
Aberrant Splicing as a Mechanism for Resistance to Cancer Therapies



Simple Summary
Abstract
1. Introduction
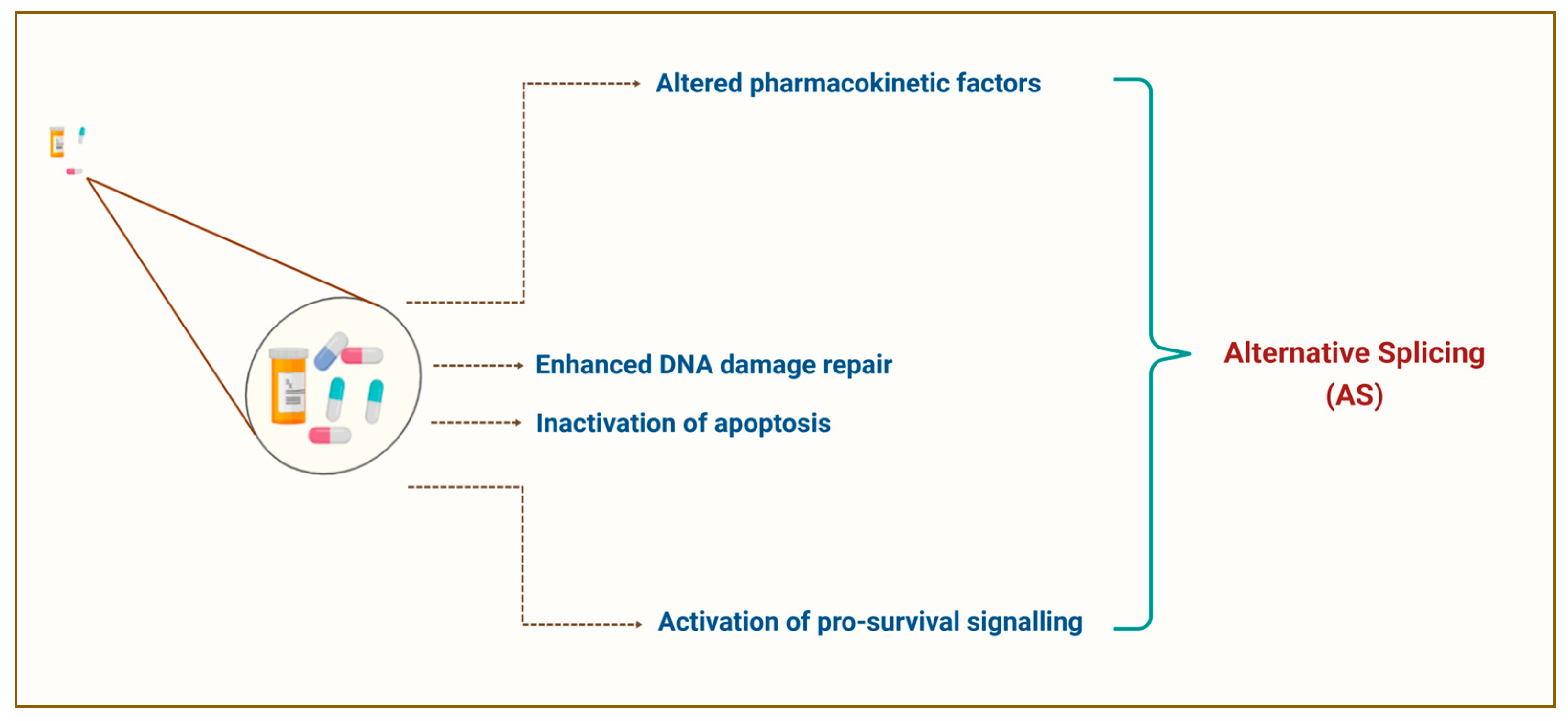
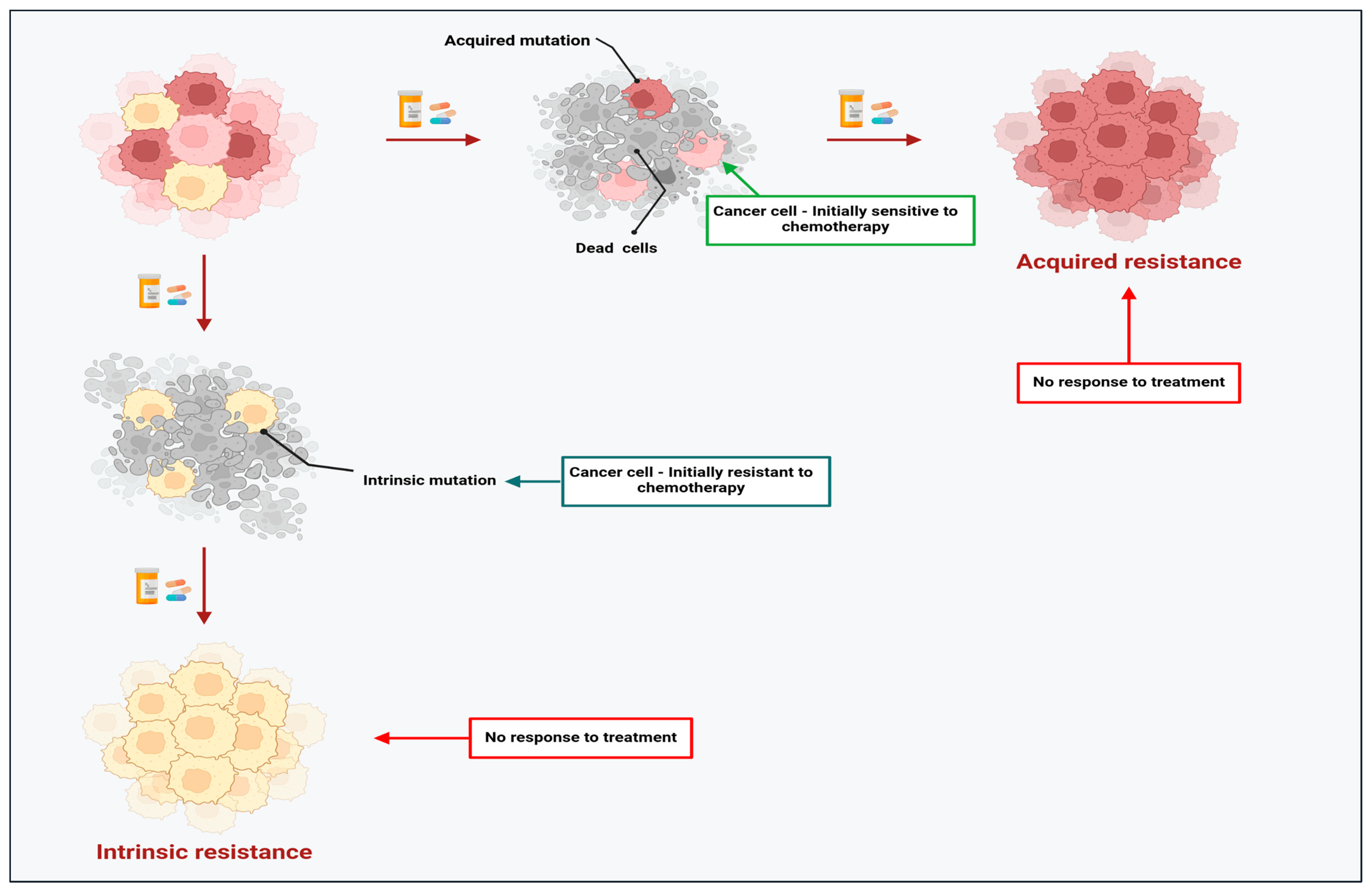
2. The Dark Side of Chemotherapy: Resistance Limits the Success of Chemotherapeutic Agents in Cancer
3. Pre-mRNA Alternative Splicing: A Promising Avenue to Novel Therapeutic Prospects in Cancer Chemoresistance
- -
- Cassette exons: an exon may be excluded or included;
- -
- Alternative 3′ splice site: a different 3′ splice junction is utilised, changing the 5′ boundary of the downstream exon;
- -
- Alternative 5′ splice site: AS at the 5′ splice site modifies the 3′ splice site of the upstream exon;
- -
- Mutually exclusive exons: different splice variants are generated from various exon combinations, but only one is spliced in the same transcript simultaneously;
- -
- Intron retention: mature mRNA transcript retains the intronic sequence that is present upon the completion of transcript processing, and it becomes a new coding sequence.
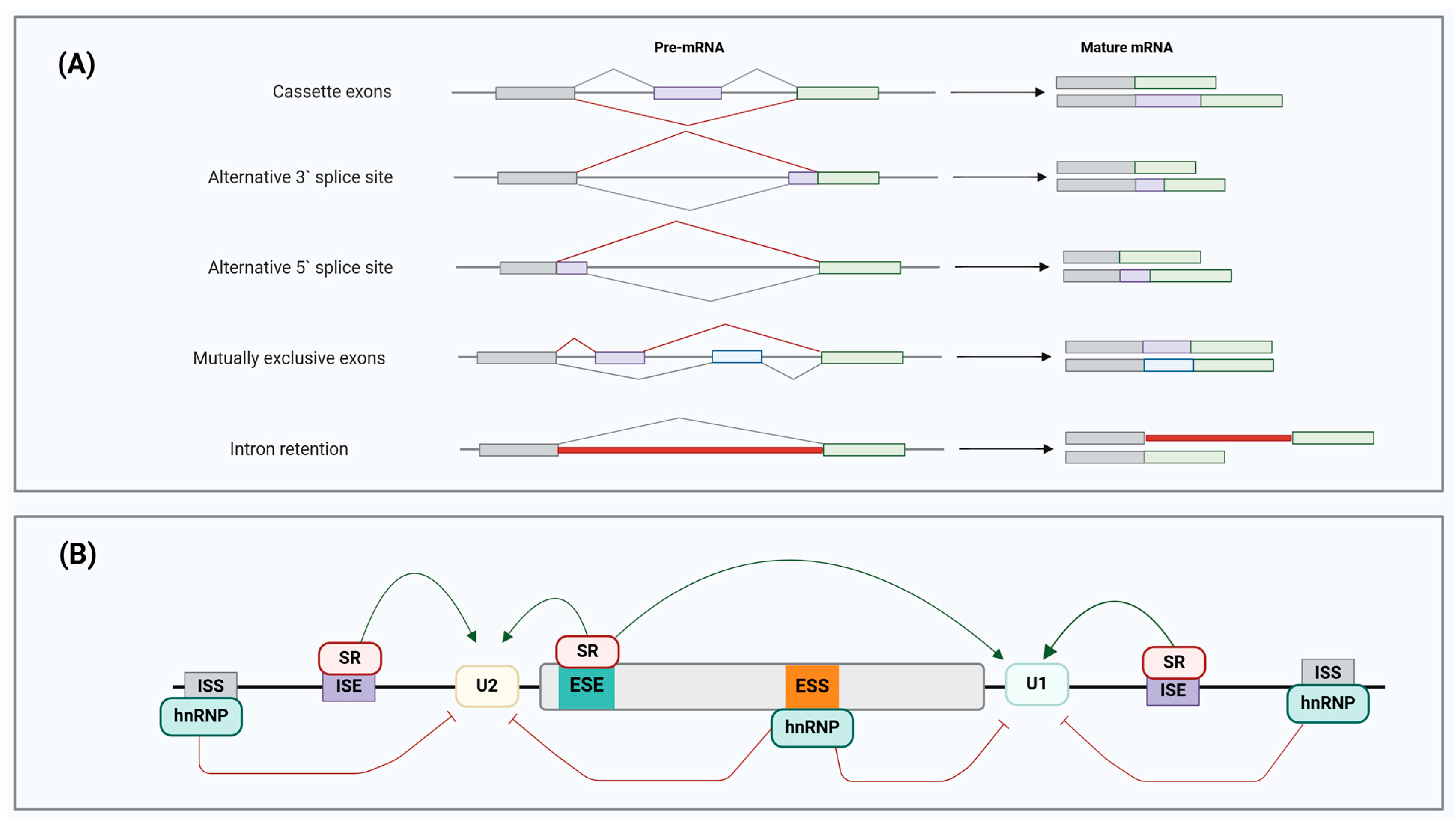
4. Aberrant Pre-mRNA Alternative Splicing as a Common Hallmark of Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Spliced Isoform | Biological | AS Pattern | Cancer | Reference |
|---|---|---|---|---|---|
| BIN1 | BIN1_12A | Anti-apoptotic | Cassette exons | NSCLC | [109] |
| CASP2 | Casp-2L | Pro-apoptotic | Cassette exons | CRC | [117] |
| CCND1 | Cyclin D1b | Pro-proliferative | Intron retention | HCC | [118] |
| CD44 | CD44s | Pro-invasive | Cassette exons | GBC | [119] |
| MENA | MenaINV | Pro-invasive | Cassette exons | BRCA | [120] |
| MKNK2 | MNK2-b | Anti-apoptotic | Cassette exons | BRCA | [121] |
| RAC1 | Rac1b | Pro-metastatic | Cassette exons | LUAD | [122] |
| RON | RONΔ165 | Pro-invasive | Cassette exons | CRC | [123] |
| MNMD4 | MDM4-FL | Anti-apoptotic | Cassette exons | BRCA | [124] |
| FAS | FASL | Pro-apoptotic | Cassette exons | NSCLC | [125] |
| PKM | PKM2 | Promote aerobic glycolysis | Mutually exclusive exons | BRCA | [126] |
| VEGF | VEGF-165b | Anti-angiogenesis | Alternative 3′ splice site | PC-3 | [112] |
5. Unravelling the ‘Rosetta Stone’ of Cancer Chemotherapy: Potential Strategies to Target Abnormal Alternative Splicing
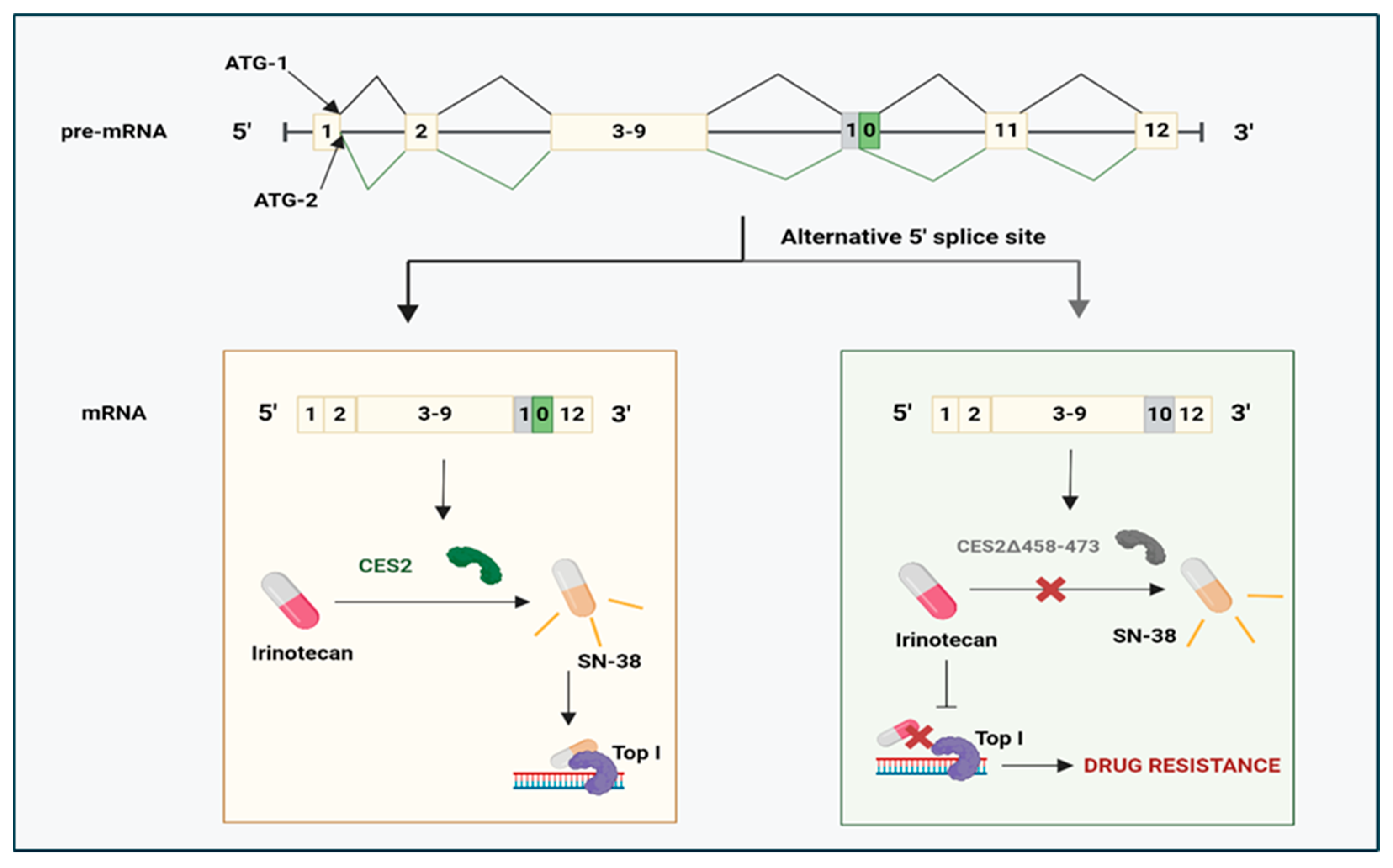
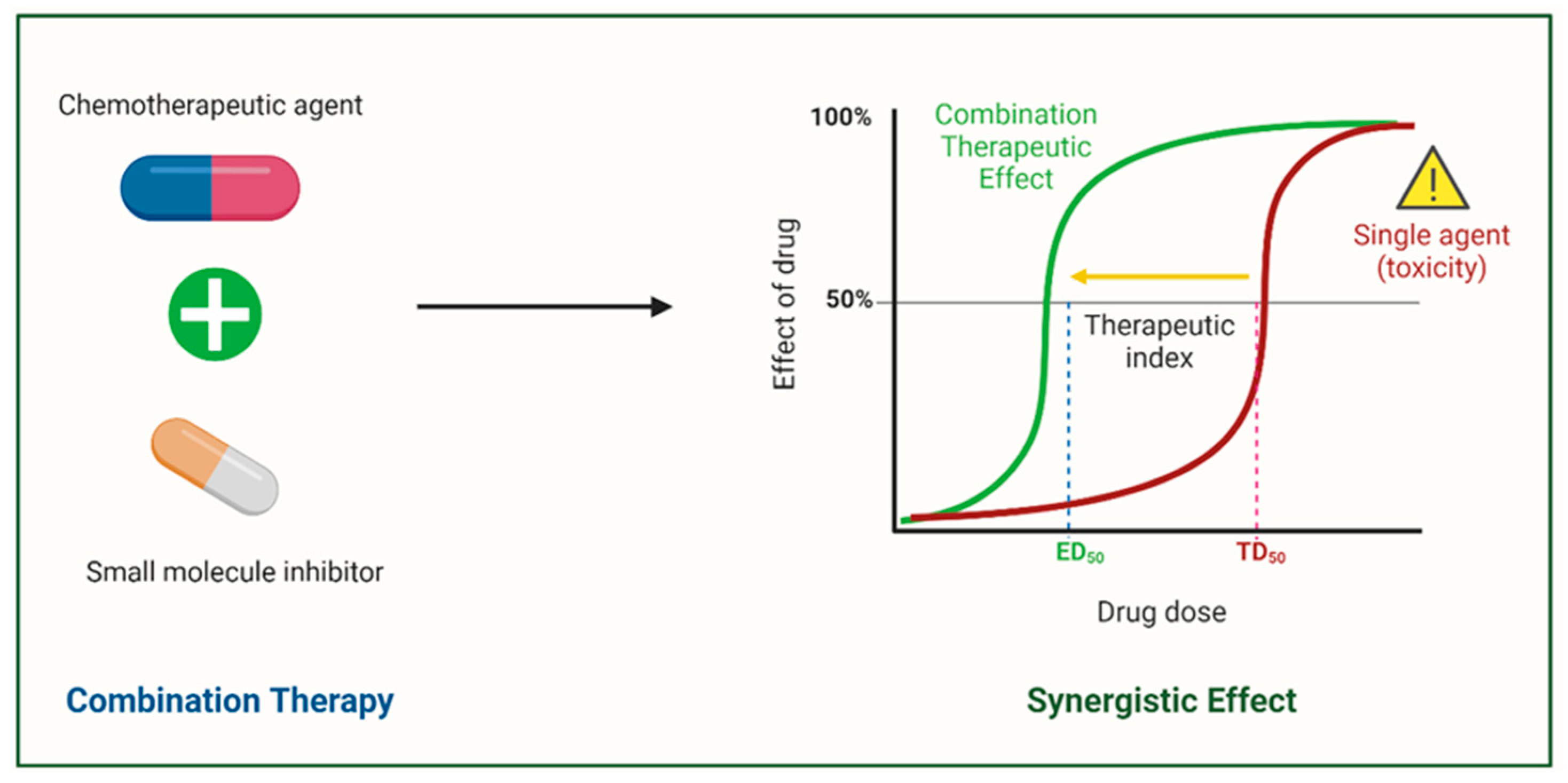
6. Rationale of Combination Cancer Therapy for Correcting Abnormal Splicing Errors
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Giaquinto, A.N.; Ahmedin, J.; Siegel, R.L. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef] [PubMed]
- Crosby, D.; Bhatia, S.; Brindle, K.M.; Coussens, L.M.; Dive, C.; Emberton, M.; Esener, S.; Fitzgerald, R.C.; Gambhir, S.S.; Kuhn, P.; et al. Early detection of cancer. Science 2022, 375, e9040. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.J.; Pillai, G.G.; Andrade, C.J.; Ligibel, J.A.; Basu, P.; Cohen, L.; Khan, I.A.; Mustian, K.M.; Puthiyedath, R.; Dhiman, K.S.; et al. Integrative oncology: Addressing the global challenges of cancer prevention and treatment. CA Cancer J. Clin. 2022, 72, 144–164. [Google Scholar] [CrossRef] [PubMed]
- Van Herck, Y.; Feyaerts, A.; Alibhai, S.; Papamichael, D.; Decoster, L.; Lambrechts, Y.; Pinchuk, M.; Bechter, O.; Herrera-Caceres, J.; Bibeau, F.; et al. Is cancer biology different in older patients? Lancet Healthy Longev. 2021, 2, e663–e677. [Google Scholar] [CrossRef]
- Thun, M.J.; DeLancey, J.O.; Center, M.M.; Jemal, A.; Ward, E.M. The global burden of cancer: Priorities for prevention. Carcinogenesis 2010, 31, 100–110. [Google Scholar] [CrossRef]
- Alfarouk, K.O.; Stock, C.M.; Taylor, S.; Walsh, M.; Muddathir, A.K.; Verduzco, D.; Bashir, A.H.H.; Mohammed, O.Y.; Elhassan, G.O.; Harguindey, S.; et al. Resistance to cancer chemotherapy: Failure in drug response from ADME to P-Gp. Cancer Cell Int. 2015, 15, 71. [Google Scholar] [CrossRef]
- Cree, I.A.; Charlton, P. Molecular chess? Hallmarks of anti-cancer drug resistance. BMC Cancer 2017, 17, 10. [Google Scholar] [CrossRef]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef]
- Urruticoechea, A.; Alemany, R.; Balart, J.; Villanueva, A.; Vinals, F.; Capella, G. Recent advances in cancer therapy: An overview. Curr. Pharm. Des. 2009, 16, 3–10. [Google Scholar] [CrossRef]
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.W. Cancer and radiation therapy: Current advances and future directions. Int. J. Med. Sci. 2012, 9, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Khalil, D.N.; Smith, E.L.; Brentjens, R.J.; Wolchok, J.D. The future of cancer treatment: Immunomodulation, CARs and combination immunotherapy. Nat. Rev. Clin. Oncol. 2016, 13, 273–290. [Google Scholar] [CrossRef]
- Haider, T.; Pandey, V.; Banjare, N.; Gupta, P.N.; Soni, V. Drug resistance in cancer: Mechanisms and tackling strategies. Pharmacol. Rep. 2020, 72, 1125–1151. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.U.; Fatima, K.; Aisha, S.; Malik, F. Unveiling the mechanisms and challenges of cancer drug resistance. Cell Commun. Signal. 2024, 22, 109. [Google Scholar] [CrossRef] [PubMed]
- Bidram, E.; Esmaeili, Y.; Ranji-Burachaloo, H.; Al-Zaubai, N.; Zarrabi, A.; Stewart, A.; Dunstan, D.E. A concise review on cancer treatment methods and delivery systems. J. Drug Deliv. Sci. Technol. 2019, 54, 101350. [Google Scholar] [CrossRef]
- Gao, Y.; Koide, K. Chemical perturbation of Mcl-1 pre-mRNA splicing to induce apoptosis in cancer cells. ACS Chem. Biol. 2013, 8, 895–900. [Google Scholar] [CrossRef]
- Miller, K.D.; Nogueira, L.; Devasia, T.; Mariotto, A.B.; Yabroff, K.R.; Jemal, A.; Kramer, J.; Siegel, R.L. Cancer treatment and survivorship statistics, 2022. CA Cancer J. Clin. 2022, 72, 409–436. [Google Scholar] [CrossRef]
- Kamran, S.C.; Efstathiou, J.A. Current state of personalized genitourinary cancer radiotherapy in the era of precision medicine. Front. Oncol. 2021, 11, 675311. [Google Scholar] [CrossRef]
- Xiang, Y.; Liu, X.; Wang, Y.; Zheng, D.; Meng, Q.; Jiang, L.; Yang, S.; Zhang, S.; Zhang, X.; Liu, Y.; et al. Mechanisms of resistance to targeted therapy and immunotherapy in non-small cell lung cancer: Promising strategies to overcoming challenges. Front. Immunol. 2024, 15, 1366260. [Google Scholar] [CrossRef]
- Zheng, H.C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef]
- Tilsed, C.M.; Fisher, S.A.; Nowak, A.K.; Lake, R.A.; Lesterhuis, W.J. Cancer chemotherapy: Insights into cellular and tumor microenvironmental mechanisms of action. Front. Oncol. 2022, 12, 960317. [Google Scholar] [CrossRef] [PubMed]
- Eslami, M.; Memarsadeghi, O.; Davarpanah, A.; Arti, A.; Nayernia, K.; Behnam, B. Overcoming chemotherapy resistance in metastatic cancer: A comprehensive review. Biomedicines 2024, 12, 183. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2019, 2, 141–160. [Google Scholar] [CrossRef]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The different mechanisms of cancer drug resistance: A brief review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef]
- Longley, D.B.; Johnston, P.G. Molecular mechanisms of drug resistance. J. Pathol. 2005, 205, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Al Saihati, H.A.; Rabaan, A.A. Cellular resistance mechanisms in cancer and the new approaches to overcome resistance mechanisms chemotherapy. Saudi Med. J. 2023, 44, 329–344. [Google Scholar] [CrossRef]
- Cetin, R.; Quandt, E.; Kaulich, M. Functional genomics approaches to elucidate vulnerabilities of intrinsic and acquired chemotherapy resistance. Cells 2021, 10, 260. [Google Scholar] [CrossRef]
- Bonnal, S.C.; López-Oreja, I.; Valcárcel, J. Roles and mechanisms of alternative splicing in cancer—Implications for care. Nat. Rev. Clin. Oncol. 2020, 17, 457–474. [Google Scholar] [CrossRef]
- Danan-Gotthold, M.; Golan-Gerstl, R.; Eisenberg, E.; Meir, K.; Karni, R.; Levanon, E.Y. Identification of recurrent regulated alternative splicing events across human solid tumors. Nucleic Acids Res. 2015, 43, 5130–5144. [Google Scholar] [CrossRef]
- Van Den Hoogenhof, M.M.G.; Pinto, Y.M.; Creemers, E.E. RNA splicing. Circ. Res. 2016, 118, 454–468. [Google Scholar] [CrossRef]
- Cambindo Botto, A.E.; Muñoz, J.C.; Giono, L.E.; Nieto-Moreno, N.; Cuenca, C.; Kornblihtt, A.R.; Muñoz, M.J. Reciprocal regulation between alternative splicing and the DNA damage response. Genet. Mol. Biol. 2020, 43, e20190111. [Google Scholar] [CrossRef]
- Wang, B.D.; Lee, N.H. Aberrant RNA splicing in cancer and drug resistance. Cancers 2018, 10, 458. [Google Scholar] [CrossRef] [PubMed]
- Reviejo, M.; Soto, M.; Lozano, E.; Asensio, M.; Martínez-Augustin, O.; Sánchez de Medina, F.; Marin, J.J.G. Impact of alternative splicing on mechanisms of resistance to anticancer drugs. Biochem. Pharmacol. 2021, 193, 114810. [Google Scholar] [CrossRef]
- Wang, J.; Seebacher, N.; Shi, H.; Kan, Q.; Duan, Z. Novel strategies to prevent the development of multi-drug resistance (MDR) in cancer. Oncotarget 2017, 8, 84559–84571. [Google Scholar] [CrossRef]
- Mellor, H.R.; Callaghan, R. Resistance to chemotherapy in cancer: A complex and integrated cellular response. Pharmacology 2008, 81, 275–300. [Google Scholar] [CrossRef]
- Pan, S.T.; Li, Z.L.; He, Z.X.; Qiu, J.X.; Zhou, S.F. Molecular mechanisms for tumor resistance to chemotherapy. Clin. Exp. Pharmacol. Physiol. 2016, 43, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Briffa, R.; Langdon, S.P.; Grech, G.; Harrison, D.J. Acquired and intrinsic resistance to colorectal cancer treatment. In Colorectal Cancer—Diagnosis, Screening and Management; IntechOpen: London, UK, 2017. [Google Scholar] [CrossRef]
- Bray, S.M.; Lee, J.; Kim, S.T.; Hur, J.Y.; Ebert, P.J.; Calley, J.N.; Wulur, I.H.; Gopalappa, T.; Wong, S.S.; Qian, H.R.; et al. Genomic characterization of intrinsic and acquired resistance to cetuximab in colorectal cancer patients. Sci. Rep. 2019, 9, 15365. [Google Scholar] [CrossRef]
- Kartal-Yandim, M.; Adan-Gokbulut, A.; Baran, Y. Molecular mechanisms of drug resistance and its reversal in cancer. Crit. Rev. Biotechnol. 2016, 36, 716–726. [Google Scholar] [CrossRef]
- Ji, X.; Lu, Y.; Tian, H.; Meng, X.; Wei, M.; Cho, W.C. Chemoresistance mechanisms of breast cancer and their countermeasures. Biomed. Pharmacother. 2019, 114, 108800. [Google Scholar] [CrossRef]
- Liu, F.W.; Tewari, K.S. New targeted agents in gynecologic cancers: Synthetic lethality, homologous recombination deficiency, and PARP inhibitors. Curr. Treat. Options Oncol. 2016, 17, 12. [Google Scholar] [CrossRef]
- Hassen, W.; Kassambara, A.; Reme, T.; Sahota, S.; Seckinger, A.; Vincent, L.; Cartron, G.; Moreaux, J.; Hose, D.; Klein, B.; et al. Drug metabolism and clearance system in tumor cells of patients with multiple myeloma. Oncotarget 2014, 6, 6431–6447. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Lei, Y.H.; Yao, N.; Wang, C.R.; Hu, N.; Ye, W.C.; Zhang, D.M.; Chen, Z.S. Autophagy and multidrug resistance in cancer. Chin. J. Cancer 2017, 36, 52. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Li, X.; Lin, M.; Yue, W.X. Meta-analysis of P53 expression and sensitivity to platinum-based chemotherapy in patients with non-small cell lung cancer. Medicine 2021, 100, e24194. [Google Scholar] [CrossRef] [PubMed]
- Tung, M.-C.; Lin, P.-L.; Wang, Y.-C.; He, T.-Y.; Lee, M.-C.; Yeh, S.-D.; Chen, C.-Y.; Lee, H. Mutant P53 confers chemoresistance in non-small cell lung cancer by upregulating Nrf2. Oncotarget 2015, 6, 41692–41705. [Google Scholar] [CrossRef]
- Martínez-Castillo, M.; Gómez-Romero, L.; Tovar, H.; Olarte-Carrillo, I.; García-Laguna, A.; Barranco-Lampón, G.; De la Cruz-Rosas, A.; Martínez-Tovar, A.; Hernández-Zavala, A.; Córdova, E.J. Genetic alterations in the BCR-ABL1 fusion gene related to imatinib resistance in chronic myeloid leukemia. Leuk. Res. 2023, 131, 107325. [Google Scholar] [CrossRef]
- Prado-Carrillo, O.; Arenas-Ramírez, A.; Llaguno-Munive, M.; Jurado, R.; Pérez-Rojas, J.; Cervera-Ceballos, E.; Garcia-Lopez, P. Ketoconazole reverses imatinib resistance in human chronic myelogenous leukemia K562 cells. Int. J. Mol. Sci. 2022, 23, 7715. [Google Scholar] [CrossRef]
- Soverini, S.; Bavaro, L.; de Benedittis, C.; Martelli, M.; Iurlo, A.; Orofino, N.; Sica, S.; Sorà, F.; Lunghi, F.; Ciceri, F.; et al. Prospective assessment of NGS-detectable mutations in CML patients with nonoptimal response: The NEXT-in-CML study. Blood 2020, 135, 534–541. [Google Scholar] [CrossRef]
- Ward, R.A.; Fawell, S.; Floc’H, N.; Flemington, V.; McKerrecher, D.; Smith, P.D. Challenges and opportunities in cancer drug resistance. Chem. Rev. 2021, 121, 3297–3351. [Google Scholar] [CrossRef]
- Kathawala, R.J.; Gupta, P.; Ashby, C.R.; Chen, Z.S. The modulation of ABC transporter-mediated multi-drug resistance in cancer: A review of the past decade. Drug Resist. Updat. 2015, 18, 1–17. [Google Scholar] [CrossRef]
- Vesel, M.; Rapp, J.; Feller, D.; Kiss, E.; Jaromi, L.; Meggyes, M.; Miskei, G.; Duga, B.; Smuk, G.; Laszlo, T.; et al. ABCB1 and ABCG2 drug transporters are differentially expressed in non-small cell lung cancers (NSCLC) and expression is modified by cisplatin treatment via altered Wnt signaling. Respir. Res. 2017, 18, 52. [Google Scholar] [CrossRef]
- Xiao, H.; Zheng, Y.; Ma, L.; Tian, L.; Sun, Q. Clinically-relevant ABC transporter for anti-cancer drug resistance. Front. Pharmacol. 2021, 12, 648407. [Google Scholar] [CrossRef]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Orellana-Serradell, O.; Herrera, D.; Castellon, E.A.; Contreras, H.R. The transcription factor ZEB1 promotes chemoresistance in prostate cancer cell lines. Asian J. Androl. 2019, 21, 460–467. [Google Scholar] [CrossRef]
- Demidenko, R.; Razanauskas, D.; Daniunaite, K.; Lazutka, J.R.; Jankevicius, F.; Jarmalaite, S. Frequent down-regulation of ABC transporter genes in prostate cancer. BMC Cancer 2015, 15, 683. [Google Scholar] [CrossRef]
- Kuo, M.T.; Huang, Y.F.; Chou, C.Y.; Chen, H.H.W. Targeting the copper transport system to improve treatment efficacies of platinum-containing drugs in cancer chemotherapy. Pharmaceuticals 2021, 14, 549. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Bernard Tchounwou, P. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Zisowsky, J.; Koegel, S.; Leyers, S.; Devarakonda, K.; Kassack, M.U.; Osmak, M.; Jaehde, U. Relevance of drug uptake and efflux for cisplatin sensitivity of tumor cells. Biochem. Pharmacol. 2007, 73, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Adeya Adella, C.; Siregar, M.F.G.; Putra, I.B.; Hasibuan, P.A.; Andrijono, A.; Bachtiar, A.; Lumbanraja, S.N.; Nasution, I.P. Promising effect of cisplatin and melatonin combination on the inhibition of cisplatin resistance in ovarian cancer. F1000Research 2024, 12, 313. [Google Scholar] [CrossRef]
- Cheng, C.; Ding, Q.; Zhang, Z.; Wang, S.; Zhong, B.; Huang, X.; Shao, Z. PTBP1 modulates osteosarcoma chemoresistance to cisplatin by regulating the expression of the copper transporter SLC31A1. J. Cell. Mol. Med. 2020, 24, 5274–5289. [Google Scholar] [CrossRef]
- Wu, G.; Peng, H.; Tang, M.; Yang, M.; Wang, J.; Hu, Y.; Li, Z.; Li, J.; Li, Z.; Song, L. ZNF711 down-regulation promotes cisplatin resistance in epithelial ovarian cancer via interacting with JHDM2A and suppressing SLC31A1 expression. eBioMedicine 2021, 71, 103558. [Google Scholar] [CrossRef]
- Cui, H.; Zhang, A.J.; McKeage, M.J.; Nott, L.M.; Geraghty, D.; Guven, N.; Liu, J.J. Copper transporter 1 in human colorectal cancer cell lines: Effects of endogenous and modified expression on oxaliplatin cytotoxicity. J. Inorg. Biochem. 2017, 177, 249–258. [Google Scholar] [CrossRef]
- Pathania, S.; Bhatia, R.; Baldi, A.; Singh, R.; Rawal, R.K. Drug metabolizing enzymes and their inhibitors’ role in cancer resistance. Biomed. Pharmacother. 2018, 105, 53–65. [Google Scholar] [CrossRef]
- Bansal, A.; Simon, M.C. Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar] [CrossRef]
- Bennaceur-Griscelli, A.; Bosq, J.; Koscielny, S.; Lefrè, F.; Turhan, A.; Brousse, N.; Hermine, O.; Ribrag, V. High level of glutathione-S-transferase π expression in mantle cell lymphomas. Clin. Cancer Res. 2004, 10, 3029–3034. [Google Scholar] [CrossRef] [PubMed]
- Sawers, L.; Ferguson, M.J.; Young, H.C.; Chakravarty, P.; Wolf, C.R.; Smith, G. Glutathione S-transferase P1 (GSTP1) directly influences platinum drug chemosensitivity in ovarian tumour cell lines. Br. J. Cancer 2014, 111, 1150–1157. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.M.; Rocha, C.R.R.; Kinker, G.S.; Pelegrini, A.L.; Menck, C.F.M. The balance between NRF2/GSH antioxidant mediated pathway and DNA repair modulates cisplatin resistance in lung cancer cells. Sci. Rep. 2019, 9, 17639. [Google Scholar] [CrossRef]
- Lushchak, V.I. Glutathione homeostasis and functions: Potential targets for medical interventions. J. Amino Acids 2012, 2012, 736837. [Google Scholar] [CrossRef]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef]
- Ju, H.Q.; Gocho, T.; Aguilar, M.; Wu, M.; Zhuang, Z.N.; Fu, J.; Yanaga, K.; Huang, P.; Chiao, P.J. Mechanisms of overcoming intrinsic resistance to gemcitabine in pancreatic ductal adenocarcinoma through the redox modulation. Mol. Cancer Ther. 2015, 14, 788–798. [Google Scholar] [CrossRef]
- Liu, Y.P.; Zheng, C.C.; Huang, Y.N.; He, M.L.; Xu, W.W.; Li, B. Molecular mechanisms of chemo-and radiotherapy resistance and the potential implications for cancer treatment. MedComm 2021, 2, 315–340. [Google Scholar] [CrossRef]
- Jurkovicova, D.; Neophytou, C.M.; Gašparović, A.Č.; Gonçalves, A.C. DNA damage response in cancer therapy and resistance: Challenges and opportunities. Int. J. Mol. Sci. 2022, 23, 14672. [Google Scholar] [CrossRef]
- Fu, X.; Li, P.; Zhou, Q.; He, R.; Wang, G.; Zhu, S.; Bagheri, A.; Kupfer, G.; Pei, H.; Li, J. Mechanism of PARP inhibitor resistance and potential overcoming strategies. Genes Dis. 2024, 11, 306–320. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Wu, W.; Li, Y.; Lin, L.; Chen, D.; Yan, H.; Xiao, X.; Chen, H.; Chen, Z.; Zhang, Y.; et al. Poly(ADP-ribosyl)ation of BRD7 by PARP1 confers resistance to DNA-damaging chemotherapeutic agents. EMBO Rep. 2019, 20, e46166. [Google Scholar] [CrossRef] [PubMed]
- Szalat, R.; Samur, M.K.; Fulciniti, M.; Lopez, M.; Nanjappa, P.; Cleynen, A.; Wen, K.; Kumar, S.; Perini, T.; Calkins, A.S.; et al. Nucleotide excision repair is a potential therapeutic target in multiple myeloma. Leukemia 2018, 32, 111–119. [Google Scholar] [CrossRef]
- Wu, X.; Zhong, Y.; Chen, Q.; Zhang, X.; Zhang, H. Enhancer of mRNA decapping protein 4 (EDC4) interacts with replication protein A (RPA) and contributes to cisplatin resistance in cervical cancer by alleviating DNA damage. Hereditas 2020, 157, 41. [Google Scholar] [CrossRef]
- Byrne, B.M.; Oakley, G.G. Replication protein A, the laxative that keeps DNA regular: The importance of RPA phosphorylation in maintaining genome stability. Semin. Cell Dev. Biol. 2019, 86, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Belanger, F.; Fortier, E.; Dube, M.; Lemay, J.F.; Buisson, R.; Masson, J.Y.; Elsherbiny, A.; Costantino, S.; Carmona, E.; Mes-Masson, A.M.; et al. Replication protein A availability during DNA replication stress is a major determinant of cisplatin resistance in ovarian cancer cells. Cancer Res. 2018, 78, 5561–5573. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Makin, G.; Dive, C. Apoptosis and cancer chemotherapy. Trends Cell Biol. 2001, 11, S22–S26. [Google Scholar] [CrossRef]
- Yao, Y.; Gong, J.-N.; Segal, D.; Riffkin, C.D.; van Delft, M.F.; Roberts, A.W.; Huang, D.C. The role of BAX/BAK-mediated apoptosis for the cytotoxic action of anti-myeloma agents. Blood 2016, 128, 5706. [Google Scholar] [CrossRef]
- Roberts, A.W. Therapeutic development and current uses of BCL-2 inhibition. Hematology 2020, 2020, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.; Wei, Z.; Yang, W.; Huang, J.; Yang, Y.; Wang, J. The role of BCL-2 family proteins in regulating apoptosis and cancer therapy. Front. Oncol. 2022, 12, 985363. [Google Scholar] [CrossRef]
- Strasser, A.; Vaux, D.L. Cell death in the origin and treatment of cancer. Mol. Cell 2020, 78, 1045–1054. [Google Scholar] [CrossRef]
- Towers, C.G.; Wodetzki, D.; Thorburn, A. Autophagy and cancer: Modulation of cell death pathways and cancer cell adaptations. J. Cell Biol. 2020, 219, e201909033. [Google Scholar] [CrossRef] [PubMed]
- Neophytou, C.M.; Trougakos, I.P.; Erin, N.; Papageorgis, P. Apoptosis deregulation and the development of cancer multi-drug resistance. Cancers 2021, 13, 4363. [Google Scholar] [CrossRef]
- Kaloni, D.; Diepstraten, S.T.; Strasser, A.; Kelly, G.L. BCL-2 protein family: Attractive targets for cancer therapy. Apoptosis 2022, 28, 20–38. [Google Scholar] [CrossRef]
- Blombery, P.; Anderson, M.A.; Gong, J.N.; Thijssen, R.; Birkinshaw, R.W.; Thompson, E.R.; Teh, C.E.; Nguyen, T.; Xu, Z.; Flensburg, C.; et al. Acquisition of the recurrent Gly101Val mutation in BCL2 confers resistance to venetoclax in patients with progressive chronic lymphocytic leukemia. Cancer Discov. 2019, 9, 342–353. [Google Scholar] [CrossRef]
- Ma, Y.; Lu, R.; Sun, X.; Wang, R. PUMA mediated afatinib-induced apoptosis in glioma cells. Arch. Med. Sci. 2022, 18. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, N.; Liu, J.; Liu, Y.; Zhang, C.; Long, S.; Luo, G.; Zhang, L.; Zhang, Y. Mutant P53 drives cancer chemotherapy resistance due to loss of function on activating transcription of PUMA. Cell Cycle 2019, 18, 3442–3455. [Google Scholar] [CrossRef]
- Hientz, K.; Mohr, A.; Bhakta-Guha, D.; Efferth, T. The role of P53 in cancer drug resistance and targeted chemotherapy. Oncotarget 2017, 8, 8921–8946. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Huang, A.; Zheng, X.; Liu, T.; Lin, Z.; Zhang, S.; Yang, Q.; Zhang, T.; Ma, H. 53BP1 loss induces chemoresistance of colorectal cancer cells to 5-fluorouracil by inhibiting the ATM–CHK2–P53 pathway. J. Cancer Res. Clin. Oncol. 2017, 143, 419–431. [Google Scholar] [CrossRef]
- Takeiwa, T.; Mitobe, Y.; Ikeda, K.; Horie-Inoue, K.; Inoue, S. Roles of splicing factors in hormone-related cancer progression. Int. J. Mol. Sci. 2020, 21, 1551. [Google Scholar] [CrossRef] [PubMed]
- Sciarrillo, R.; Wojtuszkiewicz, A.; Assaraf, Y.G.; Jansen, G.; Kaspers, G.J.L.; Giovannetti, E.; Cloos, J. The role of alternative splicing in cancer: From oncogenesis to drug resistance. Drug Resist. Updat. 2020, 53, 100728. [Google Scholar] [CrossRef] [PubMed]
- Dunham, I.; Kundaje, A.; Aldred, S.F.; Collins, P.J.; Davis, C.A.; Doyle, F.; Epstein, C.B.; Frietze, S.; Harrow, J.; Kaul, R.; et al. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Oltean, S. Modulators of alternative splicing as novel therapeutics in cancer. World J. Clin. Oncol. 2015, 6, 92–102. [Google Scholar] [CrossRef]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef]
- Biamonti, G.; Catillo, M.; Pignataro, D.; Montecucco, A.; Ghigna, C. The alternative splicing side of cancer. Semin. Cell Dev. Biol. 2014, 32, 30–36. [Google Scholar] [CrossRef]
- Yan, C.; Wan, R.; Shi, Y. Molecular mechanisms of pre-mRNA splicing through structural biology of the spliceosome. Cold Spring Harb. Perspect. Biol. 2019, 11, a032409. [Google Scholar] [CrossRef]
- Wahl, M.C.; Will, C.L.; Lührmann, R. The spliceosome: Design principles of a dynamic RNP machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef]
- Tao, Y.; Zhang, Q.; Wang, H.; Yang, X.; Mu, H. Alternative splicing and related RNA binding proteins in human health and disease. Signal Transduct. Target. Ther. 2024, 9, 26. [Google Scholar] [CrossRef] [PubMed]
- Howard, J.M.; Sanford, J.R. The RNAissance family: SR proteins as multifaceted regulators of gene expression. Wiley Interdiscip. Rev. RNA 2015, 6, 93–106. [Google Scholar] [CrossRef] [PubMed]
- West, K.O.; Scott, H.M.; Torres-Odio, S.; West, A.P.; Patrick, K.L.; Watson, R.O. The splicing factor HnRNP M is a critical regulator of innate immune gene expression in macrophages. Cell Rep. 2019, 29, 1594–1609.e5. [Google Scholar] [CrossRef] [PubMed]
- Kretova, M.; Selicky, T.; Cipakova, I.; Cipak, L. Regulation of pre-mRNA splicing: Indispensable role of post-translational modifications of splicing factors. Life 2023, 13, 604. [Google Scholar] [CrossRef]
- Ladomery, M. Aberrant alternative splicing is another hallmark of cancer. Int. J. Cell Biol. 2013, 2013, 463786. [Google Scholar] [CrossRef]
- Oltean, S.; Bates, D.O. Hallmarks of alternative splicing in cancer. Oncogene 2014, 33, 5311–5318. [Google Scholar] [CrossRef]
- Ye, X.; Abou-Rayyah, Y.; Bischoff, J.; Ritchie, A.; Sebire, N.J.; Watts, P.; Churchill, A.J.; Bates, D.O. Altered ratios of pro- and anti-angiogenic VEGF-A variants and pericyte expression of DLL4 disrupt vascular maturation in infantile haemangioma. J. Pathol. 2016, 239, 139–151. [Google Scholar] [CrossRef]
- Wang, J.; Liu, T.; Wang, M.; Lv, W.; Wang, Y.; Jia, Y.; Zhang, R.; Liu, L. SRSF1-dependent alternative splicing attenuates BIN1 expression in non-small cell lung cancer. J. Cell. Biochem. 2020, 121, 946–953. [Google Scholar] [CrossRef]
- Karsten, M.M.; Beck, M.H.; Rademacher, A.; Knabl, J.; Blohmer, J.U.; Jückstock, J.; Radosa, J.C.; Jank, P.; Rack, B.; Janni, W. VEGF-A165b levels are reduced in breast cancer patients at primary diagnosis but increase after completion of cancer treatment. Sci. Rep. 2020, 10, 59823. [Google Scholar] [CrossRef]
- Biselli-Chicote, P.M.; Biselli, J.M.; Cunha, B.R.; Castro, R.; Maniglia, J.V.; Neto, D.S.; Tajara, E.H.; de Góis Filho, J.F.; Fukuyama, E.E.; Pavarino, É.C.; et al. Overexpression of anti-angiogenic vascular endothelial growth factor isoform and splicing regulatory factors in oral, laryngeal, and pharyngeal squamous cell carcinomas. Asian Pac. J. Cancer Prev. 2017, 18, 2171–2177. [Google Scholar] [CrossRef]
- Star, E.; Stevens, M.; Gooding, C.; Smith, C.W.J.; Li, L.; Ayine, M.L.; Harper, S.J.; Bates, D.O.; Oltean, S. A drug-repositioning screen using splicing-sensitive fluorescent reporters identifies novel modulators of VEGF-A splicing with anti-angiogenic properties. Oncogenesis 2021, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, I.M.; Cheng, A.W.; Flytzanis, N.C.; Balsamo, M.; Condeelis, J.S.; Oktay, M.H.; Burge, C.B.; Gertler, F.B. An EMT–driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet. 2011, 7, e1002218. [Google Scholar] [CrossRef] [PubMed]
- Oltean, S.; Sorg, B.S.; Albrecht, T.; Bonano, V.I.; Brazas, R.M.; Dewhirst, M.W.; Garcia-Blanco, M.A. Alternative inclusion of fibroblast growth factor receptor 2 exon IIIc in Dunning prostate tumors reveals unexpected epithelial-mesenchymal plasticity. Proc. Natl. Acad. Sci. USA 2006, 103, 14116–14121. [Google Scholar] [CrossRef]
- Ranieri, D.; Rosato, B.; Nanni, M.; Magenta, A.; Belleudi, F.; Torrisi, M.R. Expression of the FGFR2 mesenchymal splicing variant in epithelial cells drives epithelial-mesenchymal transition. Oncotarget 2015, 7, 5440–5460. [Google Scholar] [CrossRef]
- Li, L.; Zheng, J.; Stevens, M.; Oltean, S. A repositioning screen using an FGFR2 splicing reporter reveals compounds that regulate epithelial-mesenchymal transitions and inhibit growth of prostate cancer xenografts. Mol. Ther. Methods Clin. Dev. 2022, 25, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Zhao, R.; Kroger, J.; Qu, M.; Wani, A.A.; Wang, Q.E. Caspase-2 short isoform interacts with membrane-associated cytoskeleton proteins to inhibit apoptosis. PLoS ONE 2013, 8, e67033. [Google Scholar] [CrossRef]
- Zeng, Z.; Tu, J.; Cheng, J.; Yao, M.; Wu, Y.; Huang, X.; Xie, X.; Zhang, X.; Lu, F.; Chen, X. Influence of CCND1 G870A polymorphism on the risk of HBV-related HCC and cyclin D1 splicing variant expression in Chinese population. Tumor Biol. 2015, 36, 6891–6900. [Google Scholar] [CrossRef]
- Miwa, T.; Nagata, T.; Kojima, H.; Sekine, S.; Okumura, T. Isoform switch of CD44 induces different chemotactic and tumorigenic ability in gallbladder cancer. Int. J. Oncol. 2017, 51, 771–780. [Google Scholar] [CrossRef]
- Oudin, M.J.; Hughes, S.K.; Rohani, N.; Moufarrej, M.N.; Jones, J.G.; Condeelis, J.S.; Lauffenburger, D.A.; Gertler, F.B. Characterization of the expression of the pro-metastatic MenaINV isoform during breast tumor progression. Clin. Exp. Metastasis 2016, 33, 249–261. [Google Scholar] [CrossRef]
- Maimon, A.; Mogilevsky, M.; Shilo, A.; Golan-Gerstl, R.; Obiedat, A.; Ben-Hur, V.; Lebenthal-Loinger, I.; Stein, I.; Reich, R.; Beenstock, J.; et al. Mnk2 alternative splicing modulates the P38-MAPK pathway and impacts Ras-induced transformation. Cell Rep. 2014, 7, 501–513. [Google Scholar] [CrossRef]
- Seiz, J.R.; Klinke, J.; Scharlibbe, L.; Lohfink, D.; Heipel, M.; Ungefroren, H.; Giehl, K.; Menke, A. Different signaling and functionality of Rac1 and Rac1b in the progression of lung adenocarcinoma. Biol. Chem. 2020, 401, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.; Kuang, Y.; Chen, L.L.; Lao, W.F.; Zhu, Y.R.; Wang, L.Q.; Wang, D. A novel RON splice variant lacking exon 2 activates the PI3K/AKT pathway via PTEN phosphorylation in colorectal carcinoma cells. Oncotarget 2017, 8, 39101–39116. [Google Scholar] [CrossRef]
- Gerhart, S.V.; Kellner, W.A.; Thompson, C.; Pappalardi, M.B.; Zhang, X.P.; Montes De Oca, R.; Penebre, E.; Duncan, K.; Boriack-Sjodin, A.; Le, B.; et al. Activation of the P53-MDM4 regulatory axis defines the anti-tumor response to PRMT5 inhibition through its role in regulating cellular splicing. Sci. Rep. 2018, 8, 28002. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-Y.; Yue, P.; Hong, K.; Lotan, R. Induction of Fas expression and augmentation of Fas/Fas ligand-mediated apoptosis by the synthetic retinoid CD437 in human lung cancer cells. Cancer Res. 2000, 60, 6537–6543. [Google Scholar] [PubMed]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef]
- Temaj, G.; Chichiarelli, S.; Saha, S.; Telkoparan-Akillilar, P.; Nuhii, N.; Hadziselimovic, R.; Saso, L. An intricate rewiring of cancer metabolism via alternative splicing. Biochem. Pharmacol. 2023, 217, 115848. [Google Scholar] [CrossRef]
- Mesrian Tanha, H.; Rahgozar, S.; Mojtabavi Naeini, M. ABCC4 functional SNP in the 3′ splice acceptor site of exon 8 (G912T) is associated with unfavorable clinical outcome in children with acute lymphoblastic leukemia. Cancer Chemother. Pharmacol. 2017, 80, 109–117. [Google Scholar] [CrossRef]
- Li, M.; Kong, X.Y.; Wang, S.M. Effects of splicing-regulatory polymorphisms in ABCC2, ABCG2, and ABCB1 on methotrexate exposure in Chinese children with acute lymphoblastic leukemia. Cancer Chemother. Pharmacol. 2023, 91, 77–87. [Google Scholar] [CrossRef]
- Cai, J.; Damaraju, V.L.; Groulx, N.; Mowles, D.; Peng, Y.; Robins, M.J.; Cass, C.E.; Gros, P. Two distinct molecular mechanisms underlying cytarabine resistance in human leukemic cells. Cancer Res. 2008, 68, 2349–2357. [Google Scholar] [CrossRef]
- Schiel, M.A.; Green, S.L.; Davis, W.I.; Sanghani, P.C.; Bosron, W.F.; Sanghani, S.P. Expression and characterization of a human carboxylesterase 2 splice variant. J. Pharmacol. Exp. Ther. 2007, 323, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Wojtuszkiewicz, A.; Raz, S.; Stark, M.; Assaraf, Y.G.; Jansen, G.; Peters, G.J.; Sonneveld, E.; Kaspers, G.J.L.; Cloos, J. Folylpolyglutamate synthetase splicing alterations in acute lymphoblastic leukemia are provoked by methotrexate and other chemotherapeutics and mediate chemoresistance. Int. J. Cancer 2016, 138, 1645–1656. [Google Scholar] [CrossRef] [PubMed]
- Wojtuszkiewicz, A.; Assaraf, Y.G.; Hoekstra, M.; Sciarrillo, R.; Jansen, G.; Peters, G.J.; Pieters, R.; Sonneveld, E.; Escherich, G.; Kaspers, G.J.L.; et al. The association of aberrant folylpolyglutamate synthetase splicing with ex vivo methotrexate resistance and clinical outcome in childhood acute lymphoblastic leukemia. Haematologica 2016, 101, e291–e294. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xu, X.; Deng, H.; Liu, L.; Xiang, Y.; Feng, J. Overcoming Cancer Drug-Resistance Calls for Novel Strategies Targeting Abnormal Alternative Splicing. Pharmacol. Ther. 2024, 261, 108697. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.J.; Henry, I.; Maylin, Z.; Smith, C.; Arunachalam, E.; Pandha, H.; Asim, M. A compendium of androgen receptor variant 7 target genes and their role in castration-resistant prostate cancer. Front. Oncol. 2023, 13, 1129140. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Luber, B.; Wang, H.; Chen, Y.; Zhu, Y.; Silberstein, J.L.; Taylor, M.N.; Maughan, B.L.; Denmeade, S.R.; et al. Clinical significance of androgen receptor splice variant-7 mRNA detection in circulating tumor cells of men with metastatic castration-resistant prostate cancer treated with first- and second-line abiraterone and enzalutamide. J. Clin. Oncol. 2017, 35, 2149–2156. [Google Scholar] [CrossRef]
- Tummala, R.; Lou, W.; Gao, A.C.; Nadiminty, N. Quercetin targets HnRNPA1 to overcome enzalutamide resistance in prostate cancer cells. Mol. Cancer Ther. 2017, 16, 2770–2779. [Google Scholar] [CrossRef]
- Poulikakos, P.I.; Persaud, Y.; Janakiraman, M.; Kong, X.; Ng, C.; Moriceau, G.; Shi, H.; Atefi, M.; Titz, B.; Gabay, M.T.; et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 2011, 480, 387–390. [Google Scholar] [CrossRef]
- Salton, M.; Kasprzak, W.K.; Voss, T.; Shapiro, B.A.; Poulikakos, P.I.; Misteli, T. Inhibition of vemurafenib-resistant melanoma by interference with pre-mRNA splicing. Nat. Commun. 2015, 6, 8103. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.W.; Abdel-Wahab, O. Therapeutic targeting of splicing in cancer. Nat. Med. 2016, 22, 976–986. [Google Scholar] [CrossRef]
- Rytelewski, M.; Tong, J.G.; Buensuceso, A.; Leong, H.S.; Maleki Vareki, S.; Figueredo, R.; Di Cresce, C.; Wu, S.Y.; Herbrich, S.M.; Baggerly, K.A.; et al. BRCA2 inhibition enhances cisplatin-mediated alterations in tumor cell proliferation, metabolism, and metastasis. Mol. Oncol. 2014, 8, 175–187. [Google Scholar] [CrossRef]
- Therapeutics, T.; Biology, C. The BRCA1-D11q alternative splice isoform bypasses germline mutations and promotes therapeutic resistance to PARP inhibition and cisplatin. Cancer Res. 2016, 76, 2778–2789. [Google Scholar] [CrossRef]
- Meyer, S.; Stevens, A.; Paredes, R.; Schneider, M.; Walker, M.J.; Williamson, A.J.; Gonzalez-Sanchez, M.-B.; Smetsers, S.; Dalal, V.; Teng, H.Y.; et al. Acquired cross-linker resistance associated with a novel spliced BRCA2 protein variant for molecular phenotyping of BRCA2 disruption. Cell Death Dis. 2017, 8, e2875. [Google Scholar] [CrossRef] [PubMed]
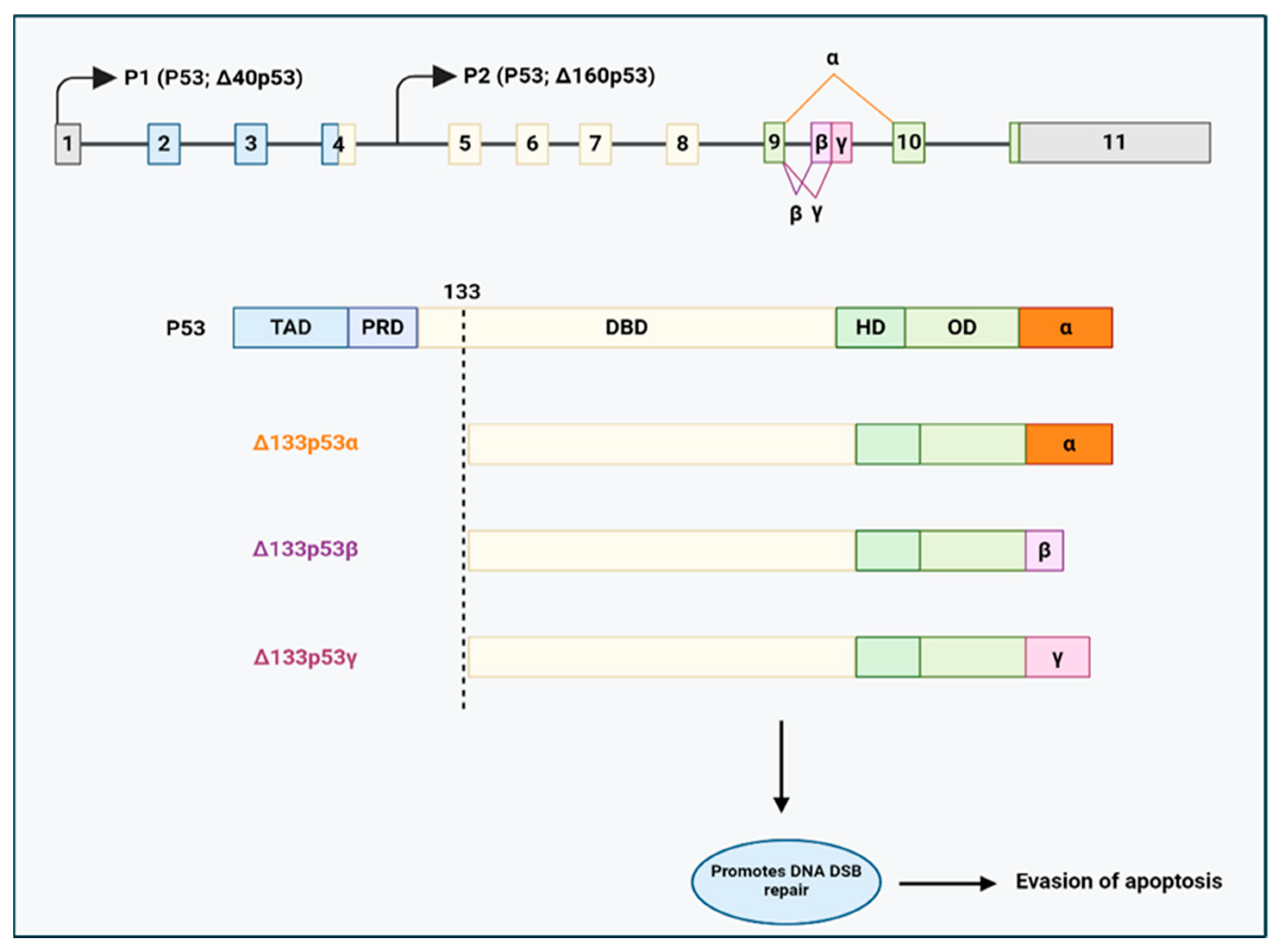
- Gadea, G.; Arsic, N.; Fernandes, K.; Diot, A.; Joruiz, S.M.; Abdallah, S.; Meuray, V.; Vinot, S.; Anguille, C.; Remenyi, J.; et al. TP53 drives invasion through expression of its Δ133p53β variant. eLife 2016, 5, e14734. [Google Scholar] [CrossRef]
- Horvat, A.; Tadijan, A.; Vlašić, I.; Slade, N. P53/P73 Protein Network in Colorectal Cancer and Other Human Malignancies. Cancers 2021, 13, 2885. [Google Scholar] [CrossRef]
- Kazantseva, M.; Eiholzer, R.A.; Mehta, S.; Taha, A.; Bowie, S.; Roth, I.; Zhou, J.; Joruiz, S.M.; Royds, J.A.; Hung, N.A.; et al. Elevation of the TP53 isoform Δ133p53β in glioblastomas: An alternative to mutant P53 in promoting tumor development. J. Pathol. 2018, 246, 77–88. [Google Scholar] [CrossRef]
- Nutthasirikul, N.V.; Hahnvajanawong, C.; Techasen, A.; Limpaiboon, T.; Wat, C.L.; Chau-In, S.; Jearanaikoon, P. Targeting the Δ133p53 isoform can restore chemosensitivity in 5-fluorouracil-resistant cholangiocarcinoma cells. Int. J. Oncol. 2015, 47, 2153–2164. [Google Scholar] [CrossRef] [PubMed]
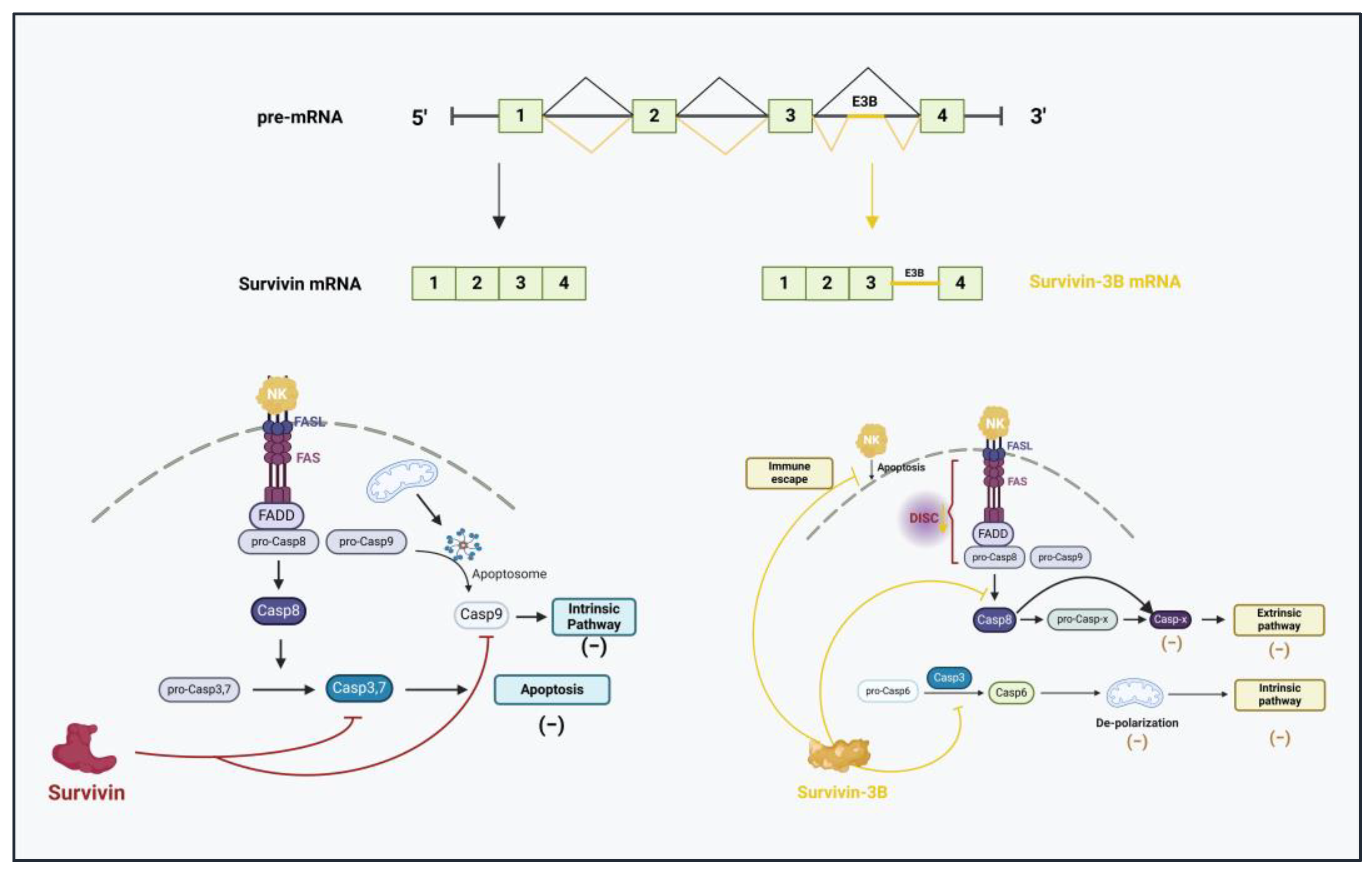
- Wang, Q.; Greene, M.I. Survivin as a therapeutic target for the treatment of human cancer. Cancers 2024, 16, 1705. [Google Scholar] [CrossRef]
- Sah, N.K.; Seniya, C. Survivin splice variants and their diagnostic significance. Tumor Biol. 2015, 36, 6623–6631. [Google Scholar] [CrossRef]
- Badran, A.; Yoshida, A.; Ishikawa, K.; Goi, T.; Yamaguchi, A.; Ueda, T.; Inuzuka, M. Identification of a novel splice variant of the human anti-apoptosis gene survivin. Biochem. Biophys. Res. Commun. 2004, 314, 902–907. [Google Scholar] [CrossRef]
- Végran, F.; Boidot, R. Survivin-3B promotes chemoresistance and immune escape by inhibiting caspase-8 and -6 in cancer cells. Oncoimmunology 2013, 2, e26328. [Google Scholar] [CrossRef]
- Végran, F.; Mary, R.; Gibeaud, A.; Mirjolet, C.; Collin, B.; Oudot, A.; Charon-Barra, C.; Arnould, L.; Lizard-Nacol, S.; Boidot, R. Survivin-3B potentiates immune escape in cancer but also inhibits the toxicity of cancer chemotherapy. Cancer Res. 2013, 73, 5391–5401. [Google Scholar] [CrossRef] [PubMed]
- Tyson-Capper, A.; Gautrey, H. Regulation of Mcl-1 alternative splicing by HnRNP F, H1, and K in breast cancer cells. RNA Biol. 2018, 15, 1448–1457. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gao, X.; Wei, C.; Guo, R.; Xu, H.; Bai, Z.; Zhou, J.; Zhu, J.; Wang, W.; Wu, Y.; et al. Modification of Mcl-1 alternative splicing induces apoptosis and suppresses tumor proliferation in gastric cancer. Aging 2020, 12, 19293–19315. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, H.; Zhao, P.; Wang, X. Targeting PKM2 promotes chemosensitivity of breast cancer cells in vitro and in vivo. Cancer Biomark. 2021, 32, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Calabretta, S.; Bielli, P.; Passacantilli, I.; Pilozzi, E.; Fendrich, V.; Capurso, G.; Delle Fave, G.; Sette, C. Modulation of PKM alternative splicing by PTBP1 promotes gemcitabine resistance in pancreatic cancer cells. Oncogene 2016, 35, 2031–2039. [Google Scholar] [CrossRef]
- Zheng, M.; Niu, Y.; Bu, J.; Liang, S.; Zhang, Z.; Liu, J.; Guo, L.; Zhang, Z.; Wang, Q. ESRP1 regulates alternative splicing of CARM1 to sensitize small cell lung cancer cells to chemotherapy by inhibiting TGF-β/Smad signaling. Aging 2021, 13, 3554–3572. [Google Scholar] [CrossRef]
- Kato, T.; Mizutani, K.; Kawakami, K.; Fujita, Y.; Ehara, H.; Ito, M. CD44v8-10 mRNA contained in serum exosomes as a diagnostic marker for docetaxel resistance in prostate cancer patients. Heliyon 2020, 6, e04138. [Google Scholar] [CrossRef]
- Zhou, J.-M.; Hu, S.-Q.; Jiang, H.; Chen, Y.-L.; Feng, J.-H.; Chen, Z.-Q.; Wen, K.-M. OCT4B1 promoted EMT and regulated the self-renewal of CSCs in CRC: Effects associated with the balance of miR-8064/PLK1. Mol. Ther. Oncolytics 2019, 14, 160–171. [Google Scholar] [CrossRef]
- Amin, E.M.; Oltean, S.; Hua, J.; Gammons, M.V.R.; Hamdollah-Zadeh, M.; Welsh, G.I.; Cheung, M.K.; Ni, L.; Kase, S.; Rennel, E.S.; et al. WT1 mutants reveal SRPK1 to be a downstream angiogenesis target by altering VEGF splicing. Cancer Cell 2011, 20, 768–780. [Google Scholar] [CrossRef]
- Siqueira, R.P.; Barbosa, É.D.A.A.; Polêto, M.D.; Righetto, G.L.; Seraphim, T.V.; Salgado, R.L.; Ferreira, J.G.; De Andrade Barros, M.V.; De Oliveira, L.L.; Laranjeira, A.B.A.; et al. Potential antileukemia effect and structural analyses of SRPK inhibition by N-(2-(piperidin-1-yl)-5-(trifluoromethyl)phenyl)isonicotinamide (SRPIN340). PLoS ONE 2015, 10, e0134882. [Google Scholar] [CrossRef]
- Mavrou, A.; Brakspear, K.; Hamdollah-Zadeh, M.; Damodaran, G.; Babaei-Jadidi, R.; Oxley, J.; Gillatt, D.A.; Ladomery, M.R.; Harper, S.J.; Bates, D.O.; et al. Serine–arginine protein kinase 1 (SRPK1) inhibition as a potential novel targeted therapeutic strategy in prostate cancer. Oncogene 2015, 34, 4311–4319. [Google Scholar] [CrossRef] [PubMed]
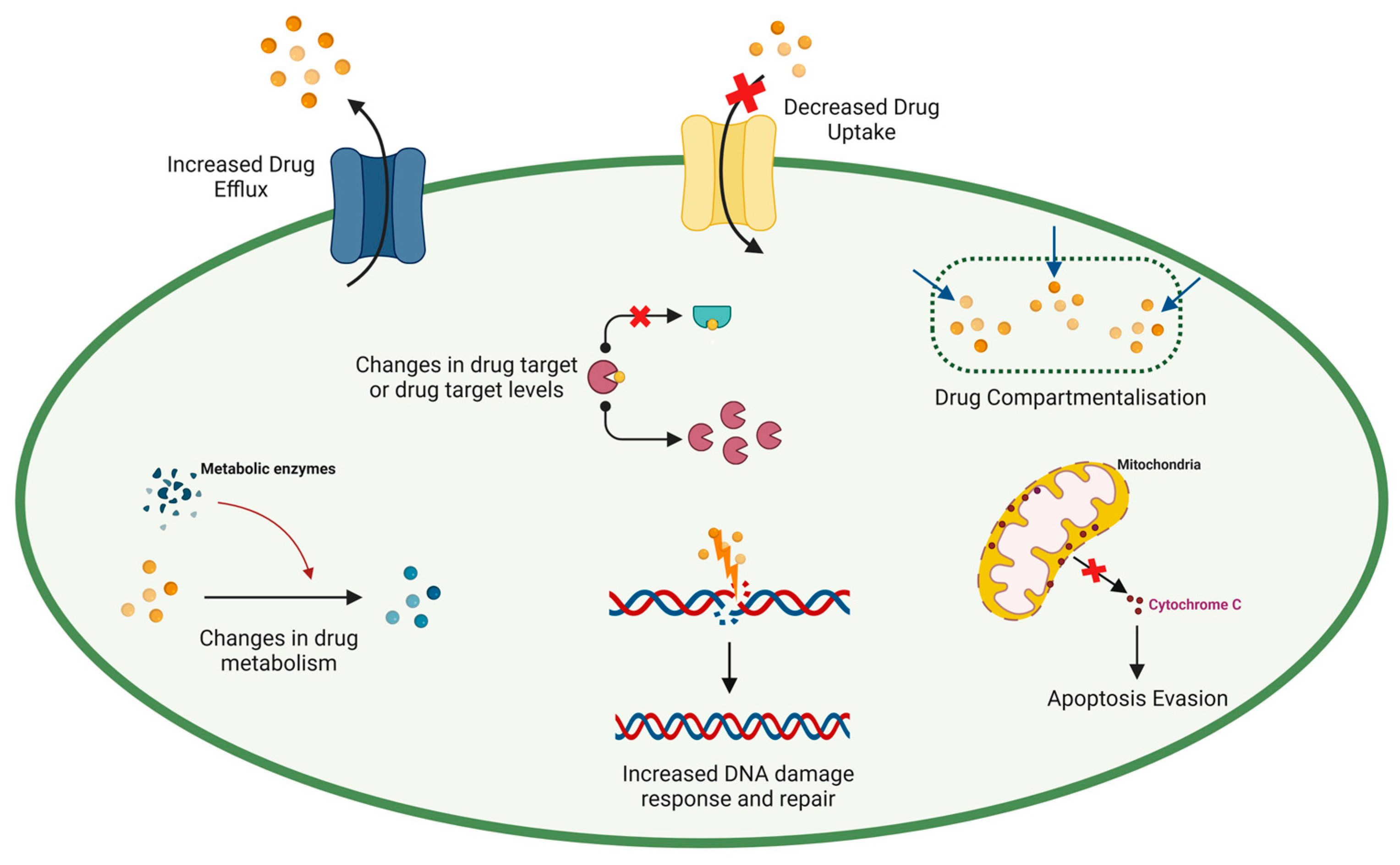
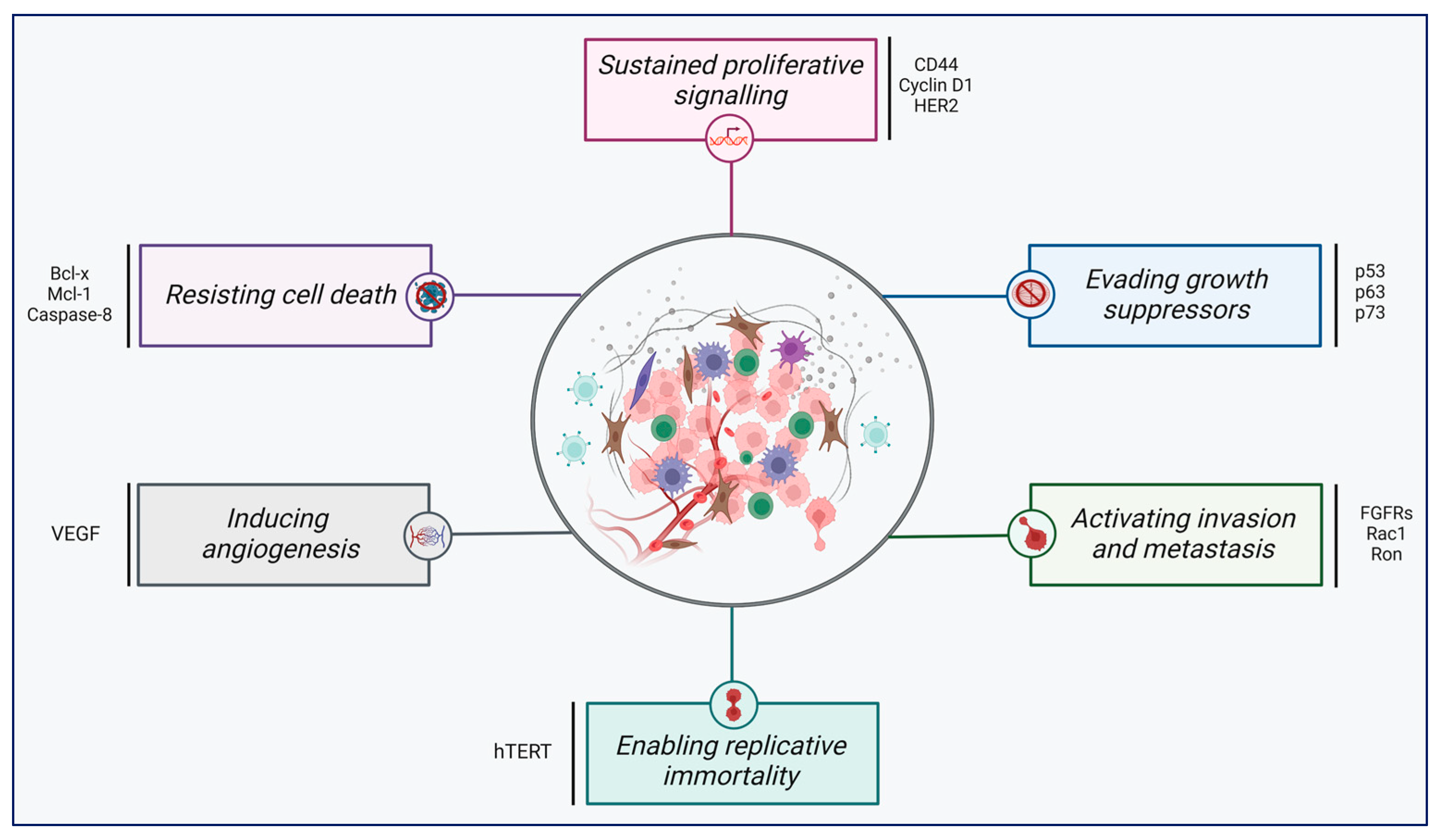
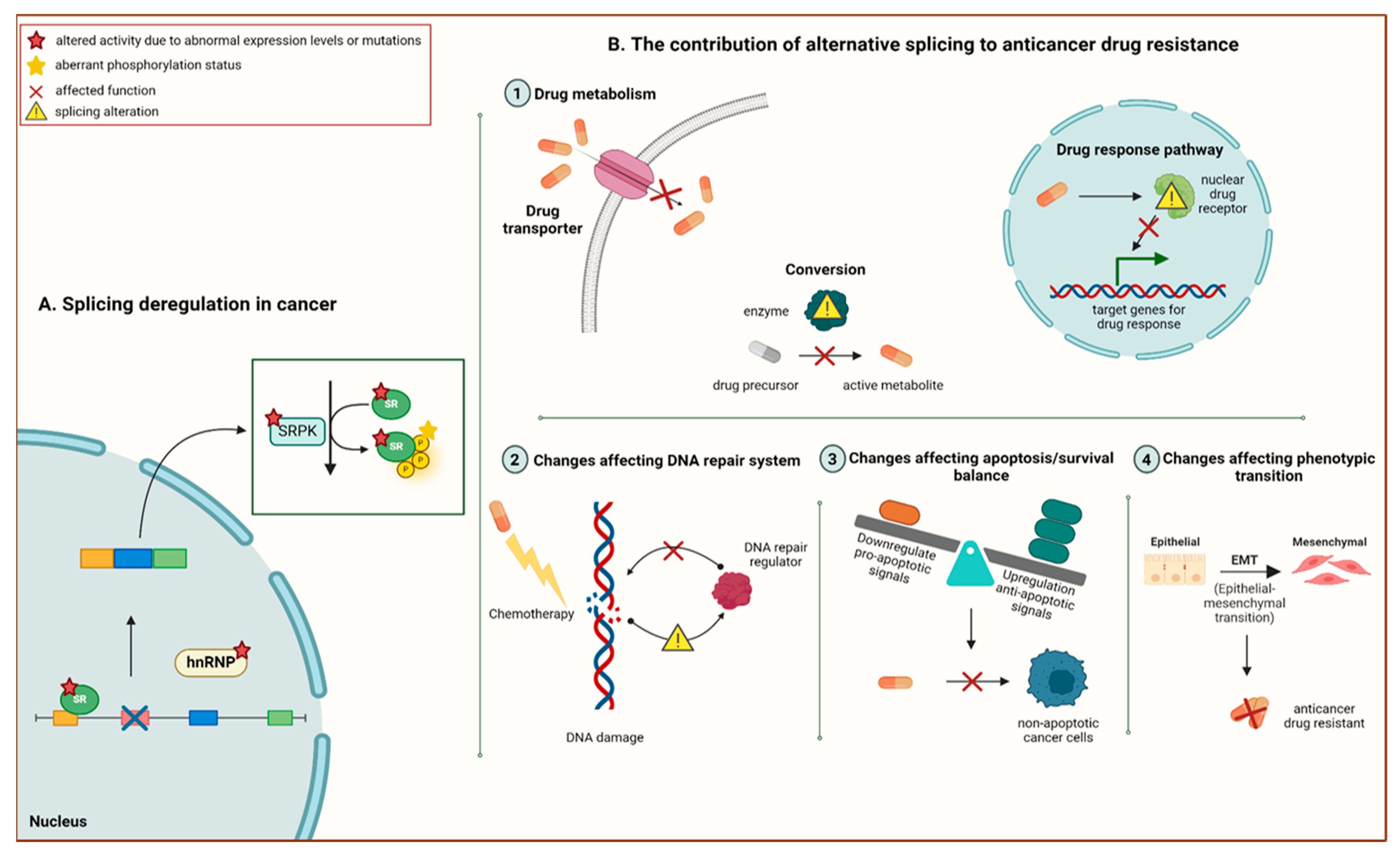
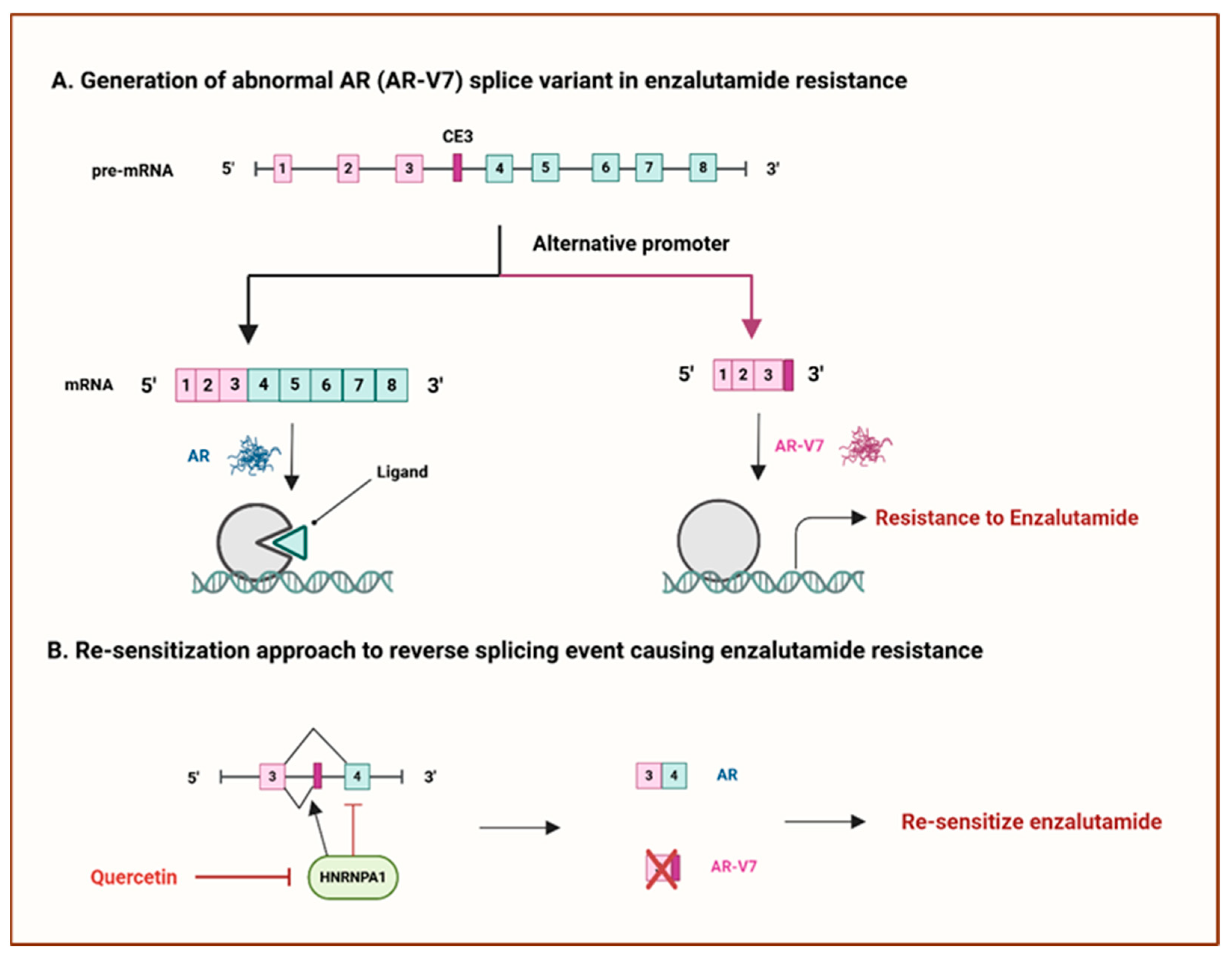
| Cancer Therapies | Definition | Limitations | References |
|---|---|---|---|
| Surgery | Removing cancer tissue |
| [16] |
| Radiation therapy | High-energy radiation is used to eliminate tumour cells |
| [17] |
| Targeted therapy | Using pharmacological agents |
| [18] |
| Immunotherapy | Targeting tumour- associated antigens to attack cancer cells |
| [19] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duzgun, D.; Oltean, S. Aberrant Splicing as a Mechanism for Resistance to Cancer Therapies. Cancers 2025, 17, 1381. https://doi.org/10.3390/cancers17081381
Duzgun D, Oltean S. Aberrant Splicing as a Mechanism for Resistance to Cancer Therapies. Cancers. 2025; 17(8):1381. https://doi.org/10.3390/cancers17081381
Chicago/Turabian StyleDuzgun, Duygu, and Sebastian Oltean. 2025. "Aberrant Splicing as a Mechanism for Resistance to Cancer Therapies" Cancers 17, no. 8: 1381. https://doi.org/10.3390/cancers17081381
APA StyleDuzgun, D., & Oltean, S. (2025). Aberrant Splicing as a Mechanism for Resistance to Cancer Therapies. Cancers, 17(8), 1381. https://doi.org/10.3390/cancers17081381