

The Molecular Basis of Pediatric Brain Tumors: A Review with Clinical Implications

 , , ,
, , ,  ,
,
Simple Summary
Abstract
1. Introduction
2. Oncologic Terms
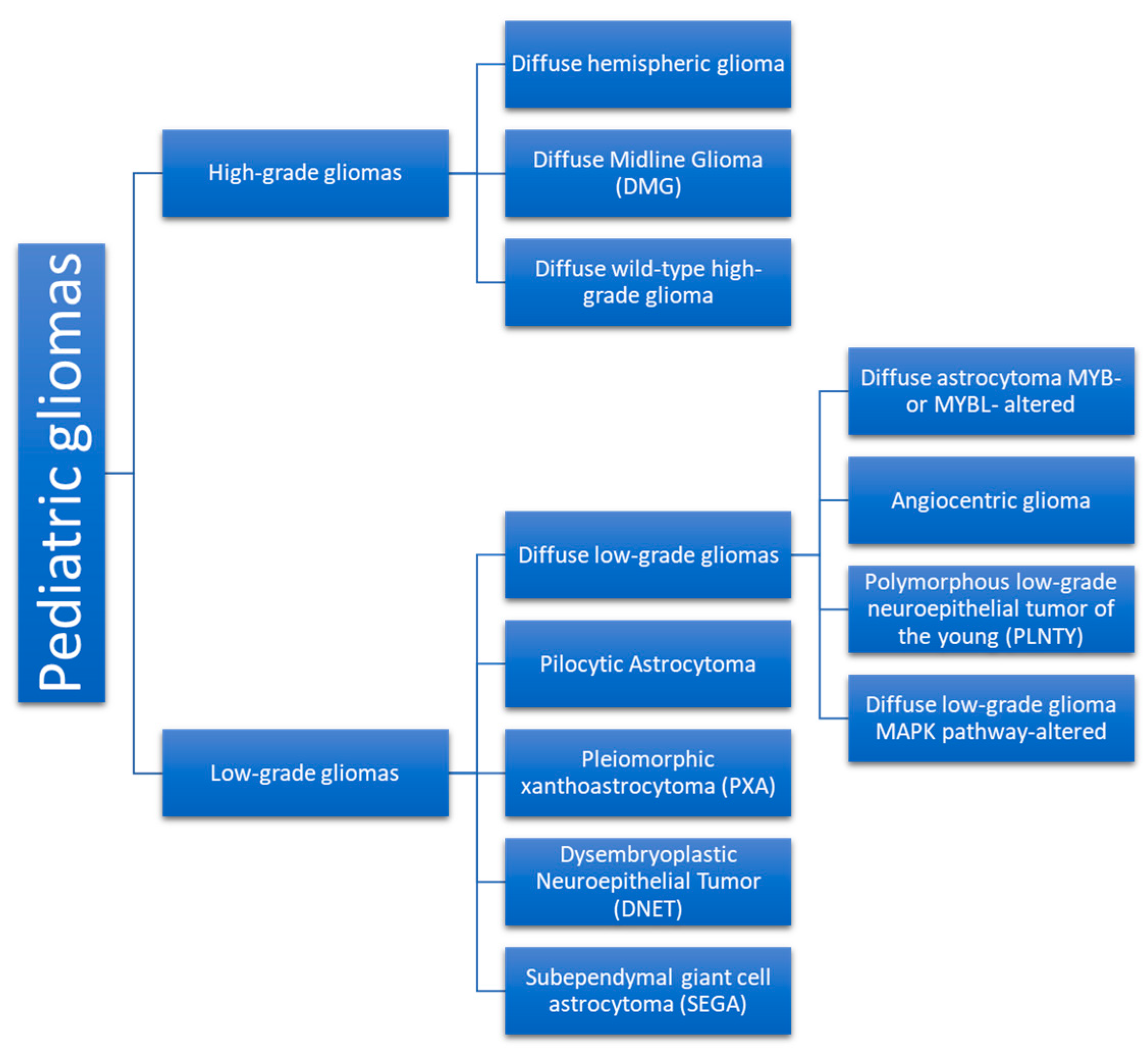
3. Neoplasm Categories
3.1. Low-Grade Gliomas
3.1.1. Molecular Events
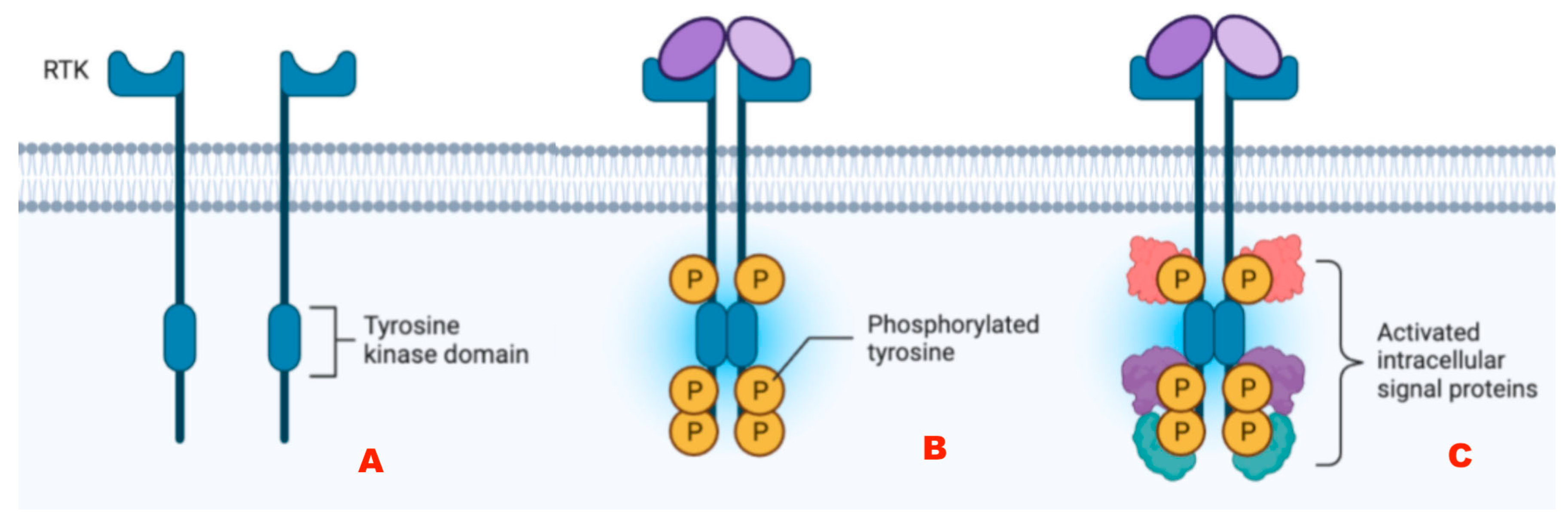
Alterations of Tyrosine Kinase Receptors
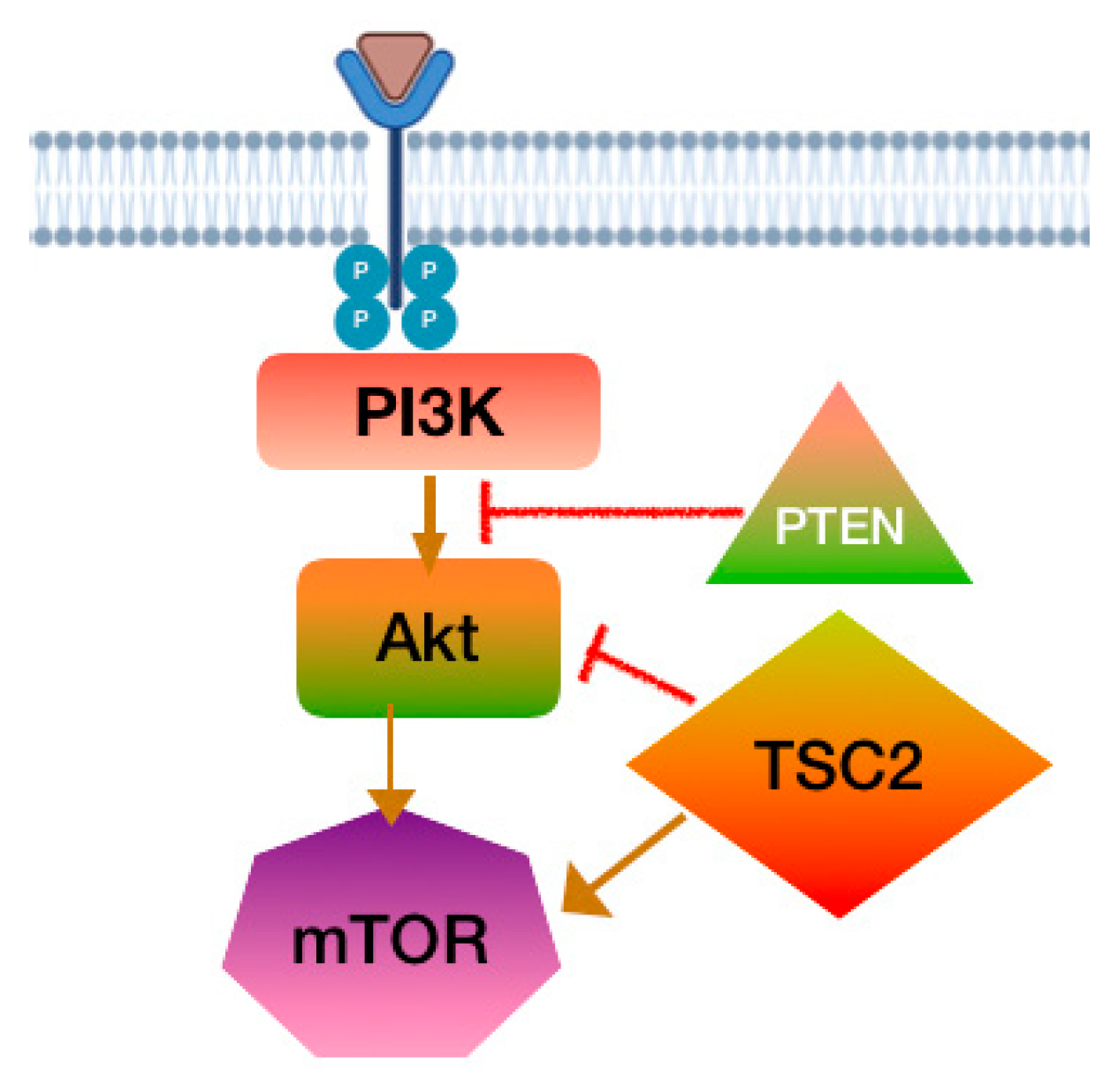
PI3K-Akt-mTOR Pathway
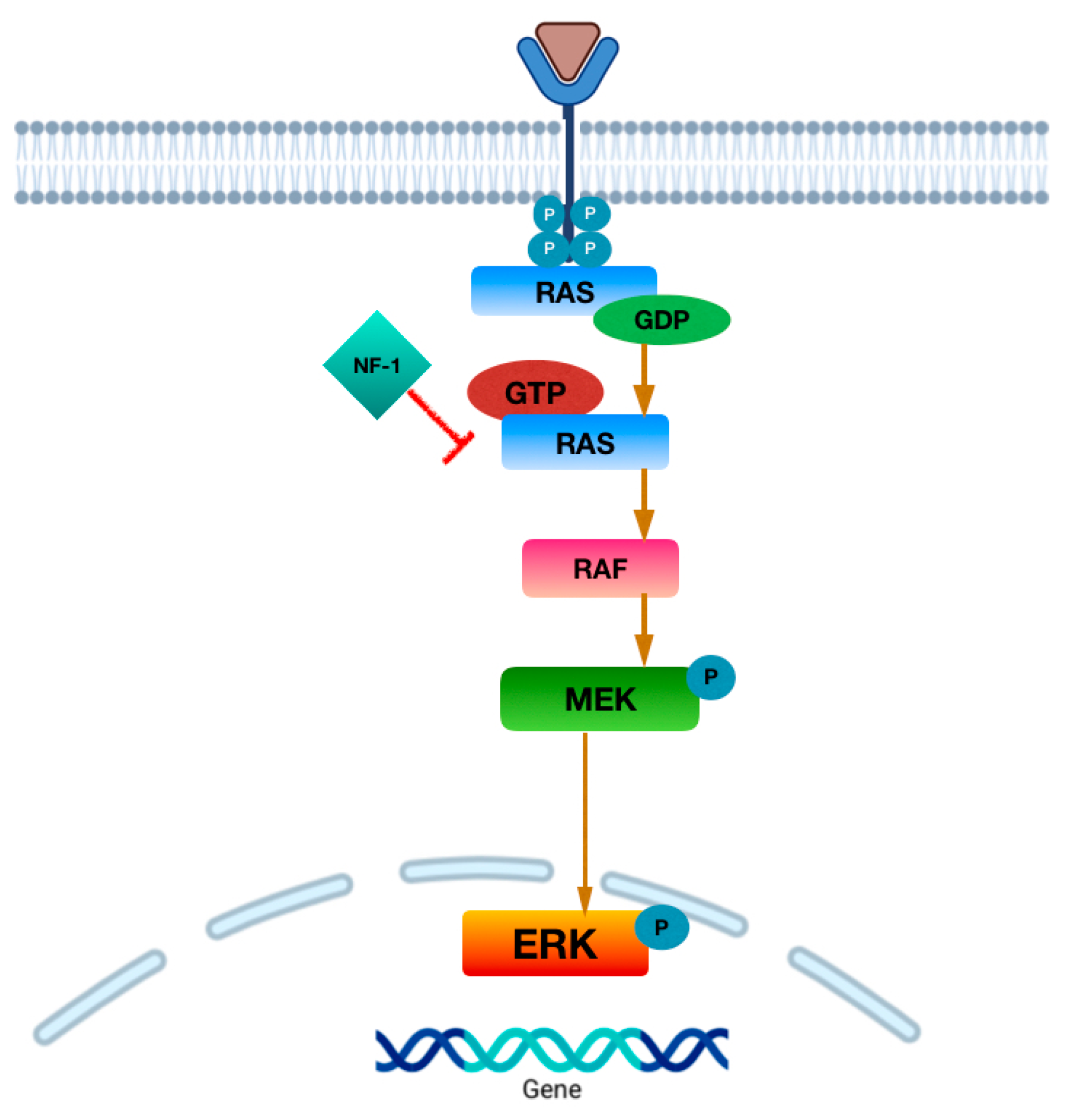
MAPK Pathway
MYB/MYBL1 Alterations
3.1.2. Types of LGGs
Diffuse Astrocytoma and MYB- or MYBL1-Altered
Angiocentric Glioma (AG)
Polymorphous Low-Grade Neuroepithelial Tumor in Youth (PLNTY)
Diffuse Low-Grade Glioma—MAPK Pathway-Altered
Pilocytic Astrocytoma (PA) (Figure 5)
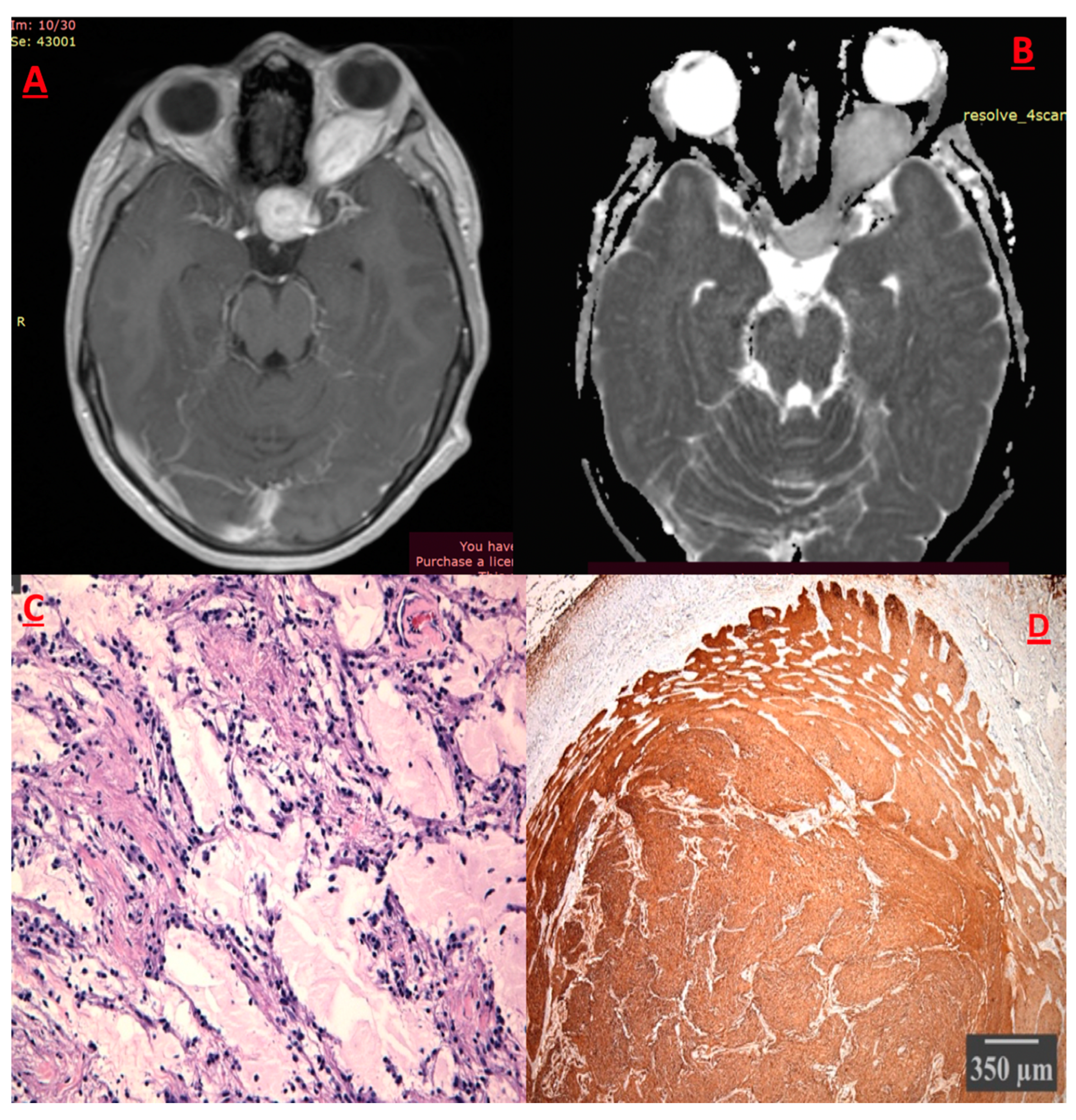
Pleiomorphic Xanthoastrocytoma (PXA)
Dysembryoblastic Neuroepithelial Tumor (DNET)
Subependymal Giant Cell Astrocytoma (SEGA)
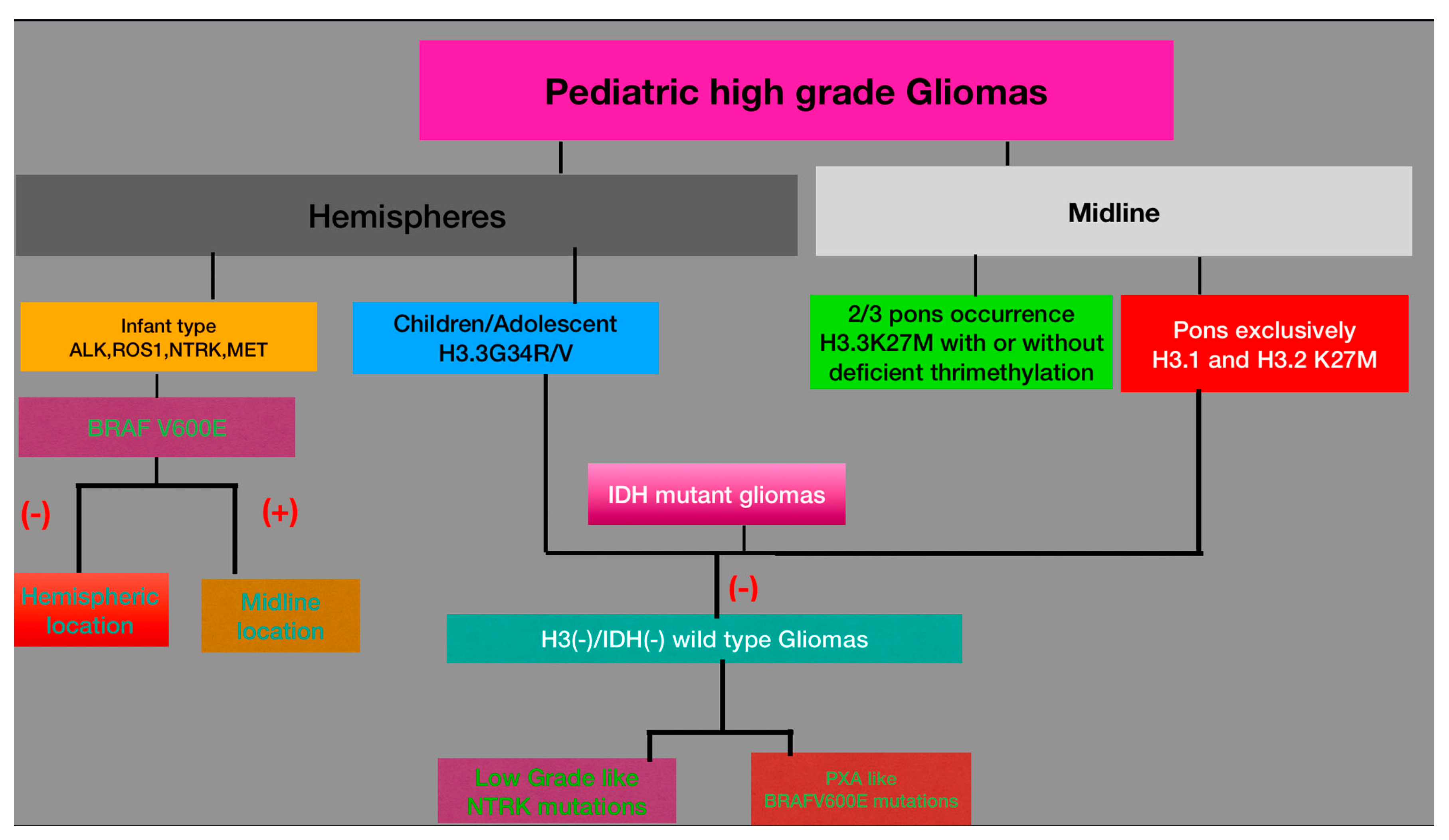
3.2. High-Grade Gliomas (HGGs)
3.2.1. Molecular Substrate
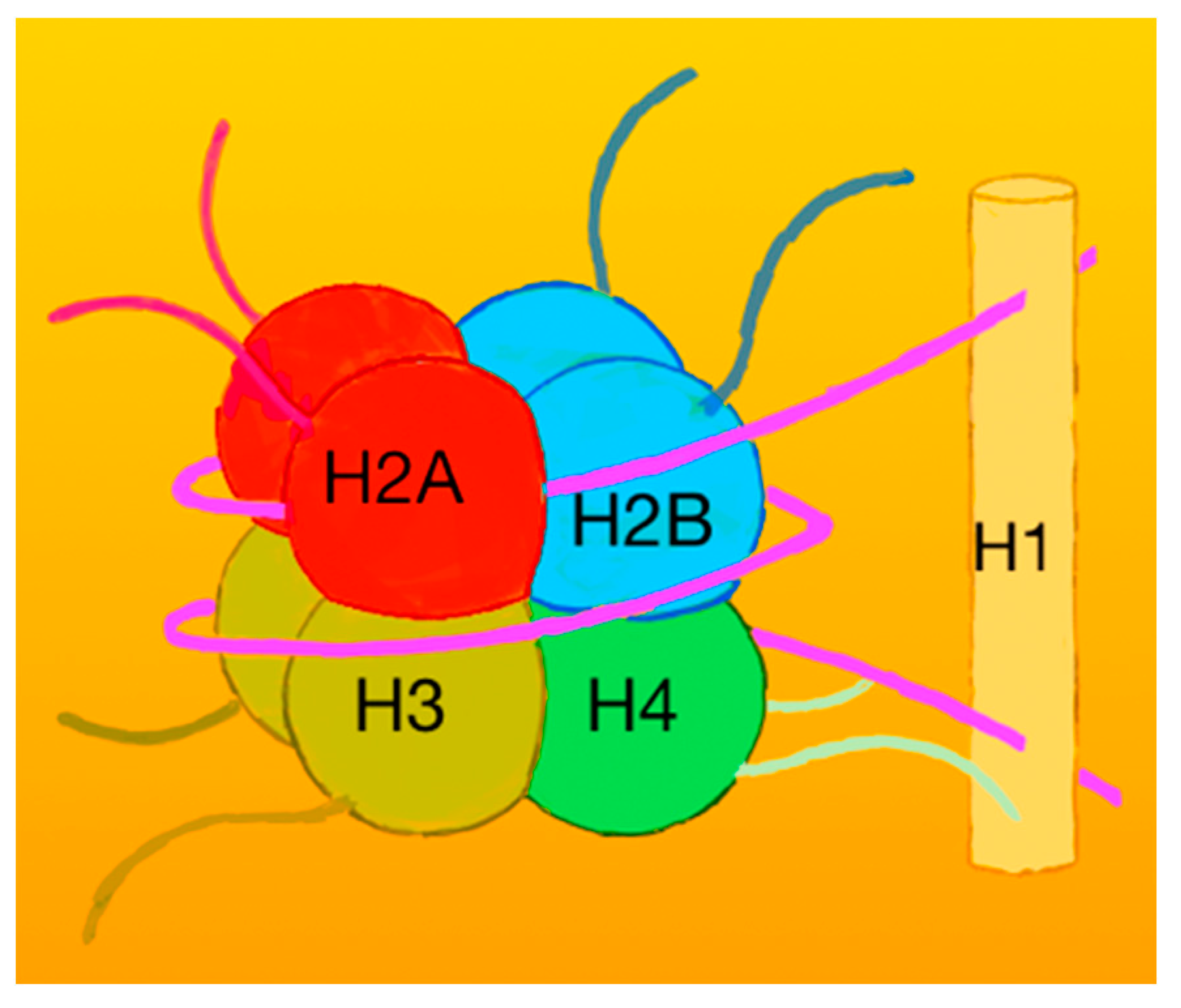
Histones and Nucleosomes
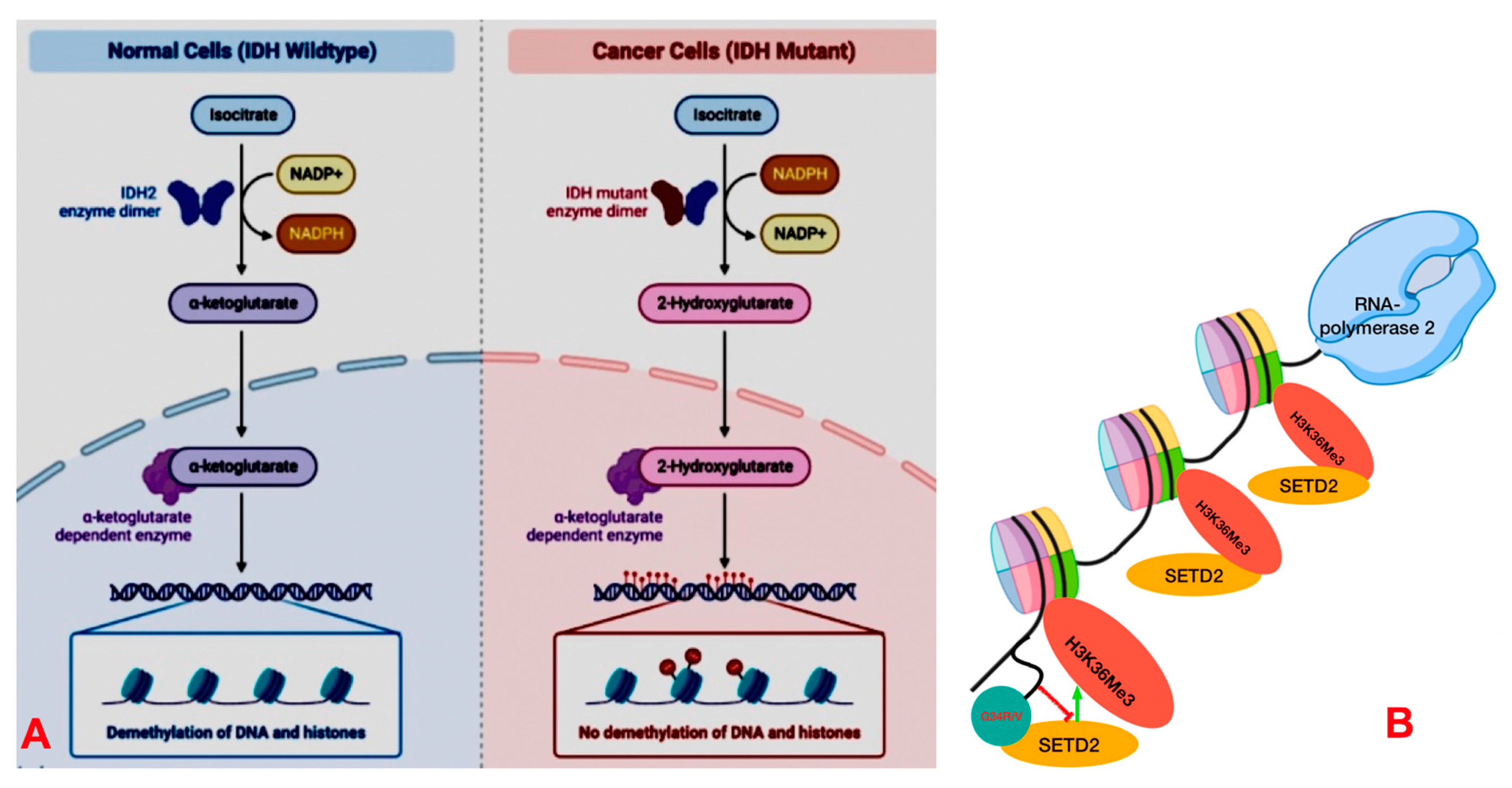
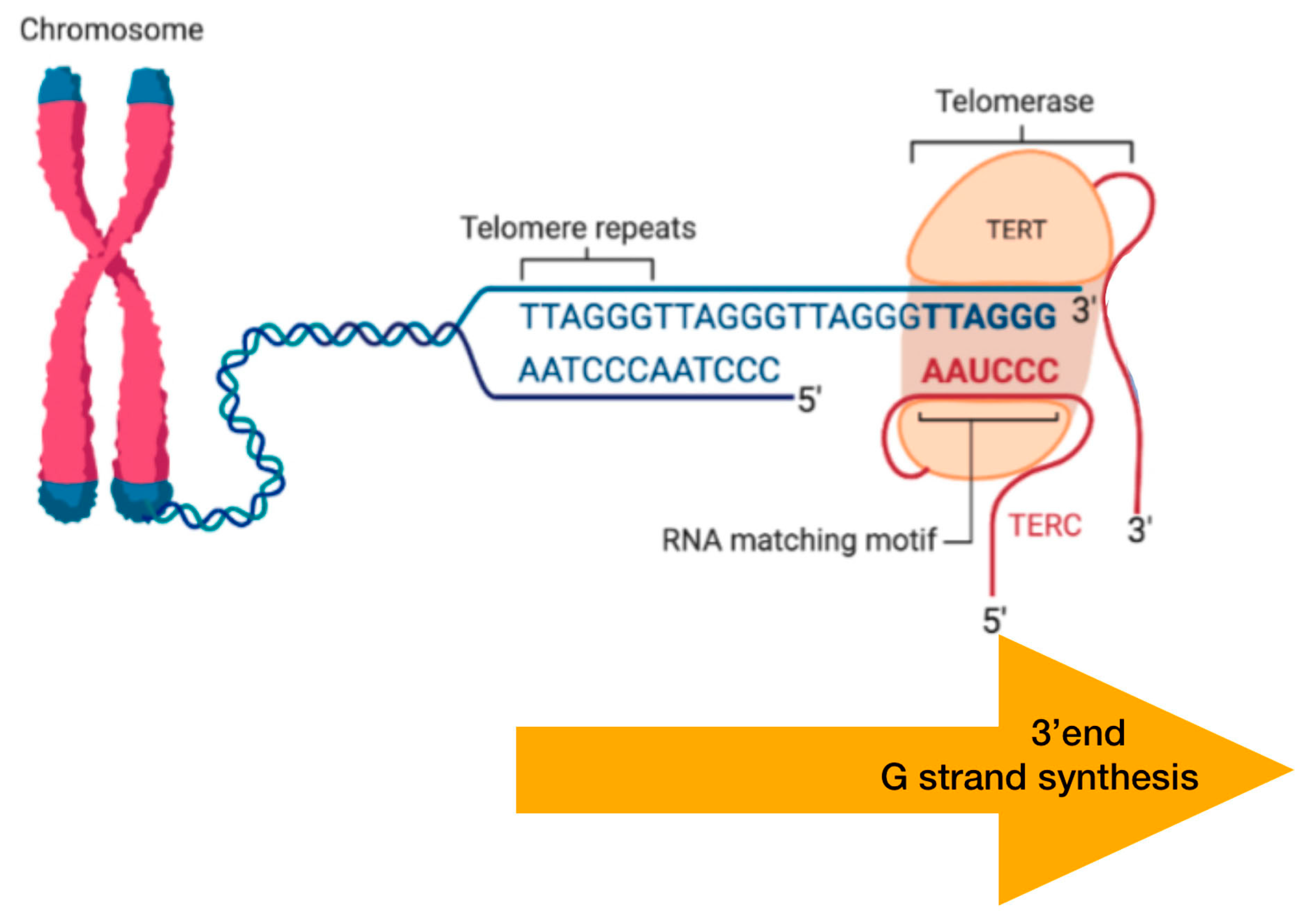
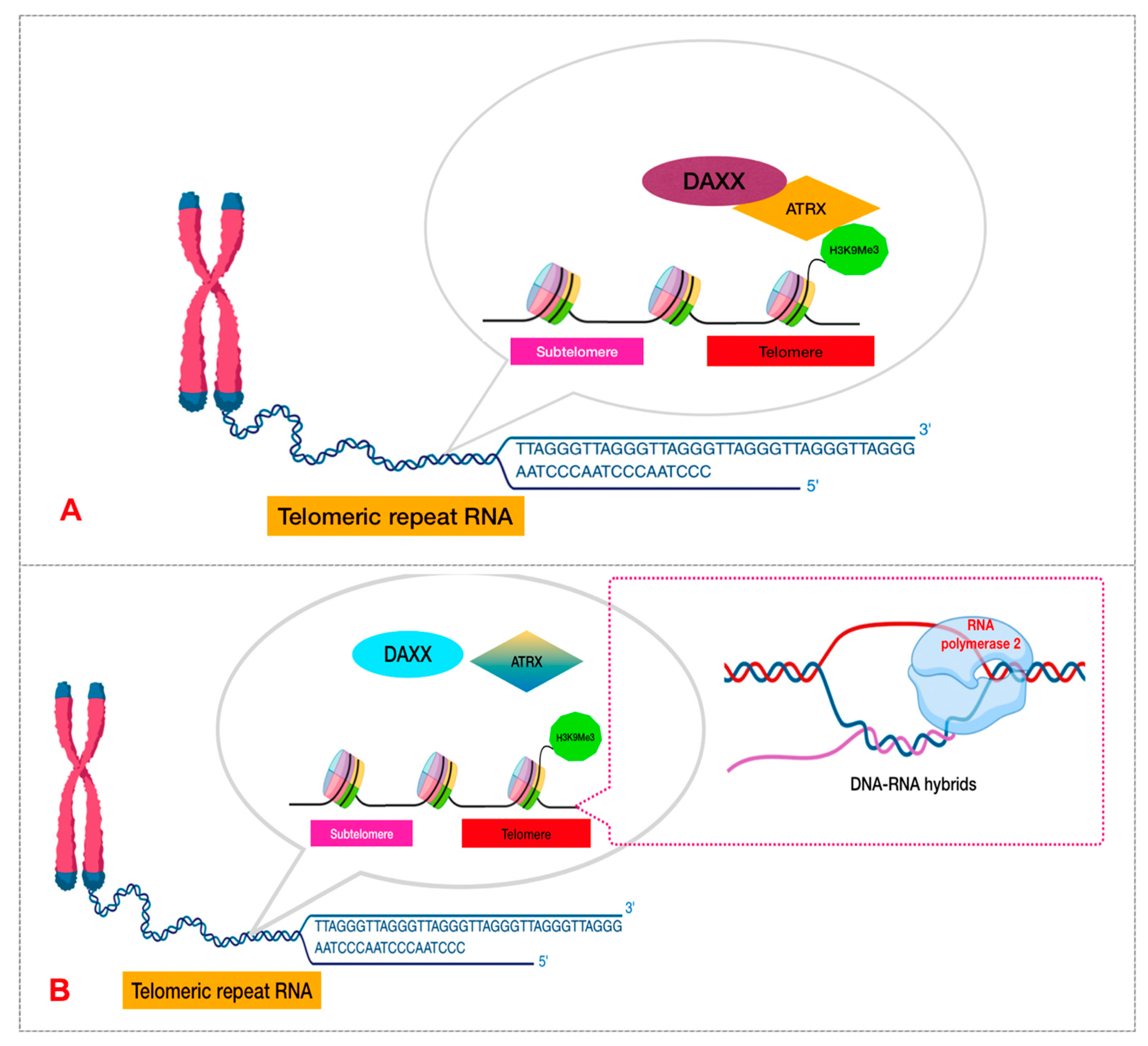
DNA Packaging and Methylation Control
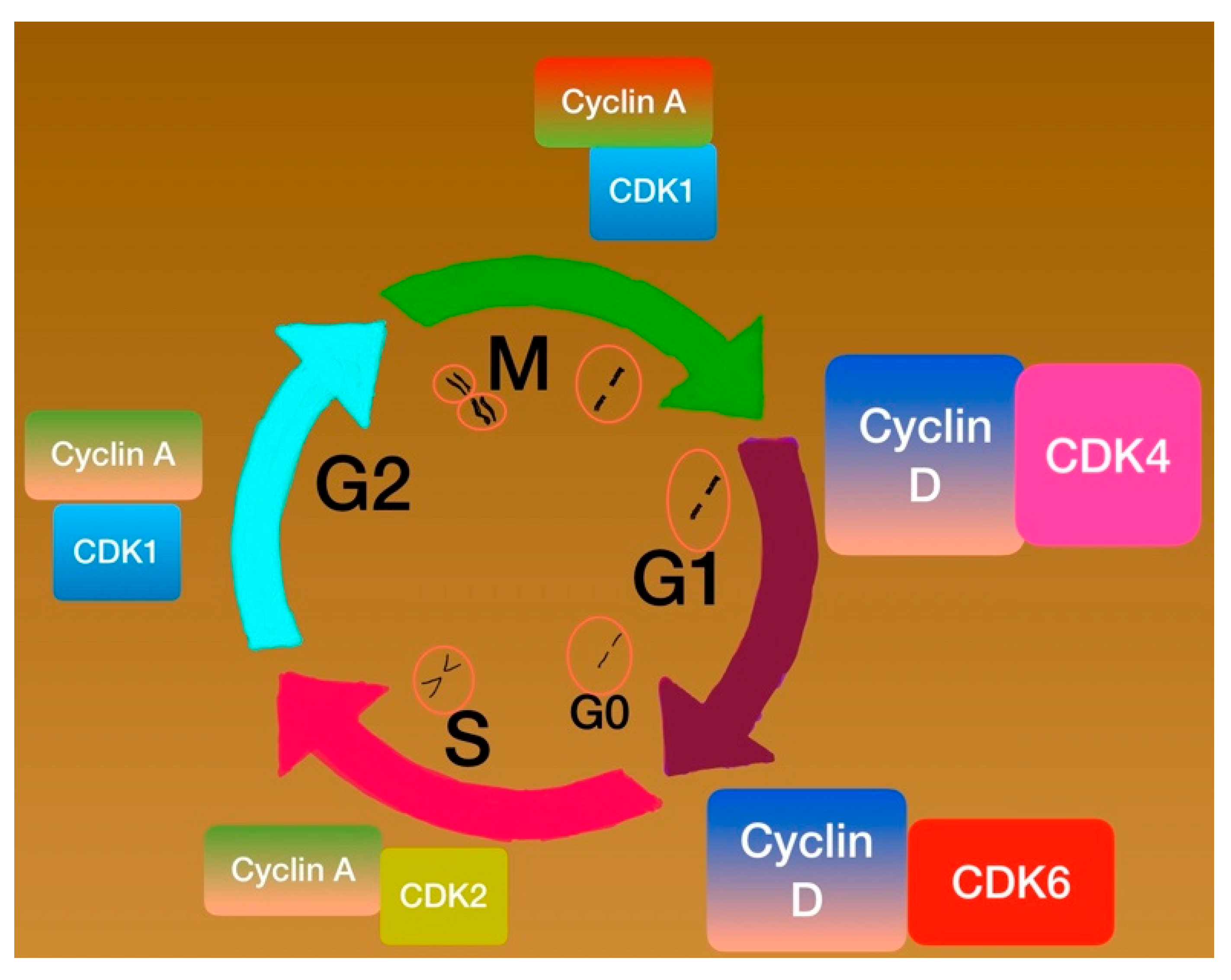
Cell Phases Control
Cyclins and Cyclin-Dependent Kinases (CDKs)
Check Point Proteins
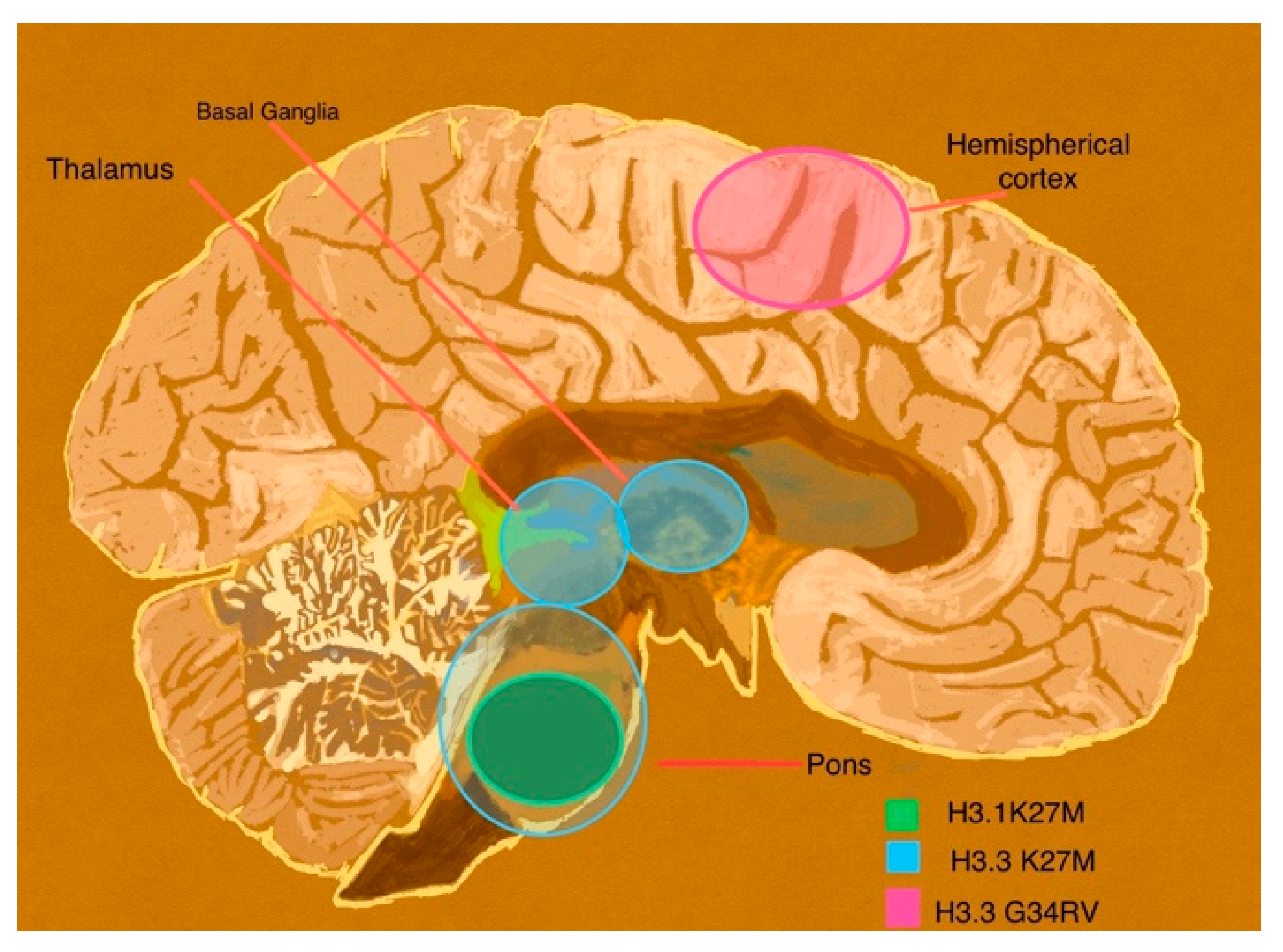
3.2.2. Diffuse Pediatric Midline Glioma (DMG)
3.2.3. Diffuse Pediatric Hemispheric Glioma
3.2.4. Diffuse Wild Type High-Grade Gliomas
3.2.5. Infant-Type Hemispheric Gliomas
3.3. Current Treatment Approaches
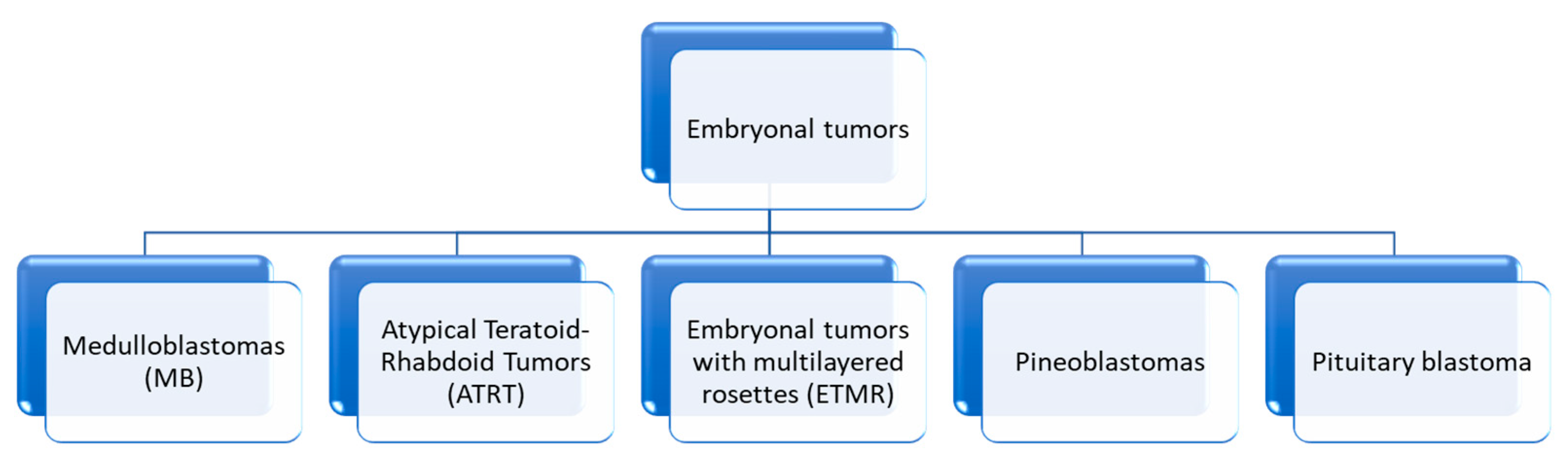
4. Embryonal Tumors
4.1. Molecular History
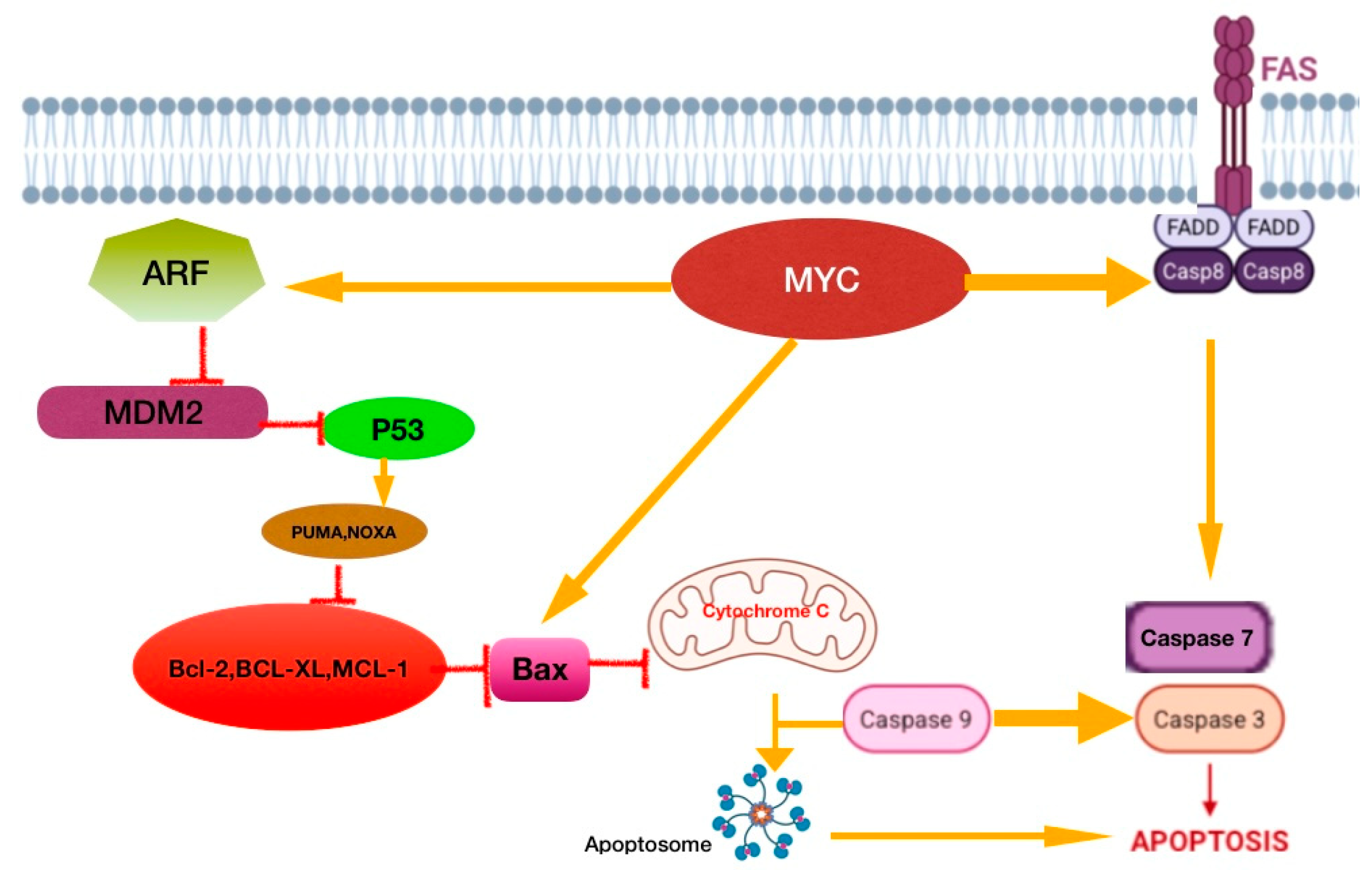
4.1.1. E-Cadherin/Beta-Catenin Complex
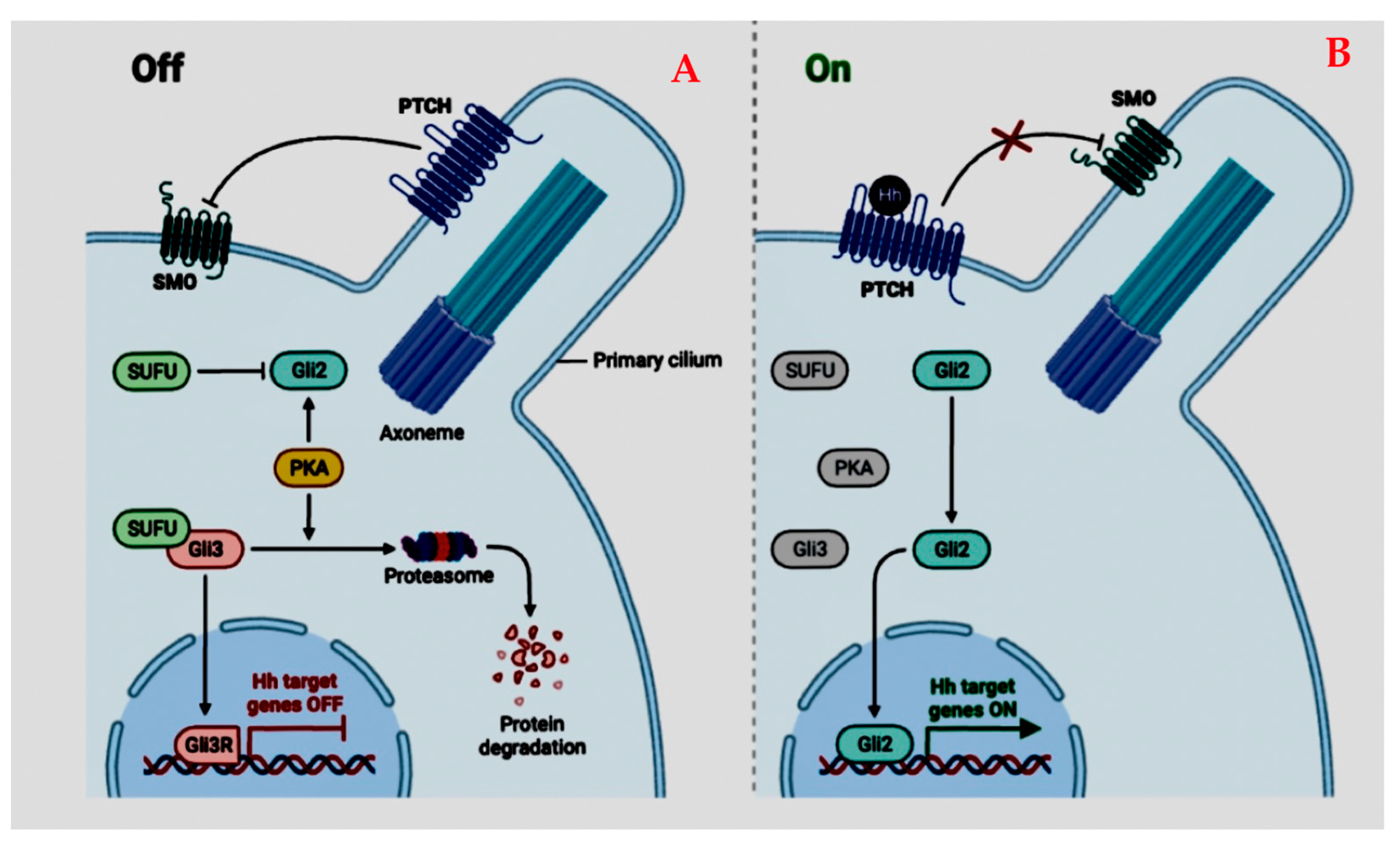
4.1.2. Sonic Hedgehog Pathway (SHH)
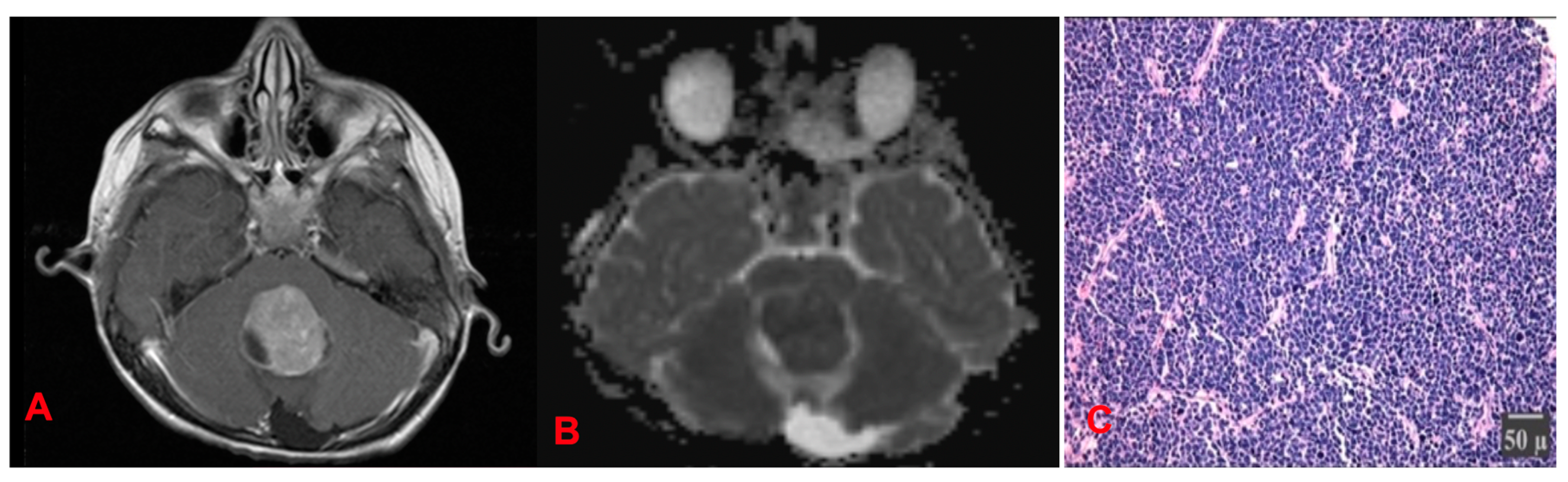
4.1.3. Medulloblastomas (MB) (Figure 17)
4.1.4. Treatment Status
Radiation Therapy
Chemotherapy in Standard-Risk Patients
4.2. Atypical Teratoid/Rhabdoid Tumors
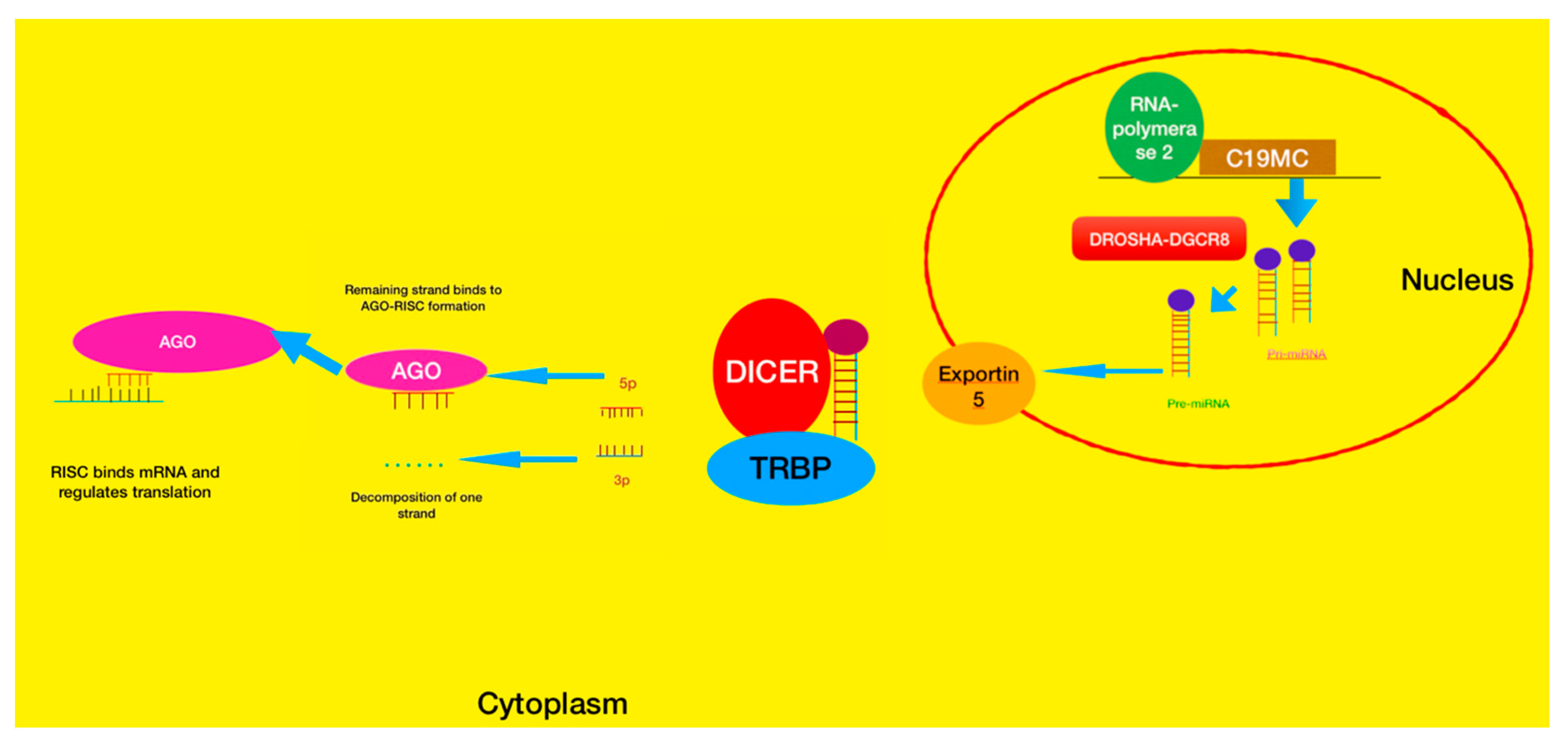
4.3. Embryonal Tumor with Multilayered Rosettes (ETMR)—C19MC-Altered) and Embryonal Tumor with Multilayered Rosettes—Not Otherwise Specified
4.3.1. ETMR Tumors
4.3.2. Pineoblastomas (PBs)
5. Pediatric Ependymomas (pEPNs)
5.1. Identity Molecules
5.1.1. NF-kB Pathway
5.1.2. Hippo Pathway (Figure 21)
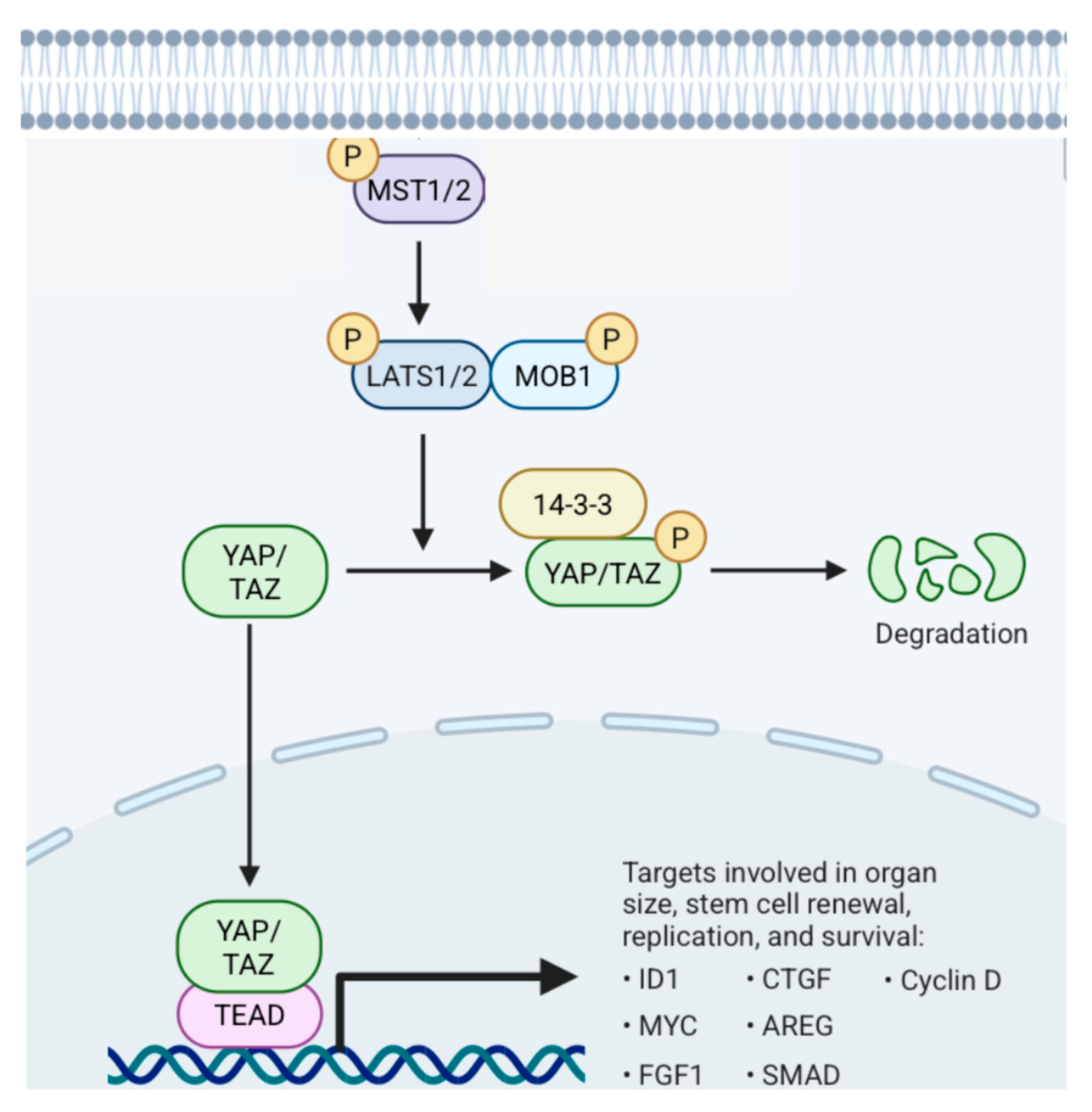
5.2. Supratentorial Ependymomas (ST EPNs)
5.3. Posterior Fossa Ependymomas (PF Ependymomas)
5.4. Spinal Ependymomas
5.5. Current Treatment Status
6. Germ Cell (GC) Tumors
6.1. Diagnostic Work-Up
6.2. Treatment
7. Future and Experimental Treatments for Pediatric Brain Cancer
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACTH | Adrenocorticotropic hormone |
| AFP | Alpha-fetoprotein |
| AG | Angiocentric glioma |
| AHPCR | Autologous hematopietic progenitor cell rescue |
| Akt1 | v-akt murine thymoma viral oncogene homolog 1 |
| ALK | Anaplastic lymphoma kinase |
| APC | Adenomatous polyposis coli |
| ARF | Alternating reading frame (tumor suppressor) |
| ATRT | Atypical teratoid/rhabdoid tumors |
| ATRX | alpha-thalassemia intellectual disability syndrome protein |
| BAF | BRG1/BRM-related factor |
| Bak | BCL2-antagonist/killer 1 |
| Bax | BCL2-associated X protein |
| BCL | B-cell leukemia/lymphoma |
| BMP | Bone morophogenetic protein |
| Bok | BCL2 ovarian killer/BCL2 family apoptosis regulator BOK |
| BRAF | v-raf murine sarcoma viral oncogene homolog B1 |
| BRG1 | hSWI/SNF brahma-related gene |
| BRM | Brahma |
| C19MC | Chromosome 19q microRNA cluster |
| CD30/TNFRSF8 | TNF receptor superfamily member 8 |
| CDK | Cyclin-dependent kinases |
| Chr | Chromosome |
| CNS | Central nervous system |
| CPA | Cerebellopontine angle |
| CSF | Cerebrospinal fluid |
| CTNNB1 | Catenin, beta-1 |
| DAXX | Death-associated protein 6 |
| DIPG | Diffuse intrinsic pontine glioma |
| DLGGs | Diffuse low-grade gliomas |
| DMG | Diffuse midline glioma |
| DNA | Deoxyribonucleic acid |
| DNET | Dysembryoplastic neuroepithelial tumor |
| DROSHA | Drosha ribonuclease III |
| Dsh | Disheveled |
| DWI | Diffusion-weighted imaging |
| EGFR | Epidermal growth factor receptor |
| EGL | External granular layer |
| EMT | Epithelial-to-mesenchymal transition |
| ERK 1/2 | Extracellular signal-regulated kinases 1/2 |
| ETANTR | Embryonal tumor with abundant neuropil and true rosettes |
| ETMR | Embryonal tumor with multilayered rosettes |
| EZH2 | Enhancer of zeste homologue 2 |
| EZHIP | Enhancer of zeste homologue inhibitory protein |
| FADD | Fas-associated via death domain |
| FGFR | Fibroblast growth factor receptor |
| FOXR2 | Forkhead box R2 |
| GBM | Glioblastoma multiforme |
| GCT | Germ cell tumors |
| GF | Growth factor |
| GFAP | Glial fibrillary acidic protein |
| Gli | Glioma-associated oncogene homolog 1 |
| GPCR | G protein-coupled receptor |
| Gy | Gray (unit of ionizing radiation) |
| HCG | Human chorionic gonadotrophin |
| HDC | High-dose chemotherapy |
| HGGs | High-grade gliomas |
| IDH | Isocitric dehydrogenase |
| INK4 | Inhibitors of CDK4 |
| IL-1 | Interleukin-1 |
| IT | Intrathecal |
| IkB | Ikappa B protein |
| KBTBD4 | Kelch repeat and BTB domain containing 4 |
| KIT | v-kit Hardy–Zuckerman 4 feline sarcoma viral oncogene homolog |
| KITLG | KIT ligand |
| LATS 1/2 | Large tumor suppressor kinases 1/2 |
| LIN28 | lin-28 homolog A |
| MAPK | Mitogen-activated protein kinases |
| MB | Medulloblastoma |
| MCL1 | Myeloid cell leukemia 1 |
| MDM2 | Mouse double minute 2 homolog |
| MEK | Mitogen-activated proteins kinase |
| MGMT | O-6-methylguanine-DNA methyltransferase |
| MKP | MAPK protein phosphatase |
| MOB | Monopolar spindle-one-binder protein 1 |
| MRI | Magnetic resonance imaging |
| MST 1/2 | Mammalian sterile-like kinase 1/2 |
| mTOR | Mammalian target of rapamycin or FK506 binding protein 12-rapamycin associated protein 1 |
| MYB | Myeloblastosis or v-myb myeloblastosis viral oncogene homolog (avian) |
| MYBL1 | v-myb myeloblastosis viral oncogene homolog (avian)-like 1 |
| MYC | MYC proto-oncogene, bHLH transcription factor |
| MYCN | v-myc myelocytomatosis viral-related oncogene, neuroblastoma-derived (avian) |
| NF1 | Neurofibromatosis type 1 |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B-cells |
| NGGCT | Non-germinomatous germ cell tumors |
| NTRK | Neurotrophic tyrosine receptor kinase |
| NOXA/PMAIP1 | Phorbol-12-myristate-13-acetate-induced protein 1 |
| OCT | Octamer binding factor |
| OS | Overall survival |
| OTX2 | Orthodenticle homeobox 2 |
| PA | Pilocytic astrocytoma |
| PBs | Pineoblastomas |
| PBAF | Polybromo-associated BAF |
| PDGFR | Platelet-derived growth factor receptor |
| pEPNs | Pediatric ependymomas |
| PF | Posterior fossa |
| PFS | Progression-free survival |
| PGC | Primordial germ cell |
| PI3K | Phosphoinositide 3-kinase |
| PIP3 | Phosphatidylinositol-3,4,5 trisphosphate |
| pLGGs | Pediatric low-grade gliomas |
| PLNTY | Polymorphus low-grade neuroepithelial tumor of the young |
| PMA | Pilomyxoid variant of pilocytic astrocytoma |
| pRb | Protein retinoblastoma |
| PRC2 | Polycomb repressive complex 2 |
| PTCH1 | Patched homolog 1 |
| PTEN | Phosphatase and tensin homolog deleted on chromosome ten |
| PUMA | p53 upregulated modulator of apoptosis |
| PXA | Pleiomorphic xanthoastrocytoma |
| RAF | Rapidly accelerated fibrosarcoma |
| RAS | Rat sarcoma |
| RelA | v-rel reticuloendotheliosis viral oncogene homolog A (avian) |
| RNA | Ribonucleic acid |
| ROS | ROS proto-oncogene 1, receptor tyrosine kinase |
| RT | Radiation therapy |
| RTK | Receptor tyrosine kinase |
| SALL4 | Spalt-like transcription factor 4 |
| SAV1 | Salvador homolog protein 1 |
| SHH | Sonic hedgehog |
| SEGA | Subependymal giant-cell astrocytoma |
| SET | Suppressor of variegation, enhancer of zeste and trithorax |
| SETD2 | SET domain containing 2, histone lysine methyltransferase |
| SMARCA4 | SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily a, member 4 |
| SMARCB1 | SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily b, member 1 |
| SMO | Smoothened, frizzled class receptor |
| SNCAIP | Synuclein alppha interacting protein |
| SOX2 | SRY (sex-determining region Y)-box 2 |
| SUFU | Supressor of fused protein |
| SWI/SNF | Switch/sucrose non-fermentable protein group |
| TACC | Transforming, acidic coiled–coil-containing protein |
| TEAD | Transcriptional enhancer factor domain |
| TERT | Telomerase reverse transcriptase |
| TKD | Tyrosine kinase domain |
| TMZ | Temozolamide |
| TNFa | Tumor necrosis factor alpha |
| TP53 | Tumor protein 53 (Li–Fraumeni syndrome) |
| TSC 1/2 | Tuberous sclerosis 1/2 |
| TTYH1 | Tweety family member 1 |
| QKI | QKI, KH domain containing RNA binding |
| WHO | World Health Organization |
| Wnt | Wingless-type MMTV integration site family member |
| YAP/TAZ | Yes-associated protein/PDZ-binding motif |
| ZFTA/C11orf95 | Zinc finger translocation associated |
References
- El-Ayadi, M.; Ansari, M.; Sturm, D.; Gielen, G.H.; Warmuth-Metz, M.; Kramm, C.M.; von Bueren, A.O. High-grade glioma in very young children: A rare and particular patient population. Oncotarget 2017, 8, 64564–64578. [Google Scholar] [CrossRef] [PubMed]
- Slegers, R.J.; Blumcke, I. Low-grade developmental and epilepsy associated brain tumors: A critical update 2020. Acta Neuropathol. Commun. 2020, 8, 27. [Google Scholar] [CrossRef] [PubMed]
- Abolhassani, H.; Wang, Y.; Hammarström, L.; Pan-Hammarström, Q. Hallmarks of cancers: Primary antibody de-ficiency versus other inborn errors of immunity. Front. Immunol. 2021, 12, 720025. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Li, C.; Wang, Z.; Xu, Y.; Shao, S.; Shao, F.; Wang, H.; Liu, J. Neutrophils in cancer: Dual roles through intercellular interactions. Oncogene 2024, 43, 1163–1177. [Google Scholar] [CrossRef]
- Zhuravleva, E.; O’rourke, C.J.; Andersen, J.B. Mutational signatures and processes in hepatobiliary cancers. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 367–382. [Google Scholar] [CrossRef]
- Trosko, J.E. The Concept of “Cancer Stem Cells” in the Context of Classic Carcinogenesis Hypotheses and Experimental Findings. Life 2021, 11, 1308. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Thompson, C.B. Metabolism of cell growth and proliferation. In The Molecular Ba-sis of Cancer, 3rd ed.; Mendelson, J., Howley, P.M., Israel, M.A., Gray, J.W., Tompson, C.B., Eds.; Elsevier Inc.: Philadelphia, PA, USA, 2008; pp. 189–203. [Google Scholar]
- Zhao, M.; Jung, Y.; Jiang, Z.; Svensson, K.J. Regulation of Energy Metabolism by Receptor Tyrosine Kinase Ligands. Front. Physiol. 2020, 11, 354. [Google Scholar] [CrossRef]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef]
- Gorodezki, D.; Schuhmann, M.U.; Ebinger, M.; Schittenhelm, J. Dissecting the Natural Patterns of Progression and Senescence in Pediatric Low-Grade Glioma: From Cellular Mechanisms to Clinical Implications. Cells 2024, 13, 1215. [Google Scholar] [CrossRef]
- Schwark, K.; Messinger, D.; Cummings, J.R.; Bradin, J.; Kawakibi, A.; Babila, C.M.; Lyons, S.; Ji, S.; Cartaxo, R.T.; Kong, S.; et al. Receptor tyrosine kinase (RTK) targeting in pediatric high-grade glioma and diffuse midline glioma: Pre-clinical models and precision medicine. Front. Oncol. 2022, 12, 922928. [Google Scholar] [CrossRef]
- Haase, S.; Nuñez, F.M.; Gauss, J.C.; Thompsonnn, S.; Brumley, E.; Lowenstein, P.; Castro, M.G. Hemispherical pediatric high-grade glioma: Molecular basis and therapeutic op-portunities. Int. J. Mol. Sci. 2020, 21, 9654. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The Biology of Cancer: Metabolic Reprogramming Fuels Cell Growth and Proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Huang, S.; Tian, W.; Liu, P.; Xie, Y.; Qiu, Y.; Li, X.; Tang, Y.; Zheng, S.; Sun, Y.; et al. PIWI-interacting RNA-YBX1 inhibits proliferation and metastasis by the MAPK signaling pathway via YBX1 in triple-negative breast cancer. Cell Death Discov. 2024, 10, 7. [Google Scholar] [CrossRef]
- Ma, Y.; Flückiger, I.; Nicolet, J.; Pang, J.; Dickinson, J.B.; De Bellis, D.; Emonet, A.; Fujita, S.; Geldner, N. Comparisons of two receptor-MAPK pathways in a single cell-type reveal mechanisms of signalling specificity. Nat. Plants 2024, 10, 1343–1362. [Google Scholar] [CrossRef]
- Kyriakis, J.M.; Avruch, J. Mammalian MAPK Signal Transduction Pathways Activated by Stress and Inflammation: A 10-Year Update. Physiol. Rev. 2012, 92, 689–737. [Google Scholar] [CrossRef]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef]
- Moses, C.; Nugent, F.; Waryah, C.B.; Garcia-Bloj, B.; Harvey, A.R.; Blancafort, P. Activating PTEN Tumor Suppressor Expression with the CRISPR/dCas9 System. Mol. Ther.—Nucleic Acids 2019, 14, 287–300. [Google Scholar] [CrossRef]
- Shorning, B.Y.; Dass, M.S.; Smalley, M.J.; Pearson, H.B. The PI3K-AKT-mTOR pathway and prostate cancer: At the crossroads of AR, MAPK, and WNT signaling. Int. J. Mol. Sci. 2020, 21, 4507. [Google Scholar] [CrossRef]
- Cicirò, Y.; Sala, A. MYB oncoproteins: Emerging players and potential therapeutic targets in human cancer. Oncogenesis 2021, 10, 19. [Google Scholar] [CrossRef]
- Andersson, M.K.; Mangiapane, G.; Nevado, P.T.; Tsakaneli, A.; Carlsson, T.; Corda, G.; Nieddu, V.; Abrahamian, C.; Chayka, O.; Rai, L.; et al. ATR is a MYB regulated gene and potential therapeutic target in adenoid cystic carcinoma. Oncogenesis 2020, 9, 5. [Google Scholar] [CrossRef]
- Mitra, P. Transcription regulation of MYB: A potential and novel therapeutic target in cancer. Ann. Transl. Med. 2018, 6, 443. [Google Scholar] [CrossRef] [PubMed]
- Hermann, B.P.; Cheng, K.; Singh, A.; Roa-De La Cruz, L.; Mutoji, K.N.; Chen, I.-C.; Gildersleeve, H.; Lehle, J.D.; Mayo, M.; Westernströer, B.; et al. The mammalian spermatogenesis single-cell transcriptome, from spermatogonial stem cells to spermatids. Cell Rep. 2018, 25, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.J.; Lee, J.M.; Lee, M.S.; Kim, Y.H.; Shin, S.Y. c-Myb negatively regulates Ras signaling through induction of dual phosphatase MKP-3 in NIH3T3 cells. Biochem. Biophys. Res. Commun. 2014, 450, 1032–1037. [Google Scholar] [CrossRef]
- Pizzimenti, C.; Fiorentino, V.; Germanò, A.; Martini, M.; Ieni, A.; Tuccari, G. Pilocytic astrocytoma: The paradigmatic entity in low grade gliomas. Oncol. Lett. 2024, 27, 146. [Google Scholar] [CrossRef]
- Ellison, D.W.; Hawkins, C.; Jones, D.T.; Onar-Thomas, A.; Pfister, S.M.; Reifenberger, G.; Louis, D.N. cIMPACT-NOW update 4: Diffuse gliomas characterized by MYB, MYBL1, or FGFR1 alterations or BRAF V600E mutation. Acta Neuropathol. 2019, 137, 683–687. [Google Scholar] [CrossRef]
- Kalelioglu, T.; Rama, B.; Cho, B.B.; Lopes, B.M.; Patel, S.H. Pediatric-type diffuse low-grade glioma with MYB/MYBL1 alteration: Report of 2 cases. Neuroradiol. J. 2022, 36, 232–235. [Google Scholar] [CrossRef]
- Quiroz Tejada, A.R.; Miranda-Lloret, P.; Llavador Ros, M.; Ramirez, E.P.; Pancucci, G.; Barber, A.R.; Simal-Julián, J.A.; Botella-Asunción, C. Gangliogliomas in the pediatric population. Childs Nerv. Syst. 2021, 37, 831–837. [Google Scholar] [CrossRef]
- Kurokawa, R.; Baba, A.; Emile, P.; Kurokawa, M.; Ota, Y.; Kim, J.; Capizzano, A.; Srinivasan, A.; Moritani, T. Neuroimaging features of angiocentric glioma: A case series and sys-tematic review. J. Neuroimaging 2022, 32, 389–399. [Google Scholar] [CrossRef]
- Bale, T.A.; Rosenblum, M.K. The 2021 WHO Classification of Tumors of the Central Nervous System: An update on pediatric low-grade gliomas and glioneuronal tumors. Brain Pathol. 2022, 32, e13060. [Google Scholar] [CrossRef]
- Huse, J.T.; Snuderl, M.; Jones, D.T.; Brathwaite, C.D.; Altman, N.; Lavi, E.; Saffery, R.; Sexton-Oates, A.; Blumcke, I.; Capper, D.; et al. Polymorphous low-grade neuroepithelial tumor of the young (PLNTY): An epilep-togenic neoplasm with oligodendroglioma-like components, aberrant CD34 expression, and genetic altera-tions involving the MAP kinase pathway. Acta Neuropathol. 2017, 133, 417–429. [Google Scholar] [CrossRef]
- Purkait, S.; Mahajan, S.; Sharma, M.C.; Sarkar, C.; Suri, V. Pediatric-type diffuse low grade gliomas: Histomolecular profile and practical approach to their integrated diagnosis according to the WHO CNS5 classification. Indian J. Pathol. Microbiol. 2022, 65, 42–49. [Google Scholar] [CrossRef]
- Chen, J.; Qi, X.; Zhang, M.; Zhang, J.; Han, T.; Wang, C.; Cai, C. Review on neuroimaging in pediatric-type diffuse low-grade gliomas. Front. Pediatr. 2023, 11, 1149646. [Google Scholar] [CrossRef] [PubMed]
- Salles, D.; Laviola, G.A.; Malinverni, C.d.M.; Stávale, J.N. Pilocytic astrocytoma: A review of general, clinical, and molecular characteristics. J. Child Neurol. 2020, 35, 852–858. [Google Scholar] [CrossRef] [PubMed]
- Collins, V.P.; Jones, D.T.; Giannini, C. Pilocytic astrocytoma: Pathology, molecular mechanisms and markers. Acta Neuropathol. 2015, 129, 775–788. [Google Scholar] [CrossRef]
- Gutmann, D.H.; McLellan, M.D.; Hussain, I.; Wallis, J.W.; Fulton, L.L.; Fulton, R.S.; Magrini, V.; Demeter, R.; Wylie, T.; Kandoth, C.; et al. Somatic neurofibromatosis type 1 (NF1) inactivation characterizes NF1-associated pilocytic astrocytoma. Genome Res. 2012, 23, 431–439. [Google Scholar] [CrossRef]
- AlShail, E.; Alahmari, A.N.; Dababo, A.A.; Alsagob, M.; Al-Hindi, H.; Khalil, H.; Al Masseri, Z.; AlSalamah, R.; Almohseny, E.; Alduhaish, A.; et al. A molecular study of pediatric pilomyxoid and pilocytic astro-cytomas: Genome-wide copy number screening, retrospective analysis of clinicopathological features and long-term clinical outcome. Front. Oncol. 2023, 13, 1034292. [Google Scholar] [CrossRef]
- Kong, X.; Mao, Y.; Xi, F.; Li, Y.; Luo, Y.; Ma, J. Development of a nomogram based on radiomics and semantic features for pre-dicting chro-mosome 7 gain/chromosome 10 loss in IDH wild-type histologically low-grade gliomas. Front. Oncol. 2023, 13, 1196614. [Google Scholar] [CrossRef]
- Tamura, R. Current understanding of neurofibromatosis type 1, 2, and schwannomatosis. Int. J. Mol. Sci. 2021, 22, 5850. [Google Scholar] [CrossRef]
- Lemberg, K.M.; Wang, J.; Pratilas, C.A. From Genes to -Omics: The Evolving Molecular Landscape of Malignant Peripheral Nerve Sheath Tumor. Genes 2020, 11, 691. [Google Scholar] [CrossRef]
- Gaudino, S.; Martucci, M.; Russo, R.; Visconti, E.; Gangemi, E.; D’argento, F.; Verdolotti, T.; Lauriola, L.; Colosimo, C. MR imaging of brain pilocytic astrocytoma: Beyond the stereotype of benign astrocytoma. Childs Nerv. Syst. 2016, 33, 35–54. [Google Scholar] [CrossRef]
- Kulac, I.; Tihan, T. Pilomyxoid astrocytomas: A short review. Brain Tumor Pathol. 2019, 36, 52–55. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Cai, J.; Lin, N.; Xu, Y.; Wang, H.; Wu, Z.; Kang, D. Identification of BRAF p. V600E-Mutant and Wild-Type by MR Imaging in Pleomorphic Xanthoastrocytoma and Anaplastic Pleomorphic Xanthoastrocytoma. Am. J. Neuroradiol. 2021, 42, 2152–2159. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, S.; Dandapath, I.; Garg, A.; Sharma, M.C.; Suri, V.; Sarkar, C. The evolution of pleomorphic xanthoastrocytoma: From genesis to molecular al-terations and mimics. Lab. Investig. 2022, 102, 670–681. [Google Scholar] [CrossRef]
- Detti, B.; Scoccianti, S.; Maragna, V.; Lucidi, S.; Ganovelli, M.; Teriaca, M.A.; Caini, S.; Desideri, I.; Agresti, B.; Greto, D.; et al. Pleomorphic Xanthoastrocytoma: A single institution retrospective analysis and a review of the literature. La Radiol. Medica 2022, 127, 1134–1141. [Google Scholar] [CrossRef]
- Alves de Castro, J.V.; D’Almeida Costa, F. Pleomorphic xanthoastrocytoma in the multiverse of epigenomics: Is it time to recognize the variants? Acta Neuropathol. Commun. 2022, 10, 85. [Google Scholar] [CrossRef]
- Vaubel, R.; Zschernack, V.; Tran, Q.T.; Jenkins, S.; Caron, A.; Milosevic, D.; Smadbeck, J.; Vasmatzis, G.; Kandels, D.; Gnekow, A.; et al. Biology and grading of pleomorphic xanthoastrocytoma—What have we learned about it? Brain Pathol. 2021, 31, 20–32. [Google Scholar] [CrossRef]
- Phillips, J.J.; Gong, H.; Chen, K.; Joseph, N.M.; van Ziffle, J.; Bastian, B.C.; Grenert, J.P.; Kline, C.N.; Mueller, S.; Banerjee, A.; et al. The genetic landscape of anaplastic pleomorphic xanthoastrocytoma. Brain Pathol. 2018, 29, 85–96. [Google Scholar] [CrossRef]
- Kim, S.H.; Hwang, K.; Lee, K.S.; Choe, G.; Kim, C.-Y. Cerebellar Pleomorphic Xanthoastrocytoma with BRAF V600E Mutation. World Neurosurg. 2020, 139, 577–581. [Google Scholar] [CrossRef]
- Shaikh, N.; Brahmbhatt, N.; Kruser, T.J.; Kam, K.L.; Appin, C.L.; Wadhwani, N.R.; Chandler, J.; Kumthekar, P.; Lukas, R.V. Pleomorphic xanthoastrocytoma: A brief review. CNS Oncol. 2019, 8, CNS39. [Google Scholar] [CrossRef]
- Moore, W.; Mathis, D.; Gargan, L.; Bowers, D.C.; Klesse, L.J.; Margraf, L.; Koral, K. Pleomorphic xanthoastrocytoma of childhood: MR imaging and dif-fusion MR imaging features. Am. J. Neuroradiol. 2014, 35, 2192–2196. [Google Scholar] [CrossRef]
- Wu, W.; Zuo, P.; Li, C.; Gong, J. Clinical Features and Surgical Results of Pediatric Pleomorphic Xanthoastrocytoma: Analysis of 17 Cases with a Literature Review. World Neurosurg. 2021, 151, e778–e785. [Google Scholar] [CrossRef] [PubMed]
- Ataseven, E.; Özcan, M.; Ölçülü, C.B.; Bolat, E.; Ertan, Y.; Kitis, Ö.; Tekgül, H.; Kantar, M. Dysembryoplastic neuroepithelial tumors of childhood: Ege Univer-sity experi-ence. Childs Nerv. Syst. 2022, 38, 1699–1706. [Google Scholar] [CrossRef] [PubMed]
- d’Amati, A.; Bargiacchi, L.; Rossi, S.; Carai, A.; Bertero, L.; Barresi, V.; Errico, M.E.; Buccoliero, A.M.; Asioli, S.; Marucci, G.; et al. Pediatric CNS tumors and 2021 WHO classification: What do oncol-ogists need from pathologists? Front. Mol. Neurosci. 2024, 17, 1268038. [Google Scholar] [CrossRef] [PubMed]
- Giannikou, K.; Zhu, Z.; Kim, J.; Winden, K.D.; Tyburczy, M.E.; Marron, D.; Parker, J.S.; Hebert, Z.; Bongaarts, A.; Taing, L.; et al. Subependymal giant cell astrocytomas are characterized by mTORC1 hyperactivation, a very low somatic mutation rate, and a unique gene expression profile. Mod. Pathol. 2021, 34, 264–279. [Google Scholar] [CrossRef]
- Davalos, V.; Esteller, M. Cancer epigenetics in clinical practice. CA Cancer J. 2023, 73, 376–424. [Google Scholar] [CrossRef]
- Ocasio, J.K.; Budd, K.M.; Roach, J.T.; Andrews, J.M.; Baker, S.J. Oncohistones and disrupted development in pediatric-type diffuse high-grade glioma. Cancer Metastasis Rev. 2023, 42, 367–388. [Google Scholar] [CrossRef]
- Baker, S.J.; Ellison, D.W.; Gutmann, D.H. Pediatric gliomas as neurodevelopmental disorders. Glia 2015, 64, 879–895. [Google Scholar] [CrossRef]
- Gogou, P.; Pakos, E.; Batistatou, A.; Panelos, I.; Briasoulis, E.; Stefanou, D.; Apostolikas, N.; Tsekeris, P. Clinicopathologic study of E-cadherin/beta-catenin complex, and topoisomerase-II in a series of 71 liposarcoma cases. World J. Surg. Oncol. 2012, 10, 28. [Google Scholar] [CrossRef]
- Sharpless, N.E. INK4a/ARF: A multifunctional tumor suppressor locus. Mut. Res. 2005, 576, 22–38. [Google Scholar] [CrossRef]
- Martire, S.; Banaszynski, L.A. The roles of histone variants in fine-tuning chromatin organization and func-tion. Nat. Rev. Mol. Cell Biol. 2020, 21, 522–541. [Google Scholar] [CrossRef]
- Sakurai, R.; Kaira, K.; Miura, Y.; Sunaga, N.; Saito, R.; Oyama, T.; Hisada, T.; Yamada, M. Clinical significance of topoisomerase-II expression in patients with advanced non-small cell lung cancer treated with amrubicin. Thorac. Cancer 2020, 11, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Xavier, M.J.; Roman, S.D.; Aitken, R.J.; Nixon, B. Transgenerational inheritance: How impacts to the epigenetic and genetic information of parents affect offspring health. Hum. Reprod. Updat. 2019, 25, 519–541. [Google Scholar] [CrossRef] [PubMed]
- Nagy, G.; Bojcsuk, D.; Tzerpos, P.; Cseh, T.; Nagy, L. Lineage-determining transcription factor-driven promoters regulate cell type-specific macrophage gene expression. Nucleic Acids Res. 2024, 52, 4234–4256. [Google Scholar] [CrossRef]
- Mortimer, T.; Wainwright, E.N.; Patel, H.; Siow, B.M.; Jaunmuktane, Z.; Brandner, S.; Scaffidi, P. Redistribution of EZH 2 promotes malignant phenotypes by rewiring de-velopmental programmes. EMBO Rep. 2019, 20, e48155. [Google Scholar] [CrossRef]
- Liu, L.; Xiao, B.; Hirukawa, A.; Smith, H.W.; Zuo, D.; Sanguin-Gendreau, V.; McCaffrey, L.; Nam, A.J.; Muller, W.J. Ezh2 promotes mammary tumor initiation through epigenetic regulation of the Wnt and mTORC1 signaling pathways. Proc. Natl. Acad. Sci. USA 2023, 120, e2303010120. [Google Scholar] [CrossRef]
- Pang, Y.; Chen, X.; Ji, T.; Cheng, M.; Wang, R.; Zhang, C.; Liu, M.; Zhang, J.; Zhong, C. The Chromatin Remodeler ATRX: Role and Mechanism in Biology and Cancer. Cancers 2023, 15, 2228. [Google Scholar] [CrossRef]
- Mittal, P.; Roberts, C.W. The SWI/SNF complex in cancer—Biology, biomarkers and therapy. Nat. Rev. Clin. Oncol. 2020, 17, 435–448. [Google Scholar] [CrossRef]
- Técher, H.; Koundrioukoff, S.; Nicolas, A.; Debatisse, M. The impact of replication stress on replication dynamics and DNA damage in vertebrate cells. Nat. Rev. Genet. 2017, 18, 535–550. [Google Scholar] [CrossRef]
- Shay, J.W.; Wright, W.E. Telomeres and telomerase: Three decades of progress. Nat. Rev. Genet. 2019, 20, 299–309. [Google Scholar] [CrossRef]
- Dratwa, M.; Wysoczańska, B.; Łacina, P.; Kubik, T.; Bogunia-Kubik, K. TERT—Regulation and roles in cancer formation. Front. Immunol. 2020, 11, 589929. [Google Scholar] [CrossRef]
- Cullen, M.M.; Floyd, W.; Dow, B.; Schleupner, B.; Brigman, B.E.; Visgauss, J.D.; Cardona, D.M.; Somarelli, J.A.; Eward, W.C. ATRX and Its Prognostic Significance in Soft Tissue Sarcoma. Sarcoma 2024, 2024, 4001796. [Google Scholar] [CrossRef] [PubMed]
- Raghunandan, M.; Yeo, J.E.; Walter, R.; Saito, K.; Harvey, A.J.; Ittershagen, S.; Lee, E.-A.; Yang, J.; Hoatlin, M.E.; Bielinsky, A.K.; et al. Functional cross talk between the Fanconi anemia and ATRX/DAXX histone chaperone pathways promotes replication fork recovery. Hum. Mol. Genet. 2020, 29, 1083–1095. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, I.; Tian, G.; Wang, J.; Hutchinson, T.E.; Kim, B.J.; Awasthee, N.; Hale, S.; Meng, C.; Moore, A.; Zhao, L.; et al. DAXX drives de novo lipogenesis and contributes to tumorigenesis. Nat. Commun. 2023, 14, 1927. [Google Scholar] [CrossRef]
- Molenaar, T.M.; van Leeuwen, F. SETD2: From chromatin modifier to multipronged regulator of the genome and beyond. Cell. Mol. Life Sci. 2022, 79, 346. [Google Scholar] [CrossRef]
- Singh, N.; Miner, A.; Hennis, L.; Mittal, S. Mechanisms of temozolomide resistance in glioblastoma-a compre-hensive review. Cancer Drug Resist. 2021, 4, 17. [Google Scholar]
- Ashkan, K.; Mirza, A.B.; Soumpasis, C.; Syrris, C.; Kalaitzoglou, D.; Sharma, C.; James, Z.J.; Khoja, A.K.; Ahmed, R.; Vastani, A.; et al. MGMT Promoter Methylation: Prognostication beyond Treatment Response. J. Pers. Med. 2023, 13, 999. [Google Scholar] [CrossRef]
- Xiao, Y.; Dong, J. The Hippo Signaling Pathway in Cancer: A Cell Cycle Perspective. Cancers 2021, 13, 6214. [Google Scholar] [CrossRef]
- Ahmadi, S.E.; Rahimi, S.; Zarandi, B.; Chegeni, R.; Safa, M. MYC: A multipurpose oncogene with prognostic and therapeutic implications in blood malignancies. J. Hematol. Oncol. 2021, 14, 1–49. [Google Scholar] [CrossRef]
- Kuang, Y.; Kang, J.; Li, H.; Liu, B.; Zhao, X.; Li, L.; Jin, X.; Li, Q. Multiple functions of p21 in cancer radiotherapy. J. Cancer Res. Clin. Oncol. 2021, 147, 987–1006. [Google Scholar] [CrossRef]
- Pluta, A.J.; Studniarek, C.; Murphy, S.; Norbury, C.J. Cyclin-dependent kinases: Masters of the eukaryotic universe. Wiley Interdiscip. Rev. RNA 2023, 15, e1816. [Google Scholar] [CrossRef]
- Magge, T.; Rajendran, S.; Brufsky, A.M.; Foldi, J. CDK4/6 inhibitors: The Devil is in the Detail. Cur. Oncol. Rep. 2024, 26, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Ziada, S.; Diharce, J.; Serillon, D.; Bonnet, P.; Aci-Sèche, S. Highlighting the Major Role of Cyclin C in Cyclin-Dependent Kinase 8 Activity through Molecular Dynamics Simulations. Int. J. Mol. Sci. 2024, 25, 5411. [Google Scholar] [CrossRef] [PubMed]
- Chin, L.; Pomerantz, J.; DePinho, R.A. The INK4a/ARF: The INK4a/ARF tumor suppressor: One gene—Two products—Two pathways. Trends. Biochem. Sci. 1998, 23, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Weidner, N.; Cote, R.J.; Suster, S.; Weiss, L. Modern Surgical Pathology E-Book, 2nd ed.; Elsevier Health Sciences: Philadelphia, PA, USA, 2009; pp. 1984–2038. [Google Scholar]
- Goel, S.; Bergholz, J.S.; Zhao, J.J. Targeting CDK4 and CDK6 in cancer. Nat. Rev. Cancer 2022, 22, 356–372. [Google Scholar] [CrossRef]
- Meel, M.H.; Schaper, S.A.; Kaspers, G.J.L.; Hulleman, E. Signaling pathways and mesenchymal transition in pediatric high-grade glioma. Cell. Mol. Life Sci. 2017, 75, 871–887. [Google Scholar] [CrossRef]
- Sturm, D.; Pfister, S.M.; Jones, D.T. Pediatric gliomas: Current concepts on diagnosis, biology, and clinical man-agement. J. Clin. Oncol. 2017, 35, 2370–2377. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014–2018. Neuro-Oncology 2021, 23, iii1–iii105. [Google Scholar] [CrossRef]
- Chai, R.-C.; Zhang, Y.-W.; Liu, Y.-Q.; Chang, Y.-Z.; Pang, B.; Jiang, T.; Jia, W.-Q.; Wang, Y.-Z. The molecular characteristics of spinal cord gliomas with or without H3 K27M mutation. Acta Neuropathol. Commun. 2020, 8, 40. [Google Scholar] [CrossRef]
- Braunstein, S.; Raleigh, D.; Bindra, R.; Mueller, S.; Haas-Kogan, D. Pediatric high-grade glioma: Current molecular landscape and therapeutic ap-proaches. J. Neuro-Oncol. 2017, 134, 541–549. [Google Scholar] [CrossRef]
- Gianno, F.; Giovannoni, I.; Cafferata, B.; Diomedi-Camassei, F.; Minasi, S.; Barresi, S.; Buttarelli, F.R.; Alesi, V.; Cardoni, A.; Antonelli, M.; et al. Paediatric-type diffuse high-grade gliomas in the 5th CNS WHO Classification. Pathologica 2022, 114, 422–435. [Google Scholar] [CrossRef]
- Mackay, A.; Burford, A.; Carvalho, D.; Izquierdo, E.; Fazal-Salom, J.; Taylor, K.R.; Bjerke, L.; Clarke, M.; Vinci, M.; Nandhabalan, M.; et al. Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 32, 520–537.e5. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.L.; Vega, J.V.; Plant, A.; MacDonald, T.J.; Becher, O.J.; Hambardzumyan, D. Tumour immune landscape of paediatric high-grade gliomas. Brain 2021, 144, 2594–2609. [Google Scholar] [CrossRef] [PubMed]
- Buccoliero, A.M.; Giunti, L.; Moscardi, S.; Castiglione, F.; Provenzano, A.; Sardi, I.; Scagnet, M.; Genitori, L.; Caporalini, C. Pediatric High-Grade Glioma Classification Criteria and Molecular Features of a Case Series. Genes 2022, 13, 624. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Lin, G.N.; Song, W.; Zhang, Y.; Lai, H.; Zhang, M.; Miao, J.; Cheng, X.; Wang, Y.; Li, W.; et al. Single-cell spatial transcriptomic analysis reveals common and divergent features of developing postnatal granule cerebellar cells and medulloblastoma. BMC Biol. 2021, 19, 135. [Google Scholar] [CrossRef]
- Vladoiu, M.C.; El-Hamamy, I.; Donovan, L.K.; Farooq, H.; Holgado, B.L.; Sundaravadanam, Y.; Ramaswamy, V.; Hendrikse, L.D.; Kumar, S.; Mack, S.C.; et al. Childhood cerebellar tumours mirror conserved fetal transcriptional programs. Nature 2019, 572, 67–73. [Google Scholar] [CrossRef]
- Mondal, G.; Lee, J.C.; Ravindranathan, A.; Villanueva-Meyer, J.E.; Tran, Q.T.; Allen, S.J.; Barreto, J.; Gupta, R.; Doo, P.; Van Ziffle, J.; et al. Pediatric bithalamic gliomas have a distinct epigenetic signature and frequent EGFR exon 20 insertions resulting in potential sensitivity to targeted kinase inhibition. Acta Neuropathol. 2020, 139, 1071–1088. [Google Scholar] [CrossRef]
- Coleman, C.; Stoller, S.; Grotzer, M.; Stucklin, A.G.; Nazarian, J.; Mueller, S. Pediatric hemispheric high-grade glioma: Targeting the future. Cancer Metastasis Rev. 2020, 39, 245–260. [Google Scholar] [CrossRef]
- Korshunov, A.; Capper, D.; Reuss, D.; Schrimpf, D.; Ryzhova, M.; Hovestadt, V.; Sturm, D.; Meyer, J.; Jones, C.; Zheludkova, O.; et al. Histologically distinct neuroepithelial tumors with histone 3 G34 mutation are molecularly similar and comprise a single nosologic entity. Acta Neuropathol. 2015, 131, 137–146. [Google Scholar] [CrossRef]
- Gianno, F.; Antonelli, M.; Di Dio, T.; Minasi, S.; Donofrio, V.; Buccoliero, A.M.; Gardiman, M.P.; Pollo, B.; Camassei, F.M.; Rossi, S.; et al. Correlation between immunohistochemistry and sequencing in H3G34-mutant gli-omas. Am. J. Surg. Pathol. 2021, 45, 200–204. [Google Scholar] [CrossRef]
- Lulla, R.R.; Saratsis, A.M.; Hashizume, R. Mutations in chromatin machinery and pediatric high-grade glioma. Sci. Adv. 2016, 2, e1501354. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Fingarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumours of the Central Nervous System: A sum-mary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Bender, K.; Kahn, J.; Perez, E.; Ehret, F.; Roohani, S.; Capper, D.; Schmid, S.; Kaul, D. Diffuse paediatric-type high-grade glioma, H3-wildtype and IDH-wildtype: Case series of a new entity. Brain Tumor Pathol. 2023, 40, 204–214. [Google Scholar] [CrossRef] [PubMed]
- DeSisto, J.; Lucas, J.T., Jr.; Xu, K.; Donson, A.; Lin, T.; Sanford, B.; Wu, G.; Tran, Q.T.; Hedges, D.; Hsu, C.-Y.; et al. Comprehensive molecular characterization of pediatric radia-tion-induced high-grade glioma. Nat. Commun. 2021, 12, 5531. [Google Scholar] [CrossRef] [PubMed]
- Nafe, R.; Hattingen, E. The Spectrum of Molecular Pathways in Gliomas-An Up-to-Date Review. Biomedicines 2023, 11, 2281. [Google Scholar] [CrossRef]
- Hong, L.; Shi, Z.-F.; Li, K.K.-W.; Wang, W.-W.; Yang, R.R.; Kwan, J.S.-H.; Chen, H.; Li, F.-C.; Liu, X.-Z.; Chan, D.T.-M.; et al. Molecular landscape of pediatric type IDH wildtype, H3 wildtype hem-ispheric glio-blastomas. Lab. Investig. 2022, 102, 731–740. [Google Scholar] [CrossRef]
- Lehmann, R.; Rayner, B.S.; Ziegler, D.S. Resistance mechanisms in BRAFV600E paediatric high-grade glioma and current therapeutic approaches. Front. Oncol. 2022, 12, 1031378. [Google Scholar] [CrossRef]
- Guerreiro Stucklin, A.S.; Ryall, S.; Fukuoka, K.; Zapotocky, M.; Lassaletta, A.; Li, C.; Bridge, T.; Kim, B.; Arnoldo, A.; Kowalski, P.E.; et al. Alterations in ALK/ROS1/NTRK/MET drive a group of infantile hemi-spheric gliomas. Nat. Commun. 2019, 10, 4343. [Google Scholar] [CrossRef]
- Lau, L.M.S.; Dagg, R.A.; Henson, J.D.; Au, A.Y.M.; Royds, J.A.; Reddel, R.R. Detection of alternative lengthening of telomeres by telomere quantitative PCR. Nucleic Acids Res. 2012, 41, e34. [Google Scholar] [CrossRef]
- Martínez-Jiménez, F.; Muiños, F.; Sentís, I.; Deu-Pons, J.; Reyes-Salazar, I.; Arnedo-Pac, C.; Mularoni, L.; Pich, O.; Bonet, J.; Kranas, H.; et al. A compendium of mutational cancer driver genes. Nat. Rev. Cancer 2020, 20, 273. [Google Scholar] [CrossRef]
- Mistry, M.; Zhukova, N.; Merico, D.; Rakopoulos, P.; Krishnatry, R.; Shago, M.; Stavropoulos, J.; Alon, N.; Pole, J.D.; Ray, P.N.; et al. BRAF Mutation and CDKN2A Deletion Define a Clinically Distinct Subgroup of Childhood Secondary High-Grade Glioma. J. Clin. Oncol. 2015, 33, 1015–1022. [Google Scholar] [CrossRef]
- Cohen, J.; Bannykh, S.; Breunig, J.; Danielpour, M. Cerebral Gliomas. In Textbook of Pediatric Neurosurgery; Di Rocco, C., Pang, D., Rutka, J., Eds.; Springer Nature: Cham, Switzerland, 2020; pp. 1853–1875. [Google Scholar]
- Wisoff, J.H.; Sanford, R.A.; Heier, L.A.; Sposto, R.; Burger, P.C.; Yates, A.J.; Holmes, E.J.; Kun, L.E. Primary neurosurgery for pediatric low-grade gliomas: A pro-spective mul-ti-institutional study from the Childrens Oncology Group. Neurosurgery 2011, 68, 1548–1555. [Google Scholar] [CrossRef] [PubMed]
- Fahmy, S.A.; Dawoud, A.; Zeinelabdeen, Y.A.; Kiriacos, C.J.; Daniel, K.A.; Eltahtawy, O.; Abdelhalim, M.M.; Braoudaki, M.; Youness, R.A. Molecular Engines, Therapeutic Targets, and Challenges in Pediatric Brain Tumors: A Special Emphasis on Hydrogen Sulfide and RNA-Based Nano-Delivery. Cancers 2022, 14, 5244. [Google Scholar] [CrossRef] [PubMed]
- Metu, C.L.N.; Sutihar, S.K.; Sohel; Zohora, F.; Hasan, A.; Miah, T.; Kar, T.R.; Hossain, A.; Rahman, H. Unraveling the signaling mechanism behind astrocytoma and possible therapeutics strategies: A comprehensive review. Cancer Rep. 2023, 6, e1889. [Google Scholar] [CrossRef] [PubMed]
- Kudus, K.; Wagner, M.W.; Namdar, K.; Bennett, J.; Nobre, L.; Tabori, U.; Hawkins, C.; Ertl-Wagner, B.B.; Khalvati, F. Beyond hand-crafted features for pretherapeutic molecular status identification of pediatric low-grade gliomas. Sci. Rep. 2024, 14, 19102. [Google Scholar] [CrossRef]
- Sait, S.F.; Giantini-Larsen, A.M.; Tringale, K.R.; Souweidane, M.M.; Karajannis, M.A. Treatment of Pediatric Low-Grade Gliomas. Curr. Neurol. Neurosci. Rep. 2023, 23, 185–199. [Google Scholar] [CrossRef]
- Maraka, S.; Janku, F. BRAF Alterations in Primary Brain Tumors. Discov. Med. 2018, 26, 51–60. [Google Scholar]
- Arafah, O.; Maher, E.; Mosaab, A.; Naguib, E.; Refaat, A.; Ahmed, S.; Taha, H.; El-Beltagy, M.; El-Ayadi, M. High-grade glioma in infants and very young children: Characteris-tics, treatment, and outcomes. Child’s Nerv. Syst. 2024, 40, 2667–2675. [Google Scholar] [CrossRef]
- Rizzo, D.; Ruggiero, A.; Martini, M.; Rizzo, V.; Maurizi, P.; Riccardi, R. Molecular biology in pediatric high-grade glioma: Impact on prog-nosis and treatment. BioMed Res. Int. 2015, 1, 215135. [Google Scholar]
- Ruffle, J.K.; Mohinta, S.; Pombo, G.; Gray, R.; Kopanitsa, V.; Lee, F.; Brandner, S.; Hyare, H.; Nachev, P. Brain tumour genetic network signatures of survival. Brain 2023, 146, 4736–4754. [Google Scholar] [CrossRef]
- The Children’s Oncology Group; Pollack, I.F.; Hamilton, R.L.; Burger, P.C.; Brat, D.J.; Rosenblum, M.K.; Murdoch, G.H.; Nikiforova, M.N.; Holmes, E.J.; Zhou, T.; et al. Akt activation is a common event in pediatric malignant gliomas and a potential adverse prognostic marker: A report from the Children’s Oncology Group. J. Neuro-Oncol. 2010, 99, 155–163. [Google Scholar] [CrossRef]
- Diaz, A.K.; Wu, G.; Paugh, B.S.; Li, Y.; Zhu, X.; Rankin, S.; Qu, C.; Chen, X.; Zhang, J.; Easton, J.; et al. Abstract PR03: The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. In Proceedings of the Abstracts: AACR Special Conference: Pediatric Cancer at the Crossroads: Translating Discovery into Improved Outcomes, San Diego, CA, USA, 3–6 November 2013; p. PR03. [Google Scholar]
- Russo, R.; Verdolotti, T.; Perna, A.; Ruscelli, L.; D’abronzo, R.; Romano, A.; Ferrara, G.; Parisi, D.; Infante, A.; Chiesa, S.; et al. Central nervous system pediatric multi-disciplinary tumor board: A single center experience. BMC Cancer 2024, 24, 1146. [Google Scholar] [CrossRef] [PubMed]
- Perwein, T.; Giese, B.; Nussbaumer, G.; von Bueren, A.O.; van Buiren, M.; Benesch, M.; Kramm, C.M. How I treat recurrent pediatric high-grade glioma (pHGG): A Europe-wide survey study. J. Neuro-Oncol. 2023, 161, 525–538. [Google Scholar] [CrossRef] [PubMed]
- Kulubya, E.S.; Kercher, M.J.; Phillips, H.W.; Antony, R.; Edwards, M.S.B. Advances in the Treatment of Pediatric Brain Tumors. Children 2022, 10, 62. [Google Scholar] [CrossRef]
- Kacker, S.; Parsad, V.; Singh, N.; Hordiichuk, D.; Alvarez, S.; Gohar, M.; Kacker, A.; Rai, S.K. Planar Cell Polarity Signaling: Coordinated Crosstalk for Cell Orientation. J. Dev. Biol. 2024, 12, 12. [Google Scholar] [CrossRef]
- Wang, X.; Wang, T.; Song, X.; Gao, J.; Xu, G.; Ma, Y.; Song, G. Current Status of Hedgehog Signaling Inhibitors. Curr. Top. Med. Chem. 2024, 24, 243–258. [Google Scholar] [CrossRef]
- Chen, L.; Li, Y.; Song, Z.; Xue, S.; Liu, F.; Chang, X.; Wu, Y.; Duan, X.; Wu, H. O-GlcNAcylation promotes cerebellum development and medulloblastoma on-cogenesis via SHH signaling. Proc. Natl. Acad. Sci. USA 2022, 119, e2202821119. [Google Scholar] [CrossRef]
- Falsini, A.; Giuntini, G.; Mori, M.; Ghirga, F.; Quaglio, D.; Cucinotta, A.; Coppola, F.; Filippi, I.; Naldini, A.; Botta, B.; et al. Hedgehog Pathway Inhibition by Novel Small Molecules Impairs Melanoma Cell Migration and Invasion under Hypoxia. Pharmaceuticals 2024, 17, 227. [Google Scholar] [CrossRef]
- Schilling, K. Specification and development of GABAergic interneurons. In Handbook of the Cerebellum and Cerebellar Dis-Orders; Manto, M.U., Gruol, D.L., Schmamann, J.D., Koibuchi, N., Sillitoe, R.V., Eds.; Springer Nature: Cham, Switzerland, 2021; pp. 235–264. [Google Scholar]
- ten Donkelaar, H.J.; Lammens, M.; Wesseling, P.; Hori, A. Development and developmental disorders of the human cerebel-lum. In Clinical Neuroembryology: Development and Developmental Disorders of the Human Central Nervous System, 2nd ed.; ten Donkelaar, H.J., Lammens, M., Hori, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 371–412. [Google Scholar]
- Zhou, Q.; Xiang, J.; Qiu, N.; Wang, Y.; Piao, Y.; Shao, S.; Tang, J.; Zhou, Z.; Shen, Y. Tumor Abnormality-Oriented Nanomedicine Design. Chem. Rev. 2023, 123, 10920–10989. [Google Scholar] [CrossRef]
- Tomecka, P.; Kunachowicz, D.; Górczyńska, J.; Gebuza, M.; Kuźnicki, J.; Skinderowicz, K.; Choromańska, A. Factors Determining Epithelial-Mesenchymal Transition in Cancer Pro-gression. Int. J. Mol. Sci. 2024, 25, 8972. [Google Scholar] [CrossRef]
- Fernando, D.; Ahmed, A.U.; Williams, B.R.G. Therapeutically targeting the unique disease landscape of pediatric high-grade gliomas. Front. Oncol. 2024, 14, 1347694. [Google Scholar] [CrossRef]
- Rees, D.; Gianferante, D.M.; Kim, J.; Stavrou, T.; Reaman, G.; Sapkota, Y.; Gramatges, M.M.; Morton, L.M.; Hudson, M.M.; Armstrong, G.T.; et al. Frequency of pathogenic germline variants in pediatric medullo-blastoma survivors. Front. Oncol. 2024, 14, 1441958. [Google Scholar] [CrossRef] [PubMed]
- Dakal, T.C.; Dhabhai, B.; Pant, A.; Moar, K.; Chaudary, K.; Yadav, V.; Ranga, V.; Sarma, N.K.; Kumar, A.; Maurya, P.K.; et al. Oncogenes and tumor suppressor genes: Functions and roles in can-cers. Med. Comm. 2024, 5, e582. [Google Scholar]
- AlRayahi, J.; Zapotocky, M.; Ramaswamy, V.; Hanagandi, P.; Branson, H.; Mubarak, W.; Raybaud, C.; Laughlin, S. Pediatric Brain Tumor Genetics: What Radiologists Need to Know. RadioGraphics 2018, 38, 2102–2122. [Google Scholar] [CrossRef]
- Aldape, K.; Brindle, K.M.; Chesler, L.; Chopra, R.; Gajjar, A.; Gilbert, M.R.; Gottardo, N.; Gutmann, D.H.; Hargrave, D.; Holland, E.C.; et al. Challenges to curing primary brain tumours. Nat. Rev. Clin. Oncol. 2019, 16, 509–520. [Google Scholar] [CrossRef]
- Blessing, M.M.; Alexandrescu, S. Embryonal tumors of the central nervous system: An update. Surg. Pathol. Clin. 2020, 13, 235–247. [Google Scholar] [CrossRef]
- Northcott, P.A.; Buchhalter, I.; Morrissy, A.S.; Hovestadt, V.; Weiscenfeldt, J.; Ehrenberger, T.; Gröbner, S.; Segura-Wang, M.; Zichner, T.; Rudneva, V.A.; et al. The whole-genome landscape of medulloblastoma sub-types. Nature 2017, 547, 311–317. [Google Scholar] [CrossRef]
- Colafati, G.S.; Voicu, I.P.; Carducci, C.; Miele, E.; Carai, A.; Di Loreto, S.; Marrazzo, A.; Cacchione, A.; Cecinati, V.; Tornesello, A.; et al. MRI features as a helpful tool to predict the molecular sub-groups of medullo-blastoma: State of the art. Ther. Adv. Neurol. Disord. 2018, 11, 1756286418775375. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Patil, N.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013–2017. Neuro-Oncology 2020, 22, iv1–iv96. [Google Scholar] [CrossRef]
- Phuong, C.; Qiu, B.; Mueller, S.; Braunstein, S.E. Precision based approach to tailoring radiotherapy in the multidisciplinary management of pediatric central nervous system tumors. J. Natl. Cancer Cent. 2023, 3, 141–149. [Google Scholar] [CrossRef]
- Kilburn, L.B.; Packer, R.J. Chemoterapy for Medulloblastoma-Childhood. In Handbook of Brain Tumor Chemotherapy, Molecular Therapeutics, and Immunotherapy, 2nd ed.; Newton, H.B., Ed.; Elsevier Academic Press: Amsterdam, The Netherlands, 2018; pp. 569–583. [Google Scholar]
- Eaton, B.R.; Esiashvili, N.; Kim, S.; Patterson, B.; Weymann, E.A.; Thornton, L.T.; Mazewski, C.; MacDonald, T.J.; Ebb, D.; MacDonald, S.M.; et al. Endocrine outcomes with proton and photon radiotherapy for standard risk medul-loblastoma. Neuro-Oncology 2016, 18, 881–887. [Google Scholar] [CrossRef]
- Fangusaro, J.; Bandopadhayay, P. Advances in the classification and treatment of pediatric brain tumors. Curr. Opin. Pediatr. 2020, 33, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.T.; Strother, D.R.; Judkins, A.R.; Burger, P.C.; Pollack, I.F.; Krailo, M.D.; Buxton, A.B.; Williams-Hughes, C.; Fouladi, M.; Mahajan, A.; et al. Efficacy of High-Dose Chemotherapy and Three-Dimensional Conformal Radiation for Atypical Teratoid/Rhabdoid Tumor: A Report From the Children’s Oncology Group Trial ACNS0333. J. Clin. Oncol. 2020, 38, 1175–1185. [Google Scholar] [CrossRef] [PubMed]
- Holdhof, D.; Johann, P.D.; Spohn, M.; Bbockmayr, M.; Safaei, S.; Joshi, P.; Masliah-Planchon, J.; Ho, B.; Andrianteranagna, M.; Bourdeaut, F.; et al. Atypical teratoid/rhabdoid tumors (ATRTs) with SMARCA4 muta-tion are mo-lecularly distinct from SMARCB1-deficient cases. Acta Neuropathol. 2021, 141, 291–301. [Google Scholar] [PubMed]
- Korshunov, A.; Sturm, D.; Ryzhova, M.; Hovestadt, V.; Gessi, M.; Jones, D.T.W.; Remke, M.; Northcott, P.; Perry, A.; Picard, D.; et al. Embryonal tumor with abundant neuropil and true rosettes (ETANTR), epen-dymoblastoma, and medulloepithelioma share molecular similarity and comprise a single clinico-pathological entity. Acta Neuropathol. 2014, 128, 279–289. [Google Scholar] [CrossRef]
- Loosen, A.M.; Kato, A.; Gu, X. Revisiting the role of computational neuroimaging in the era of integrative neuroscience. Neuropsychopharmacology 2024, 50, 103–113. [Google Scholar] [CrossRef]
- Uro-Coste, E.; Masliah-Planchon, J.; Siegfried, A.; Blanluet, M.; Lambo, S.; Kool, M.; Roujeau, T.; Boetto, S.; Palenzuela, G.; Bertozzi, A.-I.; et al. ETMR-like infantile cerebellar embryonal tumors in the extended mor-phologic spectrum of DICER1-related tumors. Acta Neuropathol. 2019, 137, 175–177. [Google Scholar] [CrossRef]
- Schmidt, C.; A Schubert, N.; Brabetz, S.; Mack, N.; Schwalm, B.; A Chan, J.; Selt, F.; Herold-Mende, C.; Witt, O.; Milde, T.; et al. Preclinical drug screen reveals topotecan, actinomycin D, and volasertib as potential new therapeutic candidates for ETMR brain tumor patients. Neuro-Oncology 2017, 19, 1607–1617. [Google Scholar] [CrossRef]
- Jaramillo, S.; Grosshans, D.R.; Philip, N.; Varan, A.; Akyüz, C.; McAleer, M.F.; Mahajan, A.; McGovern, S.L. Radiation for ETMR: Literature review and case series of patients treated with proton therapy. Clin. Transl. Radiat. Oncol. 2019, 15, 31–37. [Google Scholar] [CrossRef]
- Kommoss, F.K.; Chong, A.-S.; Chong, A.-L.; Pfaff, E.; Jones, D.T.W.; Hiemcke-Jiwa, L.S.; Kester, L.A.; Flucke, U.; Gessler, M.; Schrimpf, D.; et al. Genomic characterization of DICER1-associated neoplasms uncovers mo-lecular classes. Nat. Commun. 2023, 14, 1677. [Google Scholar] [CrossRef]
- Vasiljevic, A. Histopathology and molecular pathology of pediatric pineal parenchymal tumors. Childs Nerv. Syst. 2022, 39, 2273–2284. [Google Scholar] [CrossRef]
- Liu, A.P.; Gudenas, B.; Lin, T.; Orr, B.A.; Klimo Jr, P.; Kumar, R.; Bouffet, E.; Sridharan, G.; Crawford, J.R.; Kellie, S.J.; et al. Risk-adapted therapy and biological heterogeneity in pineoblastoma: Inte-grated clini-co-pathological analysis from the prospective, multi-center SJMB03 and SJYC07 trials. Acta Neuropathol. 2020, 139, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Schultz, K.A.P.; Williams, G.M.; Kamihara, J.; Stewart, D.R.; Harris, A.K.; Bauer, A.J.; Turner, J.; Shah, R.; Schneider, K.; Schneider, K.W.; et al. DICER1 and Associated Conditions: Identification of At-risk Individuals and Recommended Surveillance Strategies. Clin. Cancer Res. 2018, 24, 2251–2261. [Google Scholar] [CrossRef] [PubMed]
- Chung, P.E.D.; Gendoo, D.M.A.; Ghanbari-Azarnier, R.; Liu, J.C.; Jiang, Z.; Tsui, J.; Wang, D.-Y.; Xiao, X.; Li, B.; Dubuc, A.; et al. Modeling germline mutations in pineoblastoma uncovers lysosome disruption-based therapy. Nat. Commun. 2020, 11, 1825. [Google Scholar] [CrossRef]
- de Kock, L.; Priest, J.R.; Foulkes, W.D.; Alexandrescu, S. An update on the central nervous system manifestations of DICER1 syndrome. Acta Neuropathol. 2019, 139, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Priest, J.R.; Duchaine, T.F. DICER1: Mutations, microRNAs and mechanisms. Nat. Rev. Cancer 2014, 14, 662–672. [Google Scholar] [CrossRef]
- A Elsamadicy, A.; Koo, A.B.; David, W.B.; Lee, V.; Zogg, C.K.; Kundishora, A.J.; Hong, C.S.; DeSpenza, T.; Reeves, B.C.; Kahle, K.T.; et al. Comparison of epidemiology, treatments, and outcomes in pediatric versus adult ependymoma. Neuro-Oncol. Adv. 2020, 2, vdaa019. [Google Scholar] [CrossRef]
- Jünger, S.T.; Timmermann, B.; Pietsch, T. Pediatric ependymoma: An overview of a complex disease. Childs Nerv. Syst. 2021, 37, 2451–2463. [Google Scholar] [CrossRef]
- Rogovskii, V. Tumor-produced immune regulatory factors as a therapeutic target in cancer treatment. Front. Immunol. 2024, 15, 1416458. [Google Scholar] [CrossRef]
- Perrod, V.; Levy, R.; Tauziède-Espariat, A.; Roux, C.J.; Beccaria, K.; Blauwblomme, T.; Grill, J.; Dufour, C.; Guerrini-Rousseau, L.; Abbbou, S.; et al. Supra-tentorial Ependymomas with ZFTA Fusion, YAP1 Fu-sion, and As-troblastomas, MN1-altered: Characteristic Imaging Features. Clin. Neuroradiol. 2024, 34, 939–950. [Google Scholar] [CrossRef]
- Fu, M.; Hu, Y.; Lan, T.; Guan, K.L.; Luo, T.; Luo, M. The Hippo signalling pathway and its implications in human health and diseas-es. Signal Transduct. Target. Ther. 2022, 7, 376. [Google Scholar] [CrossRef]
- Apollonio, M.; Bellazzo, A.; Franco, N.; Lombardi, S.; Senigagliesi, B.; Casalis, L.; Parisse, P.; Thalhammer, A.; Baj, G.; Fania, R.D.F.; et al. The Tumor Suppressor DAB2IP Is Regulated by Cell Contact and Contributes to YAP/TAZ Inhibition in Confluent Cells. Cancers 2023, 15, 3379. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Yu, J.; Zheng, Y.; Chen, Q.; Zhang, N.; Pan, D. Spatial Organization of Hippo Signaling at the Plasma Membrane Mediated by the Tumor Suppressor Merlin/NF2. Cell 2013, 154, 1342–1355. [Google Scholar] [CrossRef] [PubMed]
- Jünger, S.T.; Zschernack, V.; Messing-Jünger, M.; Timmermann, B.; Pietsch, T. Ependymoma from Benign to Highly Aggressive Diseases: A Review. Adv. Tech. Stand. Neurosurg. 2024, 50, 31–62. [Google Scholar] [PubMed]
- Pajtler, K.W.; Witt, H.; Sill, M.; Jones, D.T.W.; Hovestadt, V.; Kratochwil, F.; Wani, K.; Tatevossian, R.; Punchihewa, C.; Johann, P.; et al. Molecular classification of ependymal tumors across all CNS compartments, histo-pathological grades, and age groups. Cancer Cell 2015, 27, 728–743. [Google Scholar] [CrossRef]
- Neumann, J.E.; Spohn, M.; Obrecht, D.; Mynarek, M.; Thomas, C.; Hasselblatt, M.; Dorostkar, M.M.; Wefers, A.K.; Frank, S.; Monoranu, C.-M.; et al. Molecular characterization of histopathological ependymoma variants. Acta Neuropathol. 2019, 139, 305–318. [Google Scholar] [CrossRef]
- Gessi, M.; Giagnacovo, M.; Modena, P.; Elefante, G.; Gianno, F.; Buttarelli, F.R.; Arcella, A.; Donofrio, V.; Camassei, F.D.; Nozza, P.; et al. Role of Immunohistochemistry in the Identification of Supratentorial C11ORF95-RELA Fused Ependymoma in Routine Neuropathology. Am. J. Surg. Pathol. 2019, 43, 56–63. [Google Scholar] [CrossRef]
- A Upadhyaya, S.; Robinson, G.W.; Onar-Thomas, A.; A Orr, B.; A Billups, C.; Bowers, D.C.; E Bendel, A.; Hassall, T.; Crawford, J.R.; Partap, S.; et al. Molecular grouping and outcomes of young children with newly diagnosed ependymoma treated on the multi-institutional SJYC07 trial. Neuro-Oncology 2019, 21, 1319–1330. [Google Scholar] [CrossRef]
- Andreiuolo, F.; Varlet, P.; Tauziède-Espariat, A.; Jünger, S.T.; Dörner, E.; Dreschmann, V.; Kuchelmeister, K.; Waha, A.; Haberler, C.; Slavc, I.; et al. Childhood supratentorial ependymomas with YAP1-MAMLD1 fusion: An entity with characteristic clinical, radiological, cytogenetic and histopathological features. Brain Pathol. 2019, 29, 205–216. [Google Scholar] [CrossRef]
- Pagès, M.; Pajtler, K.W.; Puget, S.; Castel, D.; Boddaert, N.; Tauziède-Espariat, A.; Picot, S.; Debily, M.A.; Kool, M.; Capper, D.; et al. Diagnostics of pediatric supratentorial RELA ependymomas: Integra-tion of infor-mation from histopathology, genetics, DNA methylation and imaging. Brain Pathol. 2019, 29, 325–335. [Google Scholar] [CrossRef]
- Mack, S.C.; Witt, H.; Piro, R.M.; Gu, L.; Zuyderduyn, S.D.; Stutz, A.M.; Wang, X.; Gallo, M.; Garzia, L.; Zayne, K.; et al. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 2014, 506, 445–450. [Google Scholar] [CrossRef]
- Panwalkar, P.; Clark, J.; Ramaswamy, V.; Hawes, D.; Yang, F.; Dunham, C.; Yip, S.; Hukin, J.; Sun, Y.; Schipper, M.J.; et al. Immunohistochemical analysis of H3K27me3 demonstrates global reduction in group-A childhood posterior fossa ependymoma and is a powerful predictor of outcome. Acta Neuropathol. 2017, 134, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Celano, E.; Salehani, A.; Malcolm, J.G.; Reinertsen, E.; Hadjipanayis, K.G. Spinal cord ependymoma: A review of the literature and case se-ries of ten pa-tients. J. Neuro-Oncol. 2016, 128, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, D.R.; Sill, M.; Okonechnikov, K.; Korshunov, A.; Yip, S.; Schutz, P.W.; Scheie, D.; Kruse, A.; Harter, P.N.; Kastelan, M.; et al. MYCN amplification drives an aggressive form of spinal ep-endymoma. Acta Neuropathol. 2019, 138, 1075–1089. [Google Scholar] [CrossRef]
- A Swanson, A.; Raghunathan, A.; Jenkins, R.B.; Messing-Jünger, M.; Pietsch, T.; Clarke, M.J.; Kaufmann, T.J.; Giannini, C. Spinal Cord Ependymomas With MYCN Amplification Show Aggressive Clinical Behavior. J. Neuropathol. Exp. Neurol. 2019, 78, 791–797. [Google Scholar] [CrossRef]
- Sato, M.; Gunther, J.R.; Mahajan, A.; Jo, E.; Paulino, A.C.; Adesina, A.M.; Jones, J.Y.; Ketonen, L.M.; Su, J.M.; Okcu, M.F.; et al. Progression-free survival of children with localized ependymoma treated with intensity-modulated radiation therapy or proton-beam radiation therapy. Cancer 2017, 123, 2570–2578. [Google Scholar] [CrossRef]
- Merchant, T.E. Current Clinical Challenges in Childhood Ependymoma: A Focused Review. J. Clin. Oncol. 2017, 35, 2364–2369. [Google Scholar] [CrossRef]
- Ellison, D.W.; Aldape, K.D.; Capper, D.; Fouladi, M.; Gilbert, M.R.; Gilbertson, R.J.; Hawkins, C.; Merchant, T.E.; Pajtler, K.; Venneti, S.; et al. cIMPACT-NOW update 7: Advancing the molecular classification of ependymal tumors. Brain Pathol. 2020, 30, 863–866. [Google Scholar] [CrossRef]
- Yeo, K.K.; Nagabushan, S.; Dhall, G.; Abdelbaki, M.S. Primary central nervous system germ cell tumors in children and young adults: A review of controversies in diagnostic and treatment approach. Neoplasia 2022, 36, 100860. [Google Scholar] [CrossRef]
- On behalf of The Intracranial Germ Cell Tumor Genome Analysis Consortium (The iGCTConsortium); Fukushima, S.; Yamashita, S.; Kobayashi, H.; Takami, H.; Fukuoka, K.; Nakamura, T.; Yamasaki, K.; Matsushita, Y.; Nakamura, H.; et al. Genome-wide methylation profiles in primary intracranial germ cell tumors indicate a primordial germ cell origin for germinomas. Acta Neuropathol. 2017, 133, 445–462. [Google Scholar] [CrossRef]
- Sonehara, K.; Kimura, Y.; Nakano, Y.; Ozawa, T.; Takahashi, M.; Suzuki, K.; Fujii, T.; Matsushita, Y.; Tomiyama, A.; Kishikawa, T.; et al. A common deletion at BAK1 reduces enhancer activity and con-fers risk of in-tracranial germ cell tumors. Nat. Commun. 2022, 13, 4478. [Google Scholar] [CrossRef]
- Atlasi, Y.; van Dorsten, R.T.; Sacchetti, A.; Joosten, R.; Oosterhuis, J.W.; Looijenga, L.H.J.; Fodde, R. Ectopic activation of WNT signaling in human embryonal carcinoma cells and its effects in short- and long-term in vitro culture. Sci. Rep. 2019, 9, 11928. [Google Scholar] [CrossRef] [PubMed]
- Satomi, K.; Takami, H.; Fukushima, S.; Yamashita, S.; Matsushita, Y.; Nakazato, Y.; Suzuki, T.; Tanaka, S.; Mukasa, A.; Saito, N.; et al. 12p gain is predominantly observed in non-germinomatous germ cell tumors and identifies an unfavorable subgroup of central nervous system germ cell tumors. Neuro-Oncology 2021, 24, 834–846. [Google Scholar] [CrossRef] [PubMed]
- Takami, H.; Fukuoka, K.; Fukushima, S.; Nakamura, T.; Mukasa, A.; Saito, N.; Yanagisawa, T.; Nakamura, H.; Sugiyama, K.; Kanamori, M.; et al. Integrated clinical, histopathological, and molecular data anal-ysis of 190 central nervous system germ cell tumors from the iGCT Consortium. Neuro Oncol. 2019, 21, 1565–1577. [Google Scholar] [CrossRef]
- Takami, H.; Elzawahry, A.; Mamatjan, Y.; Fukushima, S.; Fukuoka, K.; Suzuki, T.; Yanagisawa, T.; Matsushita, Y.; Nakamura, T.; Satomi, K.; et al. Transcriptome and methylome analysis of CNS germ cell tumor finds its cell-of-origin in embryogenesis and reveals shared similarities with testicular counterparts. Neuro-Oncology 2022, 24, 1246–1258. [Google Scholar] [CrossRef]
- Calaminus, G.; Frappaz, D.; Kortmann, R.D.; Krefeld, B.; Saran, F.; Pietsch, T.; Vasiljevic, A.; Garre, M.L.; Ricardi, U.; Mann, J.R.; et al. Outcome of patients with intracranial non-germinomatous germ cell tumors—Lessons from the SIOP-CNS-GCT-96 trial. Neuro-Oncology 2017, 19, 1661–1672. [Google Scholar] [CrossRef]
- Diezi, M.; Pizer, B.; Murray, M.J. Overview of current European practice for the management of patients with intracranial germ cell tumours. EJC Paediatr. Oncol. 2024, 3, 100146. [Google Scholar] [CrossRef]
- Murray, M.J.; Bartels, U.; Nishikawa, R.; Fangusaro, J.; Matsutani, M.; Nicholson, J.C. Consensus on the management of intracranial germ-cell tumours. Lancet Oncol. 2015, 16, e470–e477. [Google Scholar] [CrossRef]
- Ronsley, R.; Bouffet, E.; Dirks, P.; Drakes, J.; Kulkarni, A.; Bartels, U. Successful management of symptomatic hydrocephalus using a tempo-rary external ventricular drain with or without endoscopic third ventriculostomy in pediatric patients with ger-minoma. J. Neurosurg. 2021, 137, 807–812. [Google Scholar] [CrossRef]
- Kellie, S.J.; Boyce, H.; Dunkel, I.J.; Diez, B.; Rosenblum, M.; Brualdi, L.; Finlay, J.L. Intensive cisplatin and cyclophosphamide-based chemotherapy without radiotherapy for intracranial germinomas: Failure of a primary chemotherapy approach. Pediatr. Blood Cancer 2004, 43, 126–133. [Google Scholar] [CrossRef]
- Kretschmar, C.; Kleinberg, L.; Greenberg, M.; Burger, P.; Holmes, E.; Wharam, M. Pre-radiation chemotherapy with response-based radiation therapy in children with central nervous system germ cell tumors: A report from the Childrens Oncology Group. Pediatr. Blood Cancer 2007, 48, 285–291. [Google Scholar] [CrossRef]
- Morana, G.; Shaw, D.; Macdonald, S.M.; Alapetite, C.; Ajitkumar, T.; Bhatia, A.; Brisse, H.; Jaimes, C.; Czech, T.; Dhall, G.; et al. Imaging response assessment for CNS germ cell tumours: Consensus recom-mendations from the European Society for Paediatric Oncology Brain Tumour Group and North American Children’s Oncology Group. Lancet Oncol. 2022, 23, e218–e228. [Google Scholar] [CrossRef] [PubMed]
- Calaminus, G.; Kortmann, R.; Worch, J.; Nicholson, J.C.; Alapetite, C.; Garré, M.L.; Patte, C.; Ricardi, U.; Saran, F.; Frappaz, D.; et al. SIOP CNS GCT96: Final report of outcome of a prospective, multinational nonrandomized trial for children and adults with intracranial germinoma, comparing craniospinal irra-diation alone with chemotherapy followed by focal primary site irradiation for patients with localized disease. Neuro Oncol. 2013, 15, 788–796. [Google Scholar] [PubMed]
- Calaminus, G.; Bison, B.; Conter, C.F.; Frappaz, D.; Peyrl, A.; Gerber, N.U.; Müller, J.-E.; Ajithkumar, T.; Morana, G.; Cross, J.; et al. GCT-11. 24 Gy whole ventricular radiotherapy alone is sufficient for disease control in localised germinoma in CR after initial chemotherapy—Final of the SIOP CNS GCT II study. Neuro-Oncology 2022, 24, i56. [Google Scholar] [CrossRef]
- Dunkel, I.J.; Doz, F.; Foreman, N.K.; Hargrave, D.; Lassaletta, A.; André, N.; Hansford, J.R.; Hassall, T.; Eyrich, M.; Gururangan, S.; et al. Nivolumab with or without ipilimumab in pediatric patients with high-grade CNS malignancies: Safety, efficacy, biomarker, and pharmacokinetics—CheckMate 908. Neuro-Oncology 2023, 25, 1530–1545. [Google Scholar] [CrossRef]
- Cupit-Link, M.; Federico, S.M. Treatment of High-Risk Neuroblastoma with Dinutuximab and Chemotherapy Administered in all Cycles of Induction. Cancers 2023, 15, 4609. [Google Scholar] [CrossRef]
- Markham, A. Naxitamab: First Approval. Drugs 2021, 81, 291–296. [Google Scholar] [CrossRef]
- Filbert, E.L.; Björck, P.K.; Srivastava, M.K.; Bahjat, F.R.; Yang, X. APX005M, a CD40 agonist antibody with unique epitope specificity and Fc receptor binding profile for optimal therapeutic application. Cancer Immunol. Immunother. 2021, 70, 1853–1865. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Restifo, N.P.; Yang, J.C.; Morgan, R.A.; Dudley, M.E. Adoptive cell transfer: A clinical path to effective cancer immunotherapy. Nat. Rev. Cancer 2008, 8, 299–308. [Google Scholar] [CrossRef]
- Sagnella, S.M.; White, A.L.; Yeo, D.; Saxena, P.; van Zandwijk, N.; Rasko, J.E. Locoregional delivery of CAR-T cells in the clinic. Pharmacol. Res. 2022, 182, 106329. [Google Scholar] [CrossRef]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef]
- Thompson, E.; Landi, D.; Archer, G.; Lipp, E.; Walter, A.; Archambault, B.; Balajonda, B.; Flahiff, C.; Jaggers, D.; Herndon, J.; et al. EPCT-01. A novel peptide vaccine directed to cmv pp65 for treatment of recurrent malignant glioma and medulloblastoma in children and young adults: Preliminary results of a phase i trial. Neuro-Oncology 2021, 23, i46. [Google Scholar] [CrossRef]
- Ahluwalia, M.S.; Reardon, D.A.; Abad, A.P.; Curry, W.T.; Wong, E.T.; Figel, S.A.; Mechtler, L.L.; Peereboom, D.M.; Hutson, A.D.; Withers, H.G.; et al. Phase IIa Study of SurVaxM Plus Adjuvant Temozolomide for Newly Diagnosed Glioblastoma. J. Clin. Oncol. 2023, 41, 1453–1465. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.S.; Mitchell, D.A. Gene-modified dendritic cell vaccines for cancer. Cytotherapy 2016, 18, 1446–1455. [Google Scholar] [CrossRef] [PubMed]
- Asija, S.; Chatterjee, A.; Goda, J.S.; Yadav, S.; Chekuri, G.; Purwar, R. Oncolytic immunovirotherapy for high-grade gliomas: A novel and an evolving therapeutic option. Front. Immunol. 2023, 14, 1118246. [Google Scholar] [CrossRef]
- Friedman, G.K.; Johnston, J.M.; Bag, A.K.; Bernstock, J.D.; Li, R.; Aban, I.; Kachurak, K.; Nan, L.; Kang, K.-D.; Totsch, S.; et al. Oncolytic HSV-1 G207 Immunovirotherapy for Pediatric High-Grade Gliomas. N. Engl. J. Med. 2021, 384, 1613–1622. [Google Scholar] [CrossRef]
- Nassiri, F.; Patil, V.; Yefet, L.S.; Singh, O.; Liu, J.; Dang, R.M.A.; Yamaguchi, T.N.; Daras, M.; Cloughesy, T.F.; Colman, H.; et al. Oncolytic DNX-2401 virotherapy plus pembrolizumab in recurrent glioblastoma: A phase 1/2 trial. Nat. Med. 2023, 29, 1370–1378. [Google Scholar] [CrossRef]
- Mochizuki, A.Y.; Frost, I.M.; Mastrodimos, M.B.; Plant, A.S.; Wang, A.C.; Moore, T.B.; Prins, R.M.; Weiss, P.S.; Jonas, S.J. Precision Medicine in Pediatric Neurooncology: A Review. ACS Chem. Neurosci. 2017, 9, 11–28. [Google Scholar] [CrossRef]
- D’amico, R.S.; Aghi, M.K.; Vogelbaum, M.A.; Bruce, J.N. Convection-enhanced drug delivery for glioblastoma: A review. J. Neuro-Oncol. 2021, 151, 415–427. [Google Scholar] [CrossRef]
- Wang, W.; I Marín-Ramos, N.; He, H.; Zeng, S.; Cho, H.-Y.; Swenson, S.D.; Zheng, L.; Epstein, A.L.; Schönthal, A.H.; Hofman, F.M.; et al. NEO100 enables brain delivery of blood–brain barrier impermeable therapeutics. Neuro-Oncology 2020, 23, 63–75. [Google Scholar] [CrossRef]
- Puccetti, D.; Otto, M.; Morgenstern, D.; DeSantes, K.; Cho, S.; Hall, L.; Oliver, K.; Longcor, J. EPCT-09. CLR 131 in patients with relapsed or refractory pediatric malignancies. Neuro-Oncology 2020, 22, iii305. [Google Scholar] [CrossRef]
- Rao, R.; Patel, A.; Hanchate, K.; Robinson, E.; Edwards, A.; Shah, S.; Higgins, D.; Haworth, K.J.; Lucke-Wold, B.; Krummel, D.P.; et al. Advances in Focused Ultrasound for the Treatment of Brain Tumors. Tomography 2023, 9, 1094–1109. [Google Scholar] [CrossRef]
- Goldman, S.; Margol, A.; I Hwang, E.; Tanaka, K.; Suchorska, B.; Crawford, J.R.; Kesari, S. Safety of Tumor Treating Fields (TTFields) therapy in pediatric patients with malignant brain tumors: Post-marketing surveil-lance data. Front. Oncol. 2022, 29, 958637. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Mutation | Incidence | Major Tumor Groups |
|---|---|---|
| FGFR1-TKD Duplications | 7.4–24% | Diffuse gliomas or dysembryoblastic neuroepithelial tumor |
| FGFR1-TACC1 fusions | 3% | High-grade gliomas extra ventricular neurocytomas |
| FGFR point alterations | 9.6% | Low-grade gliomas |
| Wnt | SHH | Group 3 | Group 4 | |
|---|---|---|---|---|
| Age | Older children | Infants, older children, adults | Younger children | All age groups |
| Location | CPA | Cerebellar hemisphere | Midline or 4th ventricle | Midline or 4th ventricle |
| Survival | Very good. Independent of p53 | Intermediate. It depends on p53 | Poor | Intermediate |
| Incidence | 10% | 30% | 20% | 40% |
| M/F | 1:1 | 1:1 | 2:1 | 3:1 |
| Tumorigenesis pathway | CTNNB1 mutation, 6 monosomy | PTCHD1, SMO, SUFU, GLI 1, 2 MYC amplification LI2, MYC | Amplifications of MYC, OTX2, i17q | I17q, MYCN and OTX2 amplifications, duplication of SNCAIP |
| Modalities | Name of Medication | Mechanism | Tumor Group | |
|---|---|---|---|---|
| Biology | Monoclonal antibodies | Pembrolizumab Nivolumab [201] | PD-1/PD-L1 inhibitor | High-grade neoplasias of CNS |
| Ipilimumab [201] | CTLA-4PD-1/PD-L1 inhibitor | |||
| Dinutuximab [202] | Anti-GD2 | |||
| Naxitimab [203] | Humanized IgG1 anti-GD2 | |||
| APX005M [204] | CD40 signaling initiation | DIPG | ||
| Adoptive cell transfer | Autologous or allogenic NK or T-cell [205] Locoregional placement of chimeric antigen T-cells intraventricularly or in tumor bed [206] IL13Ra2 allogenic T-cells with chimeric antigen receptors [207] | Chimeric antigen receptors specific to cancer cell antigens (EGFR, GD2, HER2, and IL13Ra2) | Glioblastomas and high-grade gliomas | |
| Cancer vaccines | PEP-CMV [208] Survivin [209] | Tumor antigens induce epitopes which are presented by APCs to cytotoxic T-lympocytes | Recurrent malignant gliomas or medulloblastomas | |
| Dendritic cell vaccines [210] | ||||
| Oncolytic virus therapy [211] | HSV-1 G207 [212] DNX-2401 [213] (tasadenoturev or Delta-24-RGD) PVSRIPO Cytokine transgenes (IL-12, IL-15) Immune-activating molecules (OX40, GM-CSF) [212] | Direct oncolysis (apoptosis) or immune-mediated cell death | Recurrent high-grade gliomas | |
| Precision Medicine | Cellular pathways inhibitors [214] | Emurafenib Selumetinib, Trametinib Olutasidenib, Ivosidenib Lenvatinib Tovorafeib Avapritinib | BRAF inhibitors MEK inhibitors VEGF inhibitors Anti-VEGF receptor RAF kinase inhibitor KIT or PDGFRA inhibitors | Gliomas Gliomas IDH mutant gliomas IDH mutant gliomas Germinomas and high-grade gliomas |
| Novel Drug Delivery Systems for CNS Tumors | Alteration of tumor microenvironment [215] | NEO100 Intranasally [216] LAM-561 oral treatment CLR 131 [217] Ultrasound device ‘Exablate’ + doxorubicin [218] Sonocloud 9 device + carboplatin Tumor-treating fields [219] | It increases BBB permeability Modifies BBB permeability Targeted delivery of radioisotope I131 Targeted disruption of BBB Targeted disruption of BBB Locoregional, noninvasive electric fields | Primary CNS neoplasias Gliomas Refractory pediatric malignancies DIPG High-grade gliomas embryonal tumors High-grade gliomas ependymomas |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antoniades, E.; Keffes, N.; Vorri, S.; Tsitouras, V.; Gkantsinikoudis, N.; Tsitsopoulos, P.; Magras, J. The Molecular Basis of Pediatric Brain Tumors: A Review with Clinical Implications. Cancers 2025, 17, 1566. https://doi.org/10.3390/cancers17091566
Antoniades E, Keffes N, Vorri S, Tsitouras V, Gkantsinikoudis N, Tsitsopoulos P, Magras J. The Molecular Basis of Pediatric Brain Tumors: A Review with Clinical Implications. Cancers. 2025; 17(9):1566. https://doi.org/10.3390/cancers17091566
Chicago/Turabian StyleAntoniades, Elias, Nikolaos Keffes, Stamatia Vorri, Vassilios Tsitouras, Nikolaos Gkantsinikoudis, Parmenion Tsitsopoulos, and John Magras. 2025. "The Molecular Basis of Pediatric Brain Tumors: A Review with Clinical Implications" Cancers 17, no. 9: 1566. https://doi.org/10.3390/cancers17091566
APA StyleAntoniades, E., Keffes, N., Vorri, S., Tsitouras, V., Gkantsinikoudis, N., Tsitsopoulos, P., & Magras, J. (2025). The Molecular Basis of Pediatric Brain Tumors: A Review with Clinical Implications. Cancers, 17(9), 1566. https://doi.org/10.3390/cancers17091566