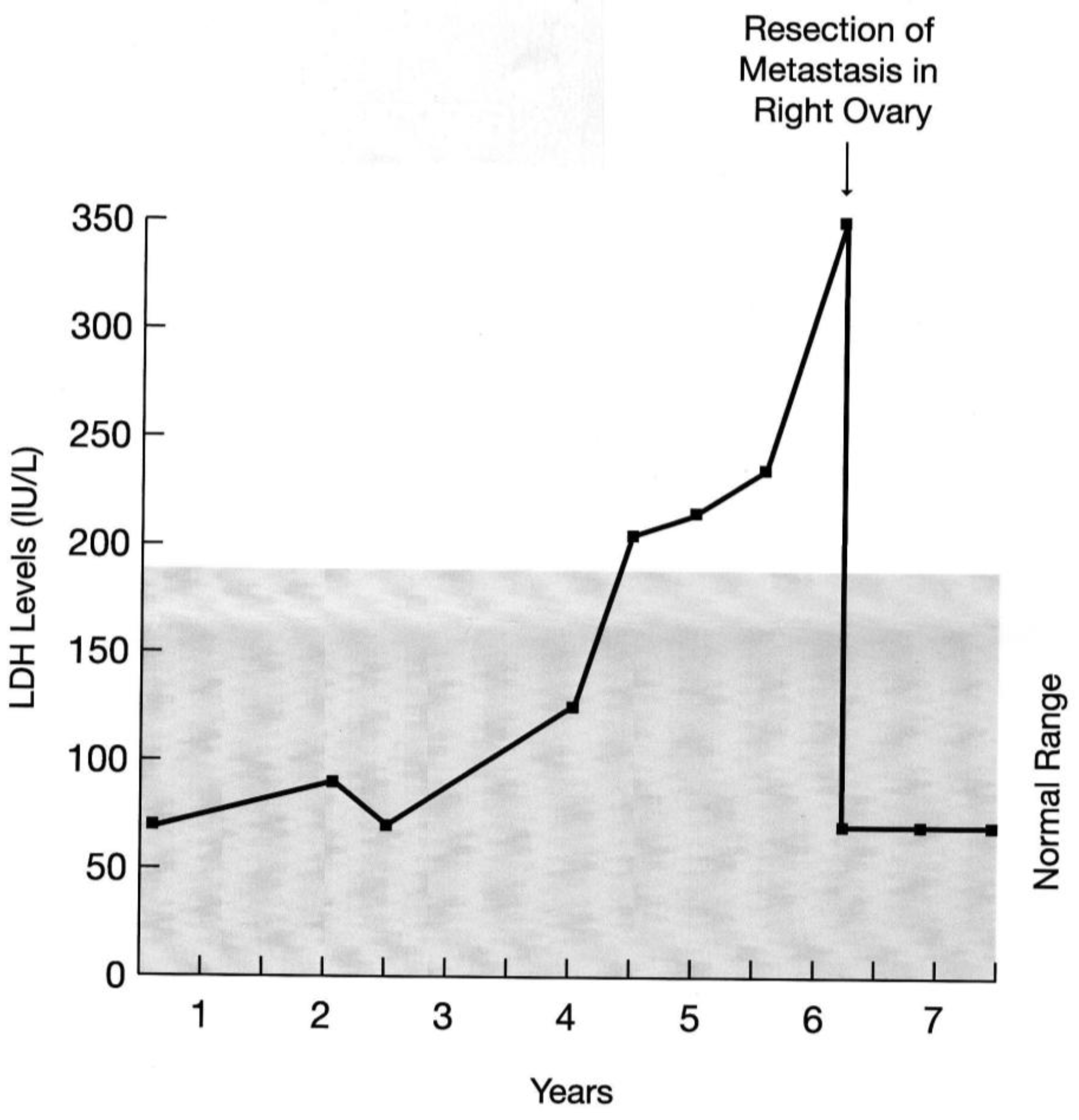
2.2. Staging

{kind=link}
{kind=link}
| STAGE | Criteria | Survival Rate | |
|---|---|---|---|
| 5-Year | 10-Year | ||
| IA | T1a : No lymph node or distant metastases | 97% | 93% |
| <1mm thickness with no ulceration | |||
| Number of mitoses <1/mm2 | |||
| IB | T1b: No lymph node or distant metastases | 94% | 87% |
| <1mm thickness with ulceration | |||
| Number of mitoses >1/mm2 | |||
| T2a: No lymph node or distant metastases | 91% | 83% | |
| 1–2 mm thickness with no ulceration | |||
| IIA | T2b: No lymph node or distant metastases | 82% | 67% |
| 1–2 mm thickness with ulceration | |||
| T3a: No lymph node or distant metastases | 79% | 66% | |
| 2–4 mm thickness with no ulceration | |||
| IIB | T3b: No lymph node or distant metastases | 68% | 55% |
| 2–4 mm thickness with ulceration | |||
| T4a: No lymph node or distant metastases | 71% | 57% | |
| >4 mm thickness with no ulceration | |||
| IIC | T4b: No lymph node or distant metastases | 55% | 39% |
| >4 mm thickness with ulceration | |||
| IIIA | N1a: micro-metastases to one regional lymph node | 78% | 68% |
| without distant metastases | |||
| N2a: micro-metastases to 2–3 regional lymph nodes | |||
| without distant metastases | |||
| IIIB | N1b: macro-metastases to one regional lymph node | 54% | 38% |
| without distant metastases | |||
| N2b: macro-metastases to 2–3 regional lymph nodes | |||
| without distant metastases | |||
| N2c: satellitosis/in transit metastases | 59% | 43% | |
| without regional lymph nodal metastases | |||
| IIIC | N3: 4 or more regional lymph nodal metastases, | 40% | 24% |
| matted regional lymph nodal metastases or | |||
| satellitosis/in transit metastases with regional nodal metastases | |||
| Median Survival | |||
| IV | M1a: metastases to skin, subcutaneous tissue or | 18 months | |
| distant lymph node | |||
| M1b: metastases to lung | 12 months | ||
| M1c: metastases to viscera and other sites | 6 months | ||
3. Clinical Management
3.1. Heterogeneity
3.2. Surgery and Chemotherapy
3.3. Cytokine Biotherapy
3.4. Antibody Biotherapy
3.5. Vaccines
3.5.1. Vaccines of Melanoma Lysates
3.5.2. Gangliosides Vaccines
3.5.3. Single Peptide Vaccines
3.5.4. Multi-Peptides Vaccine
3.5.5. Heat-Shock Peptides Vaccines
3.5.6. Dendritic cell Vaccine
3.5.7. DNA Vaccine
3.5.8. Viral vaccines
3.5.9. Allogenic Polyvalent Shed antigens Vaccine:
3.6. Melanoma Cell Vaccines
3.6.1. Allogeneic Cell Vaccines
3.6.2. Autologous Whole Cell Melanoma Vaccines
4. Targeted Therapy
4.1. Approaches
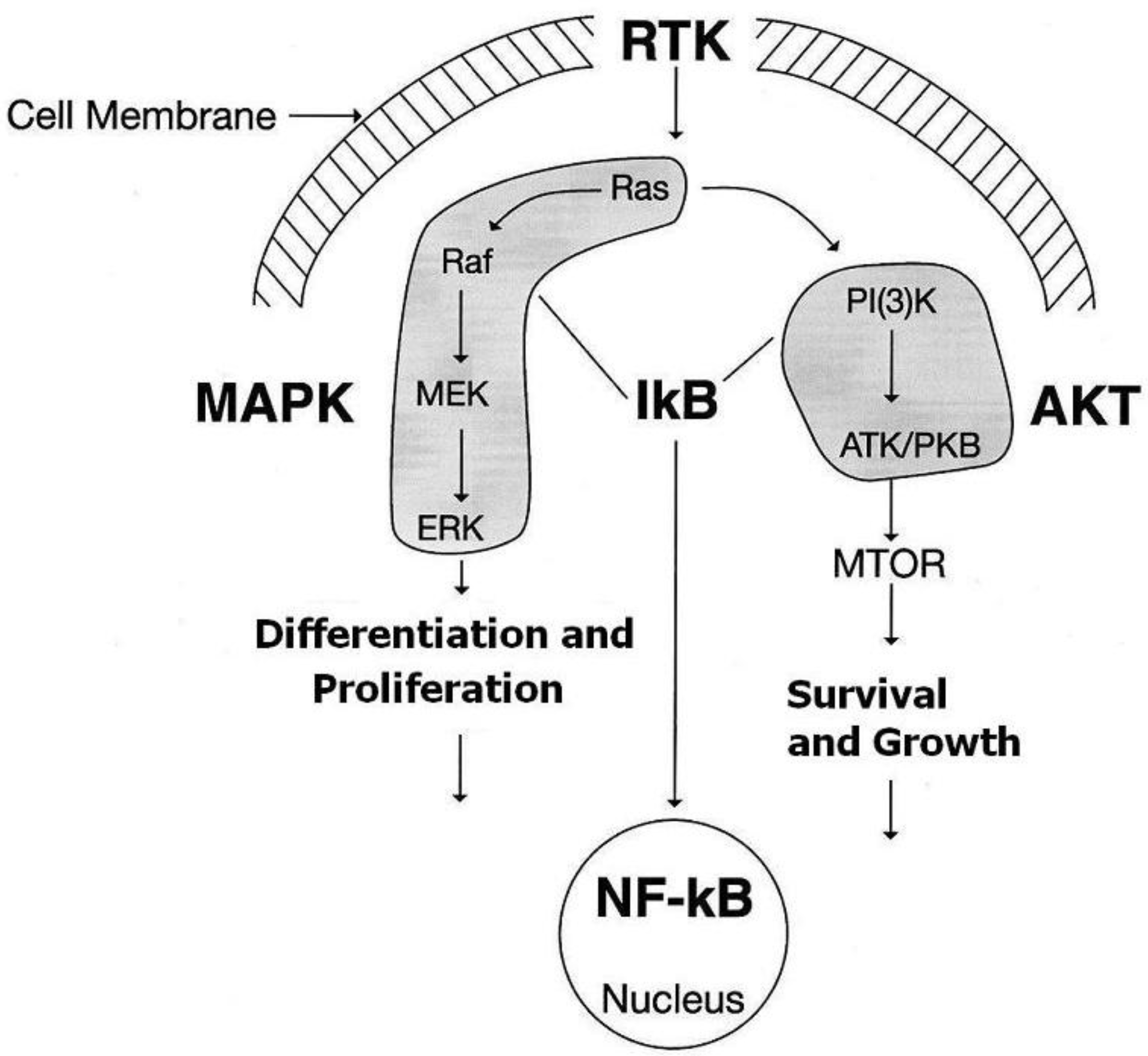
4.2. Single Targeted Agents
4.3. Multi-Targeted Agents
5. Futuristic Approaches
5.1. Adoptive Immunotherapy
5.2. RNA Interference
5.3. Melanoma Stem Cells
5.4. Curcumin
5.5. Radiosurgery
6. Conclusions
Acknowledgements
References
- Elias, E.G.; Didolkar, M.S.; Goel, I.P.; Formeister, J.F.; Valenzuela, L.A.; Pickren, J.L.; Moore, R.H. A clinicopathologic study of prognostic factors in cutaneous malignant melanoma. Surg. Gynecol. Obstet. 1977, 144, 327–334. [Google Scholar]
- Wong, J.H.; Wanek, L.; Chang, L.J.; Goradia, T.; Morton, D.L. The importance of anatomic site in prognosis in patients with cutaneous melanoma. Arch. Surg. 1991, 126, 486–489. [Google Scholar] [CrossRef]
- Blois, M.S.; Sagebiel, R.W.; Abarbanel, R.M.; Caldwell, T.M.; Tuttle, M.S. Malignant melanoma of the skin. I. The association of tumor depth and type, and patient sex, age, and site with survival. Cancer 1983, 52, 1330–1341. [Google Scholar] [CrossRef]
- Weedon, D.; Strutton, G. Skin Pathology; Churchill Livingston: Edinburgh, London, UK, 1997; pp. 254–256. [Google Scholar]
- Clark, W.H., Jr.; From, L.; Bernardino, E.A.; Mihm, M.C. The histogenesis and biologic behavior of primary human malignant melanomas of the skin. Cancer Res. 1969, 29, 705–727. [Google Scholar]
- Breslow, A. Thickness, cross-sectional areas and depth of invasion in the prognosis of cutaneous melanoma. Ann. Surg. 1970, 172, 902–908. [Google Scholar] [CrossRef]
- Clark, W.H., Jr.; Elder, D.E.; Van Horn, M. The biologic forms of malignant melanoma. Hum. Pathol. 1986, 17, 443–450. [Google Scholar] [CrossRef]
- Balch, C.M.; Buzaid, A.C.; Soong, S.J.; Atkins, M.B.; Cascinelli, N.; Coit, D.G.; Fleming, I.D.; Gershenwald, J.E.; Houghton, A., Jr.; Kirkwood, J.M.; et al. Final version of the American Joint Committee on Cancer staging system for cutaneous melanoma. J. Clin. Oncol. 2001, 19, 3635–3648. [Google Scholar]
- Balch, C.M.; Soong, S.J.; Gershenwald, J.E.; Thompson, J.F.; Reintgen, D.S.; Cascinelli, N.; Urist, M.; McMasters, K.M.; Ross, M.I.; Kirkwood, J.M.; et al. Prognostic factors analysis of 17,600 melanoma patients: validation of the American Joint Committee on Cancer melanoma staging system. J. Clin. Oncol. 2001, 19, 3622–3634. [Google Scholar]
- Edge, S.B.; Byrd, R.; Compton, C.C.; Fritz, A.G.; Greene, F.L.; Trotti, A. (Eds.) AJCC Cancer Staging Handbook, 7th ed.; Springer: New York, NY, USA, 2010; pp. 387–415.
- Mascaro, J.M.; Castro, J.; Castel, T.; Lecha, M.; Gratacos, R.; Mascaro, J.M., Jr. Why do melanomas ulcerate? J. Cutan. Pathol. 1984, 11, 269–273. [Google Scholar] [CrossRef]
- Blessing, K.; McLaren, K.M. Histological regression in primary cutaneous melanoma: recognition, prevalence and significance. Histopathology 1992, 20, 315–322. [Google Scholar] [CrossRef]
- Ronan, S.G.; Eng, A.M.; Briele, H.A.; Shioura, N.N.; Das Gupta, T.K. Thin malignant melanomas with regression and metastases. Arch. Dermatol. 1987, 123, 1326–1330. [Google Scholar] [CrossRef]
- Slingluff, C.I., Jr.; Seigler, H.E. Thin malignant melanoma: risk factors and clinical management. Ann. Plast. Surg. 1992, 28, 89–94. [Google Scholar] [CrossRef]
- Kelly, J.W.; Sagebiel, R.W.; Blois, M.S. Regression in malignant melanoma. A histologic feature without independent prognostic significance. Cancer 1985, 56, 2287–2291. [Google Scholar] [CrossRef]
- Cooper, P.H.; Wanebo, H.J.; Hagar, R.W. Regression in thin malignant melanoma. Microscopic diagnosis and prognostic importance. Arch. Dermatol. 1985, 121, 1127–1131. [Google Scholar] [CrossRef]
- Wanebo, H.J.; Cooper, P.H.; Hagar, R.W. Thin (less than or equal to 1 mm) melanomas of the extremities are biologically favorable lesions not influenced by regression. Ann. Surg. 1985, 201, 499–504. [Google Scholar] [CrossRef]
- Clark, W.H., Jr.; Elder, D.E.; Guerry, D., 4th; Braitman, L.E.; Trock, B.J.; Schultz, D.; Synnestvedt, M.; Halpern, A.C. Model predicting survival in stage I melanoma based on tumor progression. J. Natl. Cancer Inst. 1989, 81, 1893–1904. [Google Scholar] [CrossRef]
- Sumner, W.C.; Foraker, A.G. Spontaneous regression of human melanoma: clinical and experimental studies. Cancer 1960, 13, 79–81. [Google Scholar] [CrossRef]
- Elias, E.G.; Didolkar, M.S.; Goel, I.P.; Formeister, J.F.; Valenzuela, L.Z.; Pickren, J.L.; Hebel, J.R. Deeply invasive cutaneous malignant melanoma. Surg. Gynecol. Obstet. 1981, 153, 67–70. [Google Scholar]
- Bystryn, J.C.; Rigel, D.; Friedman, R.J.; Kopf, A. Prognostic significance of hypopigmentation in malignant melanoma. Arch. Dermatol. 1987, 123, 1053–1055. [Google Scholar] [CrossRef]
- Brocker, E.B.; Zwaldo, G.; Suter, L.; Brune, M.; Sorg, C. Infiltration of primary and metastatic melanomas with macrophages of the 25F9-positive phenotype. Cancer Immunol. Immunoth. 1987, 25, 81–86. [Google Scholar]
- Torisu, H.; Ono, M.; Kiryu, H.; Furue, M.; Ohmoto, Y.; Nakayama, J.; Nishioka, Y.; Sone, S.; Kuwano, M. Macrophage infiltration correlates with tumor stage and angiogenesis in human malignant melanoma: possible involvement of TNFalpha and IL-1alpha. Int. J. Cancer 2000, 85, 182–188. [Google Scholar]
- Morton, D.L.; Essner, R.; Balch, C.M. Chapter 29. Surgical excision of distant metastases. In Cutaneous Melanoma, 4th ed.; Balch, C.M., Houghton, A.N., Sober, A.J., Soong, S.J., Eds.; Quality Medical Publishing Inc: St. Louis, MO, USA, 2003; pp. 547–572. [Google Scholar]
- Lee, C.C.; Faries, M.B.; Wanek, L.A.; Morton, D.L. Improved survival after lymphadenectomy for nodal metastasis from an unknown primary melanoma. J. Clin. Oncol. 2008, 26, 535–541. [Google Scholar] [CrossRef]
- Balch, C.M.; Soong, S.J.; Murad, T.M.; Smith, J.W.; Maddox, W.A.; Durant, J.R. A multifactorial analysis of melanoma. IV. Prognostic factors in 200 melanoma patients with distant metastases (stage III). J. Clin. Oncol. 1983, 1, 126–134. [Google Scholar]
- Barth, A.; Wanek, L.A.; Morton, D.L. Prognostic factors in 1,521 melanoma patients with distant metastases. J. Am. Coll. Surg. 1995, 181, 193–201. [Google Scholar]
- Fidler, I.J.; Gersten, D.M.; Hart, I.R. The biology of cancer invasion and metastasis. Adv. Cancer Res. 1978, 28, 149–250. [Google Scholar] [CrossRef]
- Folkman, J. How is blood vessel growth regulated in normal and neoplastic tissue? G.H.A. Clowes memorial Award lecture. Cancer Res. 1986, 46, 467–473. [Google Scholar]
- Auerbach, W.; Auerbach, R. Angiogenesis inhibition: a review. Pharmacol. Ther. 1994, 63, 265–311. [Google Scholar] [CrossRef]
- Folkman, J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1995, 1, 27–31. [Google Scholar] [CrossRef]
- Elias, E.G.; Hasskamp, J.H.; Mervin, B.J. Growth factors and cytokines secreted by early passage human melanoma cell lines. In 22nd Annual Scientific Meeting of the International Society for Biological Therapy of Cance, Boston, MA, USA, November 2007; p. Abstract 45, J. Immunotherapy 2007, 30, 874.
- Elias, E.G.; Hasskamp, J.H.; Sharma, B.K.; Beam, S.L.; McCarron, E.C.; Zapas, J.L. Biological aspects of cutaneous melanoma with potential impact on target therapy. In Proceedings of the 99th American Association for Cancer Research Annual Meeting, San Diego, CA, USA, April 2008; p. Abstract 2151.
- Rietschel, P.; Wolchok, J.D.; Krown, S.; Gerst, S.; Jungbluth, A.A.; Busam, K.; Smith, K.; Orlow, I.; Panageas, K.; Chapman, P.B. Phase II study of extended-dose temozolomide in patients with melanoma. J. Clin. Oncol. 2008, 26, 2299–2304. [Google Scholar] [CrossRef]
- Hill, G.J., II; Moss, S.E.; Golomb, F.M.; Grage, T.B.; Fletcher, W.S.; Minton, J.P.; Krementz, E.T. DTIC and combination therapy for melanoma: III. DTIC (NSC 45388) Surgical Adjuvant Study COG Protocol 7040. Cancer 1981, 47, 2556–2562. [Google Scholar] [CrossRef]
- Kirkwood, J.M.; Strawderman, M.H.; Ernstoff, M.S.; Smith, T.J.; Borden, E.C.; Blum, R.H. Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group Trial EST 1684. J. Clin. Oncol. 1996, 14, 7–17. [Google Scholar]
- McMasters, K.M.; Ross, M.I.; Reintgen, D.S.; Edwards, M.J.; Noyes, R.D.; Urist, M.; Sussman, J.; Goydos, J.; Beitsch, P.; Martin, R.C.; et al. Final results of the Sunbelt Melanoma Trial. In Proceedings of the 2008 American Society of Clinical Oncology Annual Meeting, Chicago, IL, USA, May 2008; p. Abstract 9003, J. Clin. Oncol. 2008, 26, 484S; 20 May 2008 supplement.
- Rosenberg, S.A.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Parkinson, D.R.; Seipp, C.A.; Einhorn, J.H.; White, D.E. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. JAMA 1994, 271, 907–913. [Google Scholar] [CrossRef]
- Atkins, M.B.; Kunkel, L.; Sznol, M.; Rosenberg, S.A. High-dose recombinant interleukin-2 therapy in patients with metastatic melanoma: long-term survival update. Cancer J. Sci. Am. 2000, 6, S11–S14. [Google Scholar]
- Rosenberg, S.A. Keynote address: perspectives on the use of interleukin-2 in cancer treatment. Cancer J. Sci. Am. 1997, 3, S2–S6. [Google Scholar]
- Rosenberg, S.A.; Yannelli, J.R.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Parkinson, D.R.; Seipp, C.A.; Einhorn, J.H.; White, D.E. Treatment of patients with metastatic melanoma with autologous tumor–infiltrating lymphocytes and interleukin 2. J. Natl. Cancer Inst. 1994, 86, 1159–1166. [Google Scholar] [CrossRef]
- Kirkwood, J.M. Pharmacology of cancer biotherapeutics. In Cancer Principles and Practice of Oncology 2001, 6th ed.; Devita, V.Y., Hellman, S., Rosenberg, S.A., Eds.; Lippincott, Williams and Wilkins: Philadelphia, PA, USA, 2001; pp. 461–471. [Google Scholar]
- Elias, E.G.; Zapas, J.L.; Beam, S.L.; Culpepper, W.J. Perioperative adjuvant biotherapy in high–risk resected cutaneous melanoma: the results of 5 years of follow-up. Melanoma Res. 2007, 17, 310–315. [Google Scholar] [CrossRef]
- Spitler, L.E.; Grossbard, M.L.; Ernstoff, M.S.; Silver, G.; Jacobs, M.; Hayes, F.A.; Soong, S.J. Adjuvant therapy of stage III and IV malignant melanoma using granulocyte-macrophage colony-stimulating factor. J. Clin. Oncol. 2000, 18, 1614–1621. [Google Scholar]
- Elias, E.G.; Zapas, J.L.; McCarron, E.C.; Beam, S.L.; Hasskamp, J.H.; Culpepper, W.J. Sequential administration of GM-CSF (Sargramostim) and IL-2 +/- autologous vaccine as adjuvant therapy in cutaneous melanoma: an interim report of a phase II clinical trial. Cancer Biother. Radiopharm. 2008, 23, 285–291. [Google Scholar] [CrossRef]
- Hasskamp, J.H.; Elias, E.G.; Zapas, J.L. In vivo effects of sequential granulocyte-macrophage colony stimulating factor (GM-CSF) and interleukin-2 (IL-2) on circulating dendritic cells (DC) in patients with surgically resected high risk cutaneous melanoma. J. Clin. Immunol. 2006, 26, 331–338. [Google Scholar]
- Daud, A.I.; Mirza, N.; Lenox, B.; Andrews, S.; Urbas, P.; Gao, G.X.; Lee, J.H.; Sondak, V.K.; Riker, A.I.; Deconti, R.C.; et al. Phenotypic and functional analysis of dendritic cells and clinical outcome in patients with high-risk melanoma treated with adjuvant granulocyte macrophage colony-stimulating factor. J. Clin. Oncol. 2008, 26, 3235–3241. [Google Scholar] [CrossRef]
- Phan, G.Q.; Yang, J.C.; Sherry, R.M.; Hwu, P.; Topalian, S.L.; Schwartzentruber, D.J.; Restifo, N.P.; Haworth, L.R.; Seipp, C.A.; Freezer, L.J.; et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc. Natl. Acad. Sci. USA 2003, 100, 8372–8377. [Google Scholar] [CrossRef]
- Serafini, P.; Carbley, R.; Noonan, K.A.; Tan, G.; Bronte, V.; Borrello, I. High-dose granulocyte-macrophage colony-stimulating factor-producing vaccines impair the immune response through the recruitment of myeloid suppressor cells. Cancer Res. 2004, 64, 6337–6343. [Google Scholar] [CrossRef]
- Filipazzi, P.; Valenti, R.; Huber, V.; Pilla, L.; Canese, P.; Iero, M.; Castelli, C.; Mariani, L.; Parmiani, G.; Rivoltini, L. Identification of a new subset of myeloid suppressor cells in peripheral blood of melanoma patients with modulation by a granulocyte–macrophage colony-stimulation factor-based antitumor vaccine. J. Clin. Oncol. 2007, 25, 2546–2553. [Google Scholar] [CrossRef]
- Attia, P.; Phan, G.Q.; Maker, A.V.; Robinson, M.R.; Quezado, M.M.; Yang, J.C.; Sherry, R.M.; Topalian, S.L.; Kammula, U.S.; Royal, R.E.; et al. Autoimmunity correlates with tumor regression in patients with metastatic melanoma treated with anti-cytotoxic T-lymphocyte antigen-4. J. Clin. Oncol. 2005, 23, 6043–6053. [Google Scholar]
- Quezada, S.A.; Peggs, K.S.; Curran, M.A.; Allison, J.P. CTLA4 blockade and GM-CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J. Clin. Invest. 2006, 116, 1935–1945. [Google Scholar] [CrossRef]
- Wallack, M.K.; Sivanandham, M,; Balch, C.M.; Urist, M.M.; Bland, K.I.; Murray, D.; Robinson, W.A.; Flaherty, L.E.; Richards, J.M.; Bartolucci, A.A.; et al. A phase III randomized, double-blind multiinstitutional trial of vaccinia melanoma oncolysate-active specific immunotherapy for patients with stage II melanoma. Cancer 1995, 75, 34–42. [Google Scholar]
- Wallack, M.K.; Sivanandham, M.; Balch, C.M.; Urist, M.M.; Bland, K.I.; Murray, D.; Robinson, W.A.; Flaherty, L.; Richards, J.M.; Bartolucci, A.A.; et al. Surgical adjuvant active specific immunotherapy for patients with stage III melanoma: the final analysis of data from a phase III, randomized, double-blind, multicenter vaccinia melanoma oncolysate trial. J. Am. Coll. Surg. 1998, 187, 69–77. [Google Scholar] [CrossRef]
- Hersey, P.; Edwards, A.; Coates, A.; Shaw, H.; McCarthy, W.; Milton, G. Evidence that treatment with vaccinia melanoma cell lysates (VMCL) may improve survival of patients with stage II melanoma. Treatment of stage II melanoma with viral lysates. Cancer Immunol. Immunother. 1987, 25, 257–265. [Google Scholar]
- Hersey, P.; Coates, A.S.; McCarthy, W.H.; Thompson, J.F.; Sillar, R.W.; McLeod, R.; Gill, P.G.; Coventry, B.J.; McMullen, A.; Dillon, H.; et al. Adjuvant immunotherapy of patients with high-risk melanoma using vaccinia viral lysates of melanoma: results of a randomized trial. J. Clin. Oncol. 2002, 20, 4181–4190. [Google Scholar] [CrossRef]
- Sosman, J.A.; Unger, J.M.; Liu, P.Y.; Flaherty, L.E.; Park, M.S.; Kempf, R.A.; Thompson, J.A.; Terasaki, P.I.; Sondak, V.K.; Southwest Oncology Group. Adjuvant immunotherapy of resected, intermediate-thickness, node-negative melanoma with an allogeneic tumor vaccine: impact of HLA class I antigen expression on outcome. J. Clin. Oncol. 2002, 20, 2067–2075. [Google Scholar] [CrossRef]
- Portoukalian, J.; Zwingelstein, G.; Doré, J.F. Lipid composition of human malignant melanoma tumors at various levels of malignant growth. Eur. J. Biochem. 1979, 94, 19–23. [Google Scholar] [CrossRef]
- Tsuchida, T.; Saxton, R.E.; Morton, D.L.; Irie, R.F. Gangliosides of human melanoma. J. Natl. Cancer Inst. 1987, 78, 45–54. [Google Scholar]
- Livingston, P.O.; Wong, G.Y.; Adluri, S.; Tao, Y.; Padavan, M.; Parente, R.; Hanlon, C.; Calves, M.J.; Helling, F.; Ritter, G.; et al. Improved survival in stage III melanoma patients with GM2 antibodies: a randomized trial of adjuvant vaccination with GM2 ganglioside. J. Clin. Oncol. 1994, 12, 1036–1044. [Google Scholar]
- Helling, F.; Zhang, S.; Shang, A.; Adluri, S.; Calves, M.; Koganty, R.; Longenecker, B.M.; Yao, T.J.; Oettgen, H.F.; Livingston, P.O. GM2-KLH conjugate vaccine: increased immunogenicity in melanoma patients after administration with immunological adjuvant QS-21. Cancer Res. 1995, 55, 2783–2788. [Google Scholar]
- Ragupathi, G.; Livingston, P.O.; Hood, C.; Gathuru, J.; Krown, S.E.; Chapman, P.B.; Wolchok, J.D.; Williams, L.J.; Oldfield, R.C.; Hwu, W.J. Consistent antibody response against ganglioside GD2 induced in patients with melanoma by a GD2 lactone–keyhole limpet hemocyanin conjugate vaccine plus immunological adjuvant QS-21. Clin. Cancer Res. 2003, 9, 5214–5220. [Google Scholar]
- Kirkwood, J.M.; Ibrahim, J.G.; Sosman, J.A.; Sondak, V.K.; Agarwala, S.S.; Ernstoff, M.S.; Rao, U. High-dose interferon alfa-2b significantly prolongs relapse-free and overall survival compared with the GM2-KLH/QS-21 vaccine in patients with resected stage IIB-III melanoma: results of intergroup trial E1694/S9512/C509801. J. Clin. Oncol. 2001, 19, 2370–2380. [Google Scholar]
- Eggermont, A.M.; Suciu, S.; Ruka, W.; Marsden, J.; Testori, A.; Corrie, P.; Aamdal, S.; Ascierto, P.A.; Patel, P.; Spatz, A.; EORTC Melanoma Group. EORTC 18961: Post-operative adjuvant ganglioside GM2-KLH21 vaccination treatment vs observation in stage II (T3-T4N0M0) melanoma: 2nd interim analysis led to an early disclosure of the results. In Proceedings of the 2008 American Society of Clinical Oncology Annual Meeting, Chicago, IL, USA, May 2008; p. Abstract 9004, J. Clin. Oncol. 2008, 26, 484S; 20 May 2008 supplement.
- Ribas, A.; Butterfield, L.H.; Glaspy, J.A.; Economou, J.S. Current developments in cancer vaccines and cellular immunotherapy. J. Clin. Oncol. 2003, 21, 2415–2432. [Google Scholar] [CrossRef]
- Castelli, C.; Rivoltini, L.; Andreola, G.; Carrabba, M.; Renkvist, N.; Parmiani, G.J. T-cell recognition of melanoma-associated antigens. Cell Physiol. 2000, 182, 323–331. [Google Scholar] [CrossRef]
- Wang, F.; Bade, E.; Kuniyoshi, C.; Spears, L.; Jeffery, G.; Marty, V.; Groshen, S.; Weber, J. Phase I trial of a MART-1 peptide vaccine with incomplete Freund's adjuvant for resected high-risk melanoma. Clin. Cancer Res. 1999, 5, 2756–2765. [Google Scholar]
- Smith, J.W., II; Walker, E.B.; Fox, B.A.; Haley, D.; Wisner, K.P.; Doran, T.; Fisher, B.; Justice, L.; Wood, W.; Vetto, J.; et al. Adjuvant immunization of HLA-A2-positive melanoma patients with a modified gp100 peptide induces peptide-specific CD8+ T-cell responses. J. Clin. Oncol. 2003, 21, 1562–1573. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Sherry, R.M.; Morton, K.E.; Scharfman, W.J.; Yang, J.C.; Topalian, S.L.; Royal, R.E.; Kammula, U.; Restifo, N.P.; Hughes, M.S.; et al. Tumor progression can occur despite the induction of very high levels of self/tumor antigen-specific CD8+ T cells in patients with melanoma. J. Immunol. 2005, 175, 6169–6176. [Google Scholar]
- Hu, X.; Chakraborty, N.G.; Sporn, J.R.; Kurtzman, S.H.; Ergin, M.T.; Mukherji, B. Enhancement of cytolytic T lymphocyte precursor frequency in melanoma patients following immunization with the MAGE-1 peptide loaded antigen presenting cell-based vaccine. Cancer Res. 1996, 56, 2479–2483. [Google Scholar]
- Kruit, W.H.J.; van Ojik, H.H.; Brichard, V.G.; Escudier, B.; Dorval, T.; Dréno, B.; Patel, P.; van Baren, N.; Avril, M.; Piperno, S.; et al. Phase 1/2 study of subcutaneous and intradermal immunization with a recombinant MAGE-3 protein in patients with detectable metastatic melanoma. Inter. J. Cancer 2005, 117, 596–604. [Google Scholar] [CrossRef]
- Pullarkat, V.; Lee, P.P.; Scotland, R.; Rubio, V.; Groshen, S.; Gee, C.; Lau, R.; Snively, J.; Sian, S.; Woulfe, S.L.; et al. A phase I trial of SD-9427 (progenipoietin) with a multipeptide vaccine for resected metastatic melanoma. Clin. Cancer Res. 2003, 9, 1301–1312. [Google Scholar]
- Belli, F.; Testori, A.; Rivoltini, L.; Maio, M.; Andreola, G.; Sertoli, M.R.; Gallino, G.; Piris, A.; Cattelan, A.; Lazzari, I.; et al. Vaccination of metastatic melanoma patients with autologous tumor-derived heat shock protein gp96-peptide complexes: clinical and immunologic findings. J. Clin. Oncol. 2002, 20, 4169–4180. [Google Scholar] [CrossRef]
- Testori, A.; Richards, J.; Whitman, E.; Mann, G.B.; Lutzky, J.; Camacho, L.; Parmiani, G.; Tosti, G.; Kirkwood, J.M.; Hoos, A.; et al. Phase III comparison of vitespen, an autologous tumor-derived heat shock protein gp96 peptide complex vaccine, with physician's choice of treatment for stage IV melanoma: the C-100-21 Study Group. J. Clin. Oncol. 2008, 26, 955–962. [Google Scholar] [CrossRef]
- de Vries, I.J.; Bernsen, M.R.; Lesterhuis, W.J.; Scharenborg, N.M.; Strijk, S.P.; Gerritsen, M.J.; Ruiter, D.J.; Figdor, C.G.; Punt, C.J.; Adema, G.J. Immunomonitoring tumor-specific T cells in delayed-type hypersensitivity skin biopsies after dendritic cell vaccination correlates with clinical outcome. J. Clin. Oncol. 2005, 23, 5779–5787. [Google Scholar] [CrossRef]
- Bedrosian, I.; Mick, R.; Xu, S.; Nisenbaum, H.; Faries, M.; Zhang, P.; Cohen, P.A.; Koski, G.; Czerniecki, B.J. Intranodal administration of peptide-pulsed mature dendritic cell vaccines results in superior CD8+ T-cell function in melanoma patients. J. Clin. Oncol. 2003, 21, 3826–3835. [Google Scholar] [CrossRef]
- Smith, S.G.; Patel, P.M.; Porte, J.; Selby, P.J.; Jackson, A.M. Human dendritic cells genetically engineered to express a melanoma polyepitope DNA vaccine induce multiple cytotoxic T-cell responses. Clin. Cancer Res. 2001, 7, 4253–4261. [Google Scholar]
- Rosenberg, S.A.; Yang, J.C.; Schwartzentruber, D.J.; Hwu, P.; Marincola, F.M.; Topalian, S.L.; Restifo, N.P.; Dudley, M.E.; Schwarz, S.L.; Spiess , P.J.; et al. Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat. Med. 1998, 4, 321–327. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Zhai, Y.; Yang, J.C.; Schwartzentruber, D.J.; Hwu, P.; Marincola, F.M.; Topalian, S.L.; Restifo, N.P.; Seipp, C.A.; Einhorn, J.H.; et al. Immunizing patients with metastatic melanoma using recombinant adenoviruses encoding MART-1 or gp100 melanoma antigens. J. Natl. Cancer Inst. 1998, 90, 1894–1900. [Google Scholar]
- Toda, M.; Martuza, R.L.; Kojima, H.; Rabkin, S.D. In situ cancer vaccination: an IL-12 defective vector/replication-competent herpes simplex virus combination induces local and systemic antitumor activity. J. Immunol. 1998, 160, 4457–4464. [Google Scholar]
- Bystryn, J.C.; Henn, M.; Li, J.; Shroba, S. Identification of immunogenic human melanoma antigens in a polyvalent melanoma vaccine. Cancer Res. 1992, 52, 5948–5953. [Google Scholar]
- Bystryn, J.C.; Zeleniuch-Jacquotte, A.; Oratz, R.; Shapiro, R.L.; Harris, M.N.; Roses, D.F. Double-blind trial of a polyvalent, shed-antigen, melanoma vaccine. Clin. Cancer Res. 2001, 7, 1882–1887. [Google Scholar]
- Reynolds, S.R.; Zeleniuch-Jacquotte, A.; Shapiro, R.L.; Roses, D.F.; Harris, M.N.; Johnston, D.; Bystryn, J.C. Vaccine-induced CD8+ T-cell responses to MAGE-3 correlate with clinical outcome in patients with melanoma. Clin. Cancer Res. 2003, 9, 657–662. [Google Scholar]
- Elias, E.G.; Tomazic, V.J.; Buda, B.S. Adjuvant immunotherapy in melanoma: a new approach. J. Surg. Oncol. 1992, 50, 144–148. [Google Scholar] [CrossRef]
- Barth, A.; Hoon, D.S.; Foshag, L.J.; Nizze, J.A.; Famatiga, E.; Okun, E.; Morton, D.L. Polyvalent melanoma cell vaccine induces delayed-type hypersensitivity and in vitro cellular immune response. Cancer Res. 1994, 54, 3342–3345. [Google Scholar]
- Morton, D.L.; Foshag, L.J.; Hoon, D.S; Nizze, J.A; Famatiga, E; Wanek, L.A.; Chang, C.; Davtyan, D.G.; Gupta, R.K.; Elashoff, R.; et al. Prolongation of survival in metastatic melanoma after active specific immunotherapy with a new polyvalent melanoma vaccine. Ann. Surg. 1992, 216, 463–482. [Google Scholar] [CrossRef]
- Morton, D.L.; Barth, A. Vaccine therapy for malignant melanoma. CA Cancer J. Clin. 1996, 46, 225–244. [Google Scholar] [CrossRef]
- Morton, D.L.; Hsueh, E.C.; Essner, R.; Foshag, L.J.; O'Day, S.J.; Bilchik, A.; Gupta, R.K.; Hoon, D.S.; Ravindranath, M.; Nizze, J.A.; et al. Prolonged survival of patients receiving active immunotherapy with Canvaxin therapeutic polyvalent vaccine after complete resection of melanoma metastatic to regional lymph nodes. Ann. Surg. 2002, 236, 438–448. [Google Scholar] [CrossRef]
- Elias, E.G. Melanoma vaccines. A review. G. Ital. Dermatol. Venereol 2006, 141, 33–43. [Google Scholar] [CrossRef]
- Elias, E.G.; Suter, C.M.; Fabian, D.S. Adjuvant immunotherapy in melanoma with irradiated autologous tumor cells and low dose cyclophosphamide. J. Surg. Oncol. 1997, 64, 17–22. [Google Scholar] [CrossRef]
- Berd, D.; Maguire, H.C., Jr.; Schuchter, L.M.; Hamilton, R.; Hauck, W.W.; Sato, T.; Mastrangelo, M.J. Autologous hapten-modified melanoma vaccine as postsurgical adjuvant treatment after resection of nodal metastases. J. Clin. Oncol. 1997, 15, 2359–2370. [Google Scholar]
- Berd, D.; Sato, T.; Maguire, H.C., Jr.; Kairys, J.; Mastrangelo, M.J. Immunopharmacologic analysis of an autologous, hapten-modified human melanoma vaccine. J. Clin. Oncol. 2004, 22, 403–415. [Google Scholar]
- Chin, L.; Garraway, L.A.; Fisher, D.E. Malignant melanoma: genetics and therapeutics in the genomic era. Genes Dev. 2006, 20, 2149–2182. [Google Scholar] [CrossRef]
- Lobbezoo, M.W.; Giaccone, G.; Van Kalken, C. Signal transduction modulators for cancer therapy: from promise to practice? Oncologist 2003, 8, 210–213. [Google Scholar] [CrossRef]
- Collins, I.; Workman, P. Design and development of signal transduction inhibitors for cancer treatment: experience and challenges with kinase targets. Curr. Signal Transduct. Ther. 2006, 1, 13–23. [Google Scholar] [CrossRef]
- Lázár-Molnár, E.; Hegyesi, H.; Tóth, S.; Falus, A. Autocrine and paracrine regulation by cytokines and growth factors in melanoma. Cytokine 2000, 12, 547–554. [Google Scholar] [CrossRef]
- Gray-Schopfer, V.; Wellbrock, C.; Marais, R. Melanoma biology and new targeted therapy. Nature 2007, 445, 851–857. [Google Scholar] [CrossRef]
- Meier, F.; Busch, S.; Lasithiotakis, K.; Kulms, D.; Garbe, C.; Maczey, E.; Herlyn, M.; Schittek, B. Combined targeting of MAPK and AKT signalling pathways is a promising strategy for melanoma treatment. Br. J. Dermatol. 2007, 156, 1204–1213. [Google Scholar] [CrossRef]
- Ferrara, N.; Davis-Smyth, T. The biology of vascular endothelial growth factor. Endocr. Rev. 1997, 18, 4–25. [Google Scholar] [CrossRef]
- Waksal, H.W. Role of an anti-epidermal growth factor receptor in treating cancer. Cancer Metastasis Rev. 1999, 18, 427–436. [Google Scholar] [CrossRef]
- Selzer, E.; Schlagbauer-Wadl, H.; Okamoto, I.; Pehamberger, H.; Pötter, R.; Jansen, B. Expression of Bcl-2 family members in human melanocytes, in melanoma metastases and in melanoma cell lines. Melanoma Res. 1998, 8, 197–203. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Sabatini, D.M. Growing roles for the mTOR pathway. Curr. Opin. Cell Biol. 2005, 17, 596–603. [Google Scholar] [CrossRef]
- Thompson, C.B.; Allison, J.P. The emerging role of CTLA-4 as an immune attenuator. Immunity 1997, 7, 445–450. [Google Scholar] [CrossRef]
- Lux, M.L.; Rubin, B.P.; Biase, T.L.; Chen, C.J.; Maclure, T.; Demetri, G.; Xiao, S.; Singer, S.; Fletcher, C.D.; Fletcher, J.A. KIT extracellular and kinase domain mutations in gastrointestinal stromal tumors. Am. J. Pathol. 2000, 156, 791–795. [Google Scholar] [CrossRef]
- Druker, B.J.; Talpaz, M.; Resta, D.J.; Peng, B.; Buchdunger, E.; Ford, J.M.; Lydon, N.B.; Kantarjian, H.; Capdeville, R.; Ohno-Jones, S.; et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 2001, 344, 1031–1037. [Google Scholar] [CrossRef]
- McDermott, D.F.; Sosman, J.A.; Gonzalez, R.; Hodi, F.S.; Linette, G.P.; Richards, J.; Jakub, J.W.; Beeram, M.; Tarantolo, S.; Agarwala, S.; et al. Double-blind randomized phase II study of the combination of sorafenib and dacarbazine in patients with advanced melanoma: a report from the 11715 Study Group. J. Clin. Oncol. 2008, 26, 2178–2185. [Google Scholar] [CrossRef]
- Kantarjian, H.; Giles, F.; Wunderle, L.; Bhalla, K.; O'Brien, S.; Wassmann, B.; Tanaka, C.; Manley, P.; Rae, P.; Mietlowski, W.; et al. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N. Engl. J. Med. 2006, 354, 2542–2551. [Google Scholar] [CrossRef]
- Talpaz, M.; Shah, N.P.; Kantarjian, H.; Donato, N.; Nicoll, J.; Paquette, R.; Cortes, J.; O'Brien, S.; Nicaise, C.; Bleickardt, E.; et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N. Engl. J. Med. 2006, 354, 2531–2541. [Google Scholar] [CrossRef]
- Juarez, J.C.; Betancourt, O., Jr.; Pirie-Shepherd, S.R.; Guan, X.; Price, M.L.; Shaw, D.E.; Mazar, A.P.; Doñate, F. Copper binding by tetrathiomolybdate attenuates angiogenesis and tumor cell proliferation through the inhibition of superoxide dismutase 1. Clin. Cancer Res. 2006, 12, 4974–4982. [Google Scholar] [CrossRef]
- Hinrichs, C.S.; Borman, Z.A.; Cassard, L.; Gattinoni, L.; Spolski, R.; Yu, Z.; Sanchez-Perez, L.; Muranski, P.; Kern, S.J.; Logun, C.; et al. Adoptively transferred effector cells derived from naive rather than central memory CD8+ T cells mediate superior antitumor immunity. Proc. Natl. Acad. Sci. USA 2009, 106, 17469–17474. [Google Scholar] [CrossRef]
- Johnson, L.A.; Morgan, R.A.; Dudley, M.E.; Cassard, L.; Yang, J.C.; Hughes, M.S.; Kammula, U.S.; Royal, R.E.; Sherry, R.M.; Wunderlich, J.R.; et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 2009, 114, 535–546. [Google Scholar] [CrossRef]
- Tao, J.; Tu, Y.T.; Huang, C.Z.; Feng, A.P.; Wu, Q.; Lian, Y.J.; Zhang, L.X.; Zhang, X.P.; Shen, G.X. Inhibiting the growth of malignant melanoma by blocking the expression of vascular endothelial growth factor using an RNA interference approach. Br. J. Dermatol. 2005, 153, 715–724. [Google Scholar] [CrossRef]
- Elbashir, S.M.; Lendeckel, W.; Tuschl, T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001, 15, 188–200. [Google Scholar] [CrossRef]
- Akhtar, S.; Benter, I. Toxicogenomics of non-viral drug delivery systems for RNAi: potential impact on siRNA-mediated gene silencing activity and specificity. Adv. Drug Deliv. Rev. 2007, 59, 164–182. [Google Scholar] [CrossRef]
- Zabierowski, S.E.; Herlyn, M. Melanoma stem cells: the dark seed of melanoma. J. Clin. Oncol. 2008, 26, 2890–2894. [Google Scholar] [CrossRef]
- Klein, W.M.; Wu, B.P.; Zhao, S.; Wu, H.; Klein-Szanto, A.J.; Tahan, S.R. Increased expression of stem cell markers in malignant melanoma. Mod. Pathol. 2007, 20, 102–107. [Google Scholar] [CrossRef]
- Sharma, B.K.; Manglik, V.; Elias, E.G. Identification of melanoma stem cell markers in human cutaneous melanoma. J. Surg. Res. 2010, 158, 400. [Google Scholar] [CrossRef]
- Hasskamp, J.H.; Elias, E.G.; Avergas, A.; Sharma, B.K.; Zapas, J.L. Inhibition of proliferation of early passage human melanoma cell lines by curcumin. In Proceedings of the 99th American Association for Cancer Research Annual Meeting, San Diego, CA, USA, April 2008; p. Abstract 617.
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Elias, E.G.; Hasskamp, J.H.; Sharma, B.K. Biology of Human Cutaneous Melanoma. Cancers 2010, 2, 165-189. https://doi.org/10.3390/cancers2010165
Elias EG, Hasskamp JH, Sharma BK. Biology of Human Cutaneous Melanoma. Cancers. 2010; 2(1):165-189. https://doi.org/10.3390/cancers2010165
Chicago/Turabian StyleElias, Elias G., Joanne H. Hasskamp, and Bhuvnesh K. Sharma. 2010. "Biology of Human Cutaneous Melanoma" Cancers 2, no. 1: 165-189. https://doi.org/10.3390/cancers2010165
APA StyleElias, E. G., Hasskamp, J. H., & Sharma, B. K. (2010). Biology of Human Cutaneous Melanoma. Cancers, 2(1), 165-189. https://doi.org/10.3390/cancers2010165