
Chromosomal Instability and Clonal Heterogeneity in Breast Cancer: From Mechanisms to Clinical Applications

 , and
, and
Simple Summary
Abstract
1. Introduction
2. CIN and CH: Cellular Mechanisms and Their Implication for Cancer
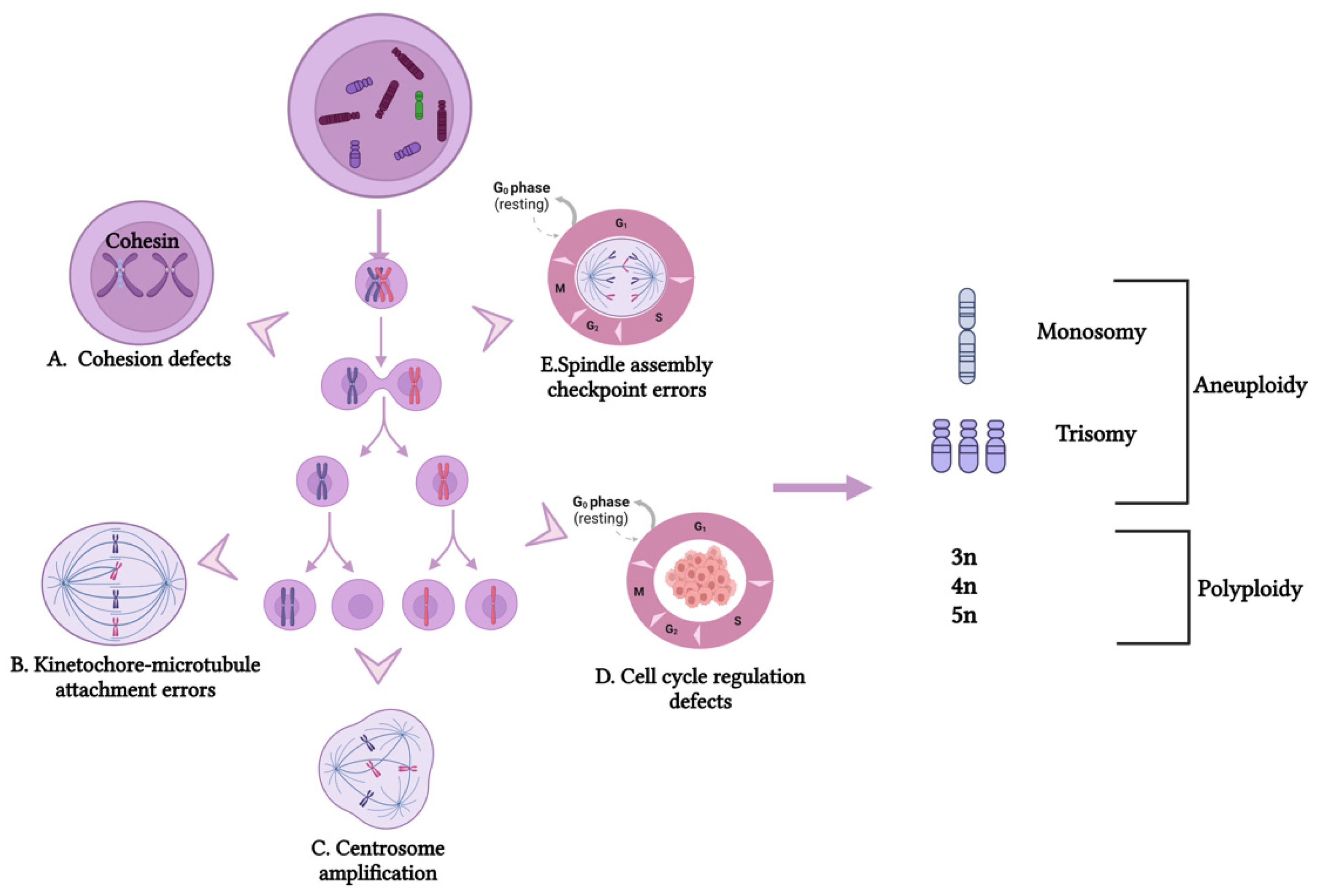
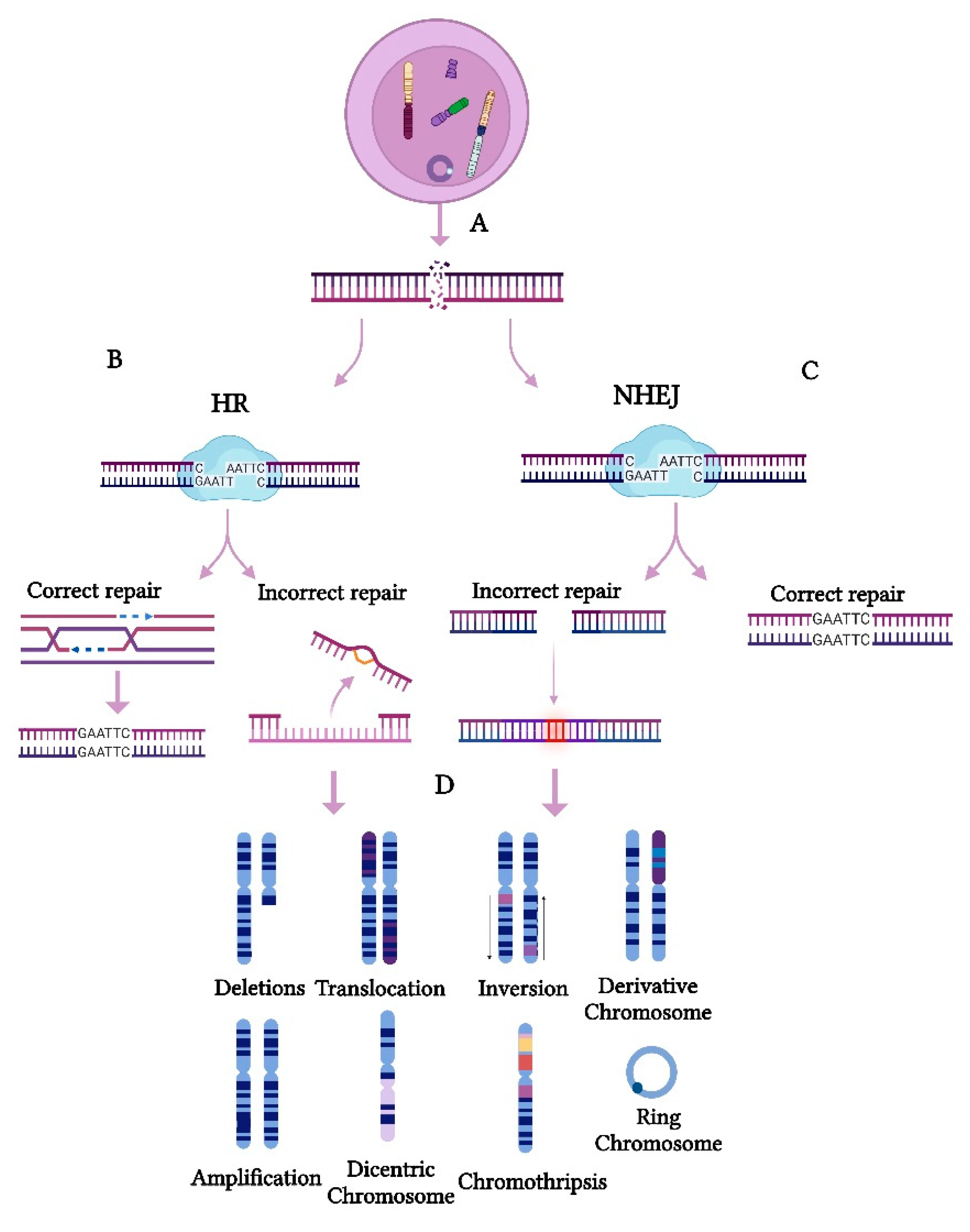
2.1. Cellular Mechanism of CIN
2.2. CIN in Cancer Progression
2.3. Polyploid Giant Cancer Cells (PGCCs) and CIN
2.4. Karyotype Coding: The Two-Phased Cancer Evolution Model and CIN
2.5. CH in Cancer Progression
2.6. The Impact of CIN and CH on Therapeutic Resistance and Patient Outcomes
3. CIN Phenotype and CH: Detection Methods
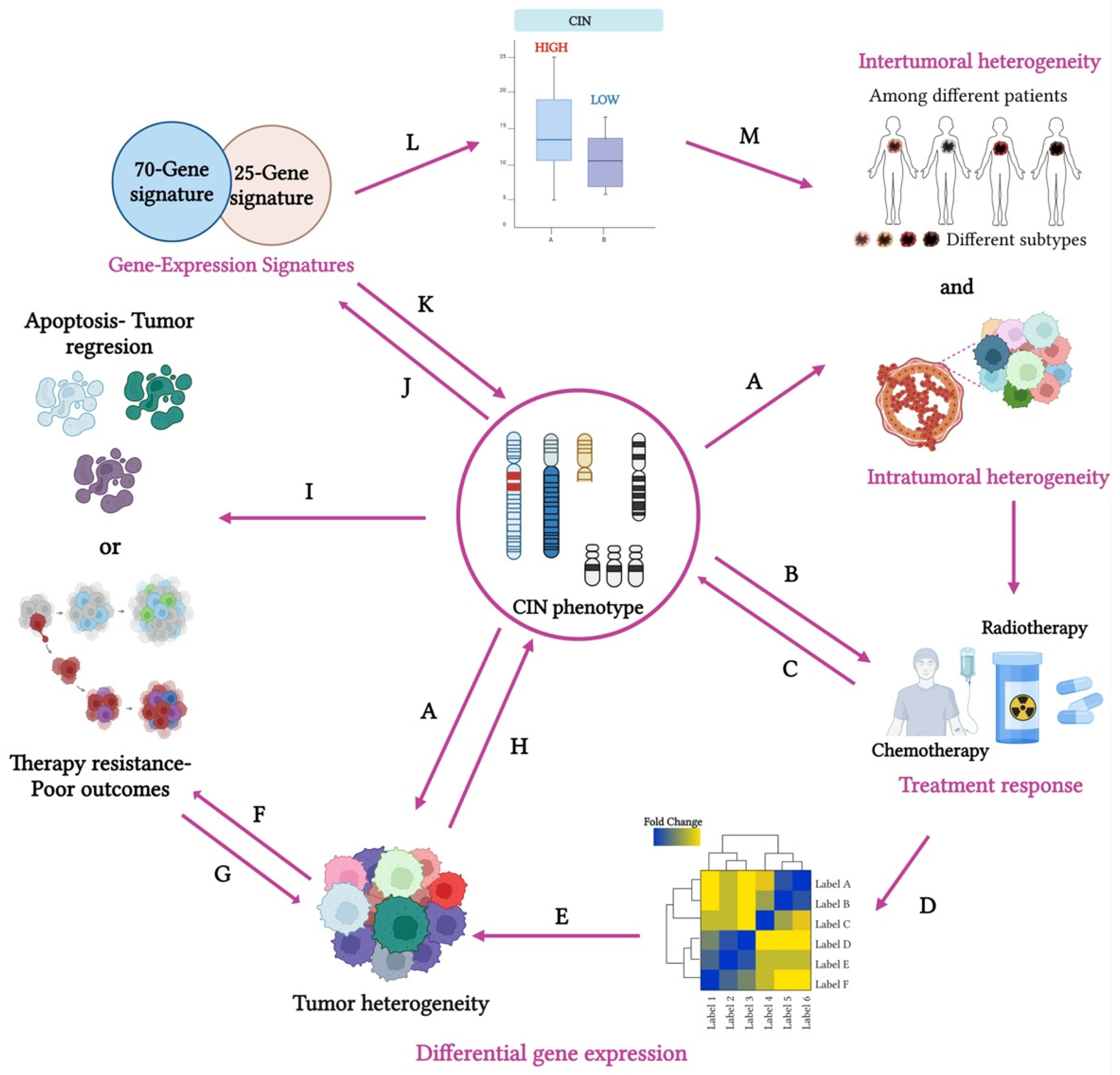
3.1. Gene Expression Signatures as Indicators of the CIN Phenotype and CH
The Role of CIN25 and CIN70 Gene Signatures in Predicting CIN and Their Clinical Implications
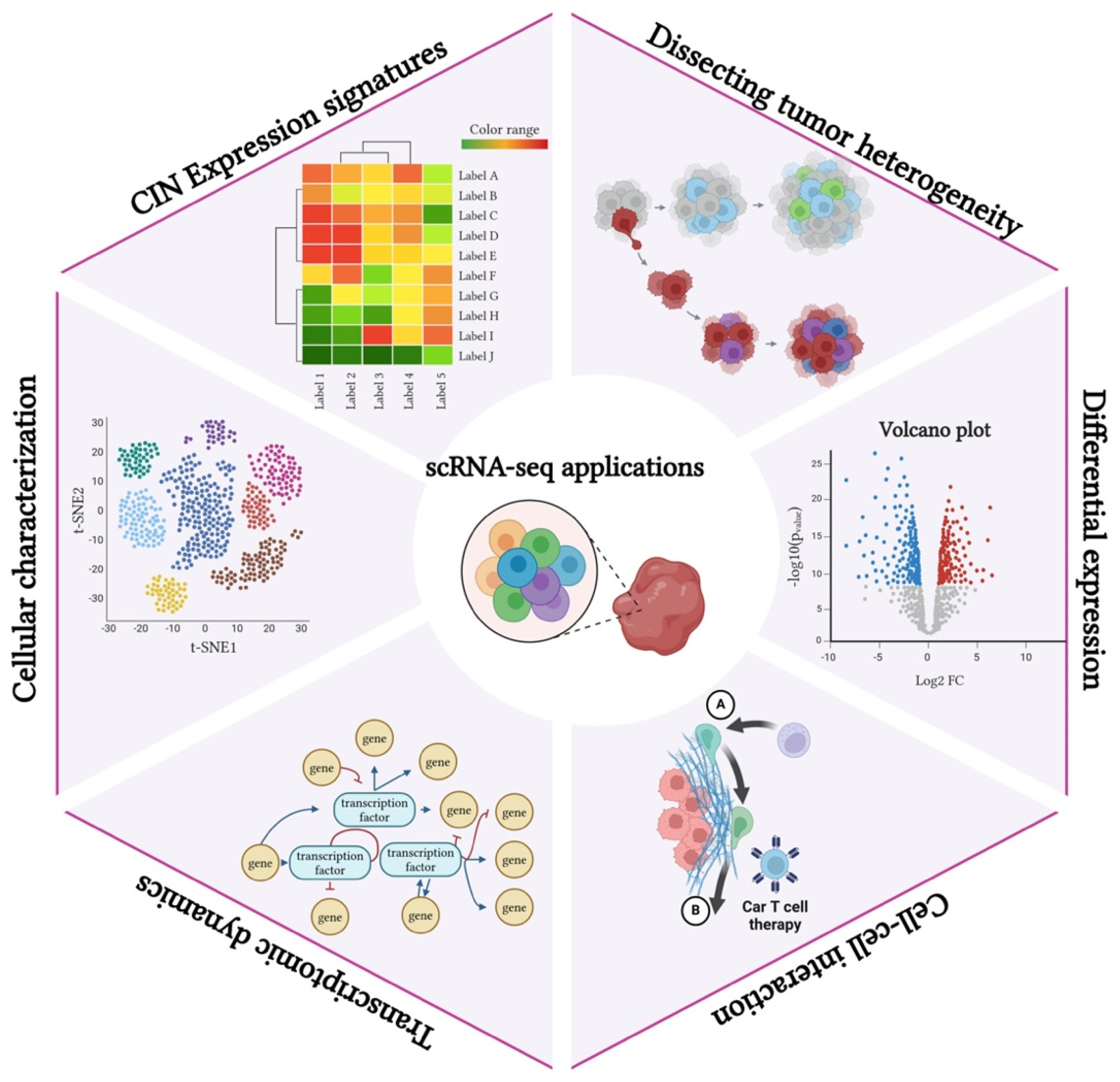
3.2. Single-Cell RNA Sequencing in the Study of the CIN Phenotype and CH
3.2.1. Overview of scRNA-seq Technology and Its Relevance to Cancer Research
3.2.2. Applications of scRNA-seq in Mapping the CIN Phenotype and CH
3.2.3. Insights Gained from Single-Cell Analyses Regarding the CIN Phenotype and Tumor Diversity
4. CIN and CH Targeting and Therapeutic Potential
4.1. CIN and Aneuploidy as Therapeutic Targets
4.2. Overview of Potential Therapeutic Strategies Targeting Aneuploidy, the CIN Phenotype, and CH
4.3. Clinical Implications of CIN in Current Cancer Therapies
5. Future Perspectives
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [PubMed]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar]
- Bakhoum, S.F.; Compton, D.A. Chromosomal instability and cancer: A complex relationship with therapeutic potential. J. Clin. Investig. 2012, 122, 1138–1143. [Google Scholar] [CrossRef] [PubMed]
- Sansregret, L.; Vanhaesebroeck, B.; Swanton, C. Determinants and clinical implications of chromosomal instability in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 139–150. [Google Scholar] [PubMed]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar]
- Gerstung, M.; Jolly, C.; Leshchiner, I.; Dentro, S.C.; Gonzalez, S.; Rosebrock, D.; Mitchell, T.J.; Rubanova, Y.; Anur, P.; Yu, K.; et al. The evolutionary history of 2,658 cancers. Nature 2020, 578, 122–128. [Google Scholar]
- Andor, N.; Graham, T.A.; Jansen, M.; Xia, L.C.; Aktipis, C.A.; Petritsch, C.; Ji, H.P.; Maley, C.C. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat. Med. 2016, 22, 105–113. [Google Scholar]
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef]
- Gao, R.; Davis, A.; McDonald, T.O.; Sei, E.; Shi, X.; Wang, Y.; Tsai, P.C.; Casasent, A.; Waters, J.; Zhang, H.; et al. Punctuated copy number evolution and clonal stasis in triple-negative breast cancer. Nat. Genet. 2016, 48, 1119–1130. [Google Scholar]
- Roylance, R.; Endesfelder, D.; Gorman, P.; Burrell, R.A.; Sander, J.; Tomlinson, I.; Hanby, A.M.; Speirs, V.; Richardson, A.L.; Birkbak, N.J.; et al. Relationship of extreme chromosomal instability with long-term survival in a retrospective analysis of primary breast cancer. Cancer Epidemiol. Biomark. Prev. 2011, 20, 2183–2194. [Google Scholar] [CrossRef]
- Liao, Y.Y.; Cao, W.M. The progress in our understanding of CIN in breast cancer research. Front. Oncol. 2023, 13, 1067735. Available online: https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2023.1067735/full (accessed on 25 November 2024).
- Birkbak, N.J.; Eklund, A.C.; Li, Q.; McClelland, S.E.; Endesfelder, D.; Tan, P.; Tan, I.B.; Richardson, A.L.; Szallasi, Z.; Swanton, C. Paradoxical relationship between chromosomal instability and survival outcome in cancer. Cancer Res. 2011, 71, 3447–3452. [Google Scholar] [CrossRef] [PubMed]
- Jamal-Hanjani, M.; A’Hern, R.; Birkbak, N.J.; Gorman, P.; Grönroos, E.; Ngang, S.; Nicola, P.; Rahman, L.; Thanopoulou, E.; Kelly, G.; et al. Extreme chromosomal instability forecasts improved outcome in ER-negative breast cancer: A prospective validation cohort study from the TACT trial. Ann. Oncol. 2015, 26, 1340–1346. [Google Scholar] [PubMed]
- Mo, H.; Wang, X.; Ma, F.; Qian, Z.; Sun, X.; Yi, Z.; Guan, X.; Li, L.; Liu, B.; Xu, B. Genome-wide chromosomal instability by cell-free DNA sequencing predicts survival in patients with metastatic breast cancer. Breast 2020, 53, 111–118. [Google Scholar] [CrossRef]
- Asbaghi, Y.; Thompson, L.L.; Lichtensztejn, Z.; McManus, K.J. KIF11 silencing and inhibition induces chromosome instability that may contribute to cancer. Genes Chromosomes Cancer 2017, 56, 668–680. [Google Scholar]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef]
- Smith, L.; Plug, A.; Thayer, M. Delayed replication timing leads to delayed mitotic chromosome condensation and chromosomal instability of chromosome translocations. Proc. Natl. Acad. Sci. USA 2001, 98, 13300–13305. [Google Scholar]
- Davoli, T.; Uno, H.; Wooten, E.C.; Elledge, S.J. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 2017, 355, eaaf8399. [Google Scholar] [CrossRef]
- Schubert, M.; Hong, C.; Jilderda, L.J.; Rueda, M.R.; Tijhuis, A.E.; Simon, J.E.; Bakker, P.L.; Cooper, J.L.; Damaskou, A.; Wardenaar, R.; et al. Cancer tolerance to chromosomal instability is driven by Stat1 inactivation in vivo. bioRxiv 2021. [Google Scholar] [CrossRef]
- van den Brink, A.; Suárez Peredo Rodríguez, M.F.; Foijer, F. Chromosomal instability and inflammation: A catch-22 for cancer cells. Chromosome Res. 2023, 31, 19. [Google Scholar]
- Bayani, J.; Selvarajah, S.; Maire, G.; Vukovic, B.; Al-Romaih, K.; Zielenska, M.; Squire, J.A. Genomic mechanisms and measurement of structural and numerical instability in cancer cells. Semin. Cancer Biol. 2007, 17, 5–18. [Google Scholar] [PubMed]
- Tanaka, K.; Hirota, T. Chromosomal instability: A common feature and a therapeutic target of cancer. Biochim. Biophys. Acta 2016, 1866, 64–75. [Google Scholar] [PubMed]
- Rangel, N.; Forero-Castro, M.; Rondón-Lagos, M. New Insights in the Cytogenetic Practice: Karyotypic Chaos, Non-Clonal Chromosomal Alterations and Chromosomal Instability in Human Cancer and Therapy Response. Genes 2017, 8, 155. [Google Scholar] [CrossRef] [PubMed]
- Heng, H.H.Q.; Regan, S.M.; Liu, G.; Ye, C.J. Why it is crucial to analyze non clonal chromosome aberrations or NCCAs? Mol. Cytogenet. 2016, 9, 15. [Google Scholar]
- Heng, H.H.Q.; Bremer, S.W.; Stevens, J.; Ye, K.J.; Miller, F.; Liu, G.; Ye, C.J. Cancer progression by non-clonal chromosome aberrations. J. Cell. Biochem. 2006, 98, 1424–1435. [Google Scholar]
- Thompson, S.L.; Bakhoum, S.F.; Compton, D.A. Mechanisms of Chromosomal Instability. Curr. Biol. 2010, 20, R285–R295. [Google Scholar]
- Li, Y.; Roberts, N.D.; Wala, J.A.; Shapira, O.; Schumacher, S.E.; Kumar, K.; Khurana, E.; Waszak, S.; Korbel, J.O.; Haber, J.E.; et al. Patterns of somatic structural variation in human cancer genomes. Nature 2020, 578, 112–121. [Google Scholar]
- Thompson, S.L.; Compton, D.A. Examining the link between chromosomal instability and aneuploidy in human cells. J Cell Biol. 2008, 180, 665–672. [Google Scholar]
- Moynahan, M.E.; Jasin, M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 2010, 11, 196–207. [Google Scholar]
- Geigl, J.B.; Obenauf, A.C.; Schwarzbraun, T.; Speicher, M.R. Defining “chromosomal instability”. Trends Genet. 2008, 24, 64–69. [Google Scholar]
- Bunting, S.F.; Callén, E.; Wong, N.; Chen, H.T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010, 141, 243–254. [Google Scholar] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [PubMed]
- Hosea, R.; Hillary, S.; Naqvi, S.; Wu, S.; Kasim, V. The two sides of chromosomal instability: Drivers and brakes in cancer. Sig. Transduct. Target. Ther. 2024, 9, 1–30. [Google Scholar]
- ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar]
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhang, C.Z.; Wala, J.; Mermel, C.H.; et al. Pan-cancer patterns of somatic copy-number alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar]
- Holland, A.J.; Cleveland, D.W. Boveri revisited: Chromosomal instability, aneuploidy and tumorigenesis. Nat. Rev. Mol. Cell Biol. 2009, 10, 478–487. [Google Scholar]
- Janiszewska, M. The microcosmos of intratumor heterogeneity: The space-time of cancer evolution. Oncogene 2020, 39, 2031–2039. [Google Scholar]
- Lee, A.J.X.; Endesfelder, D.; Rowan, A.J.; Walther, A.; Birkbak, N.J.; Futreal, P.A.; Downward, J.; Szallasi, Z.; Tomlinson, I.P.M.; Howell, M.; et al. Chromosomal Instability Confers Intrinsic Multidrug Resistance. Cancer Res. 2011, 71, 1858–1870. [Google Scholar]
- van Dijk, E.; van den Bosch, T.; Lenos, K.J.; El Makrini, K.; Nijman, L.E.; van Essen, H.F.B.; Lansu, N.; Boekhout, M.; Hageman, J.H.; Fitzgerald, R.C.; et al. Chromosomal copy number heterogeneity predicts survival rates across cancers. Nat. Commun. 2021, 12, 3188. [Google Scholar]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; McGranahan, N.; Dewhurst, S.M.; Burrell, R.A.; Tomlinson, I.; Swanton, C. Cancer: Evolution within a lifetime. Annu. Rev. Genet. 2014, 48, 215–236. [Google Scholar] [CrossRef] [PubMed]
- Drews, R.M.; Hernando, B.; Tarabichi, M.; Haase, K.; Lesluyes, T.; Smith, P.S.; Morrill Gavarró, L.; Couturier, D.L.; Liu, L.; Schneider, M.; et al. A pan-cancer compendium of chromosomal instability. Nature 2022, 606, 976–983. [Google Scholar] [CrossRef] [PubMed]
- Houlahan, K.E.; Mangiante, L.; Sotomayor-Vivas, C.; Adimoelja, A.; Park, S.; Khan, A.; Pribus, S.J.; Ma, Z.; Caswell-Jin, J.L.; Curtis, C. Complex rearrangements fuel ER+ and HER2+ breast tumours. Nature 2025, 638, 510–518. [Google Scholar] [CrossRef]
- Kuzmin, E.; Baker, T.M.; Lesluyes, T.; Monlong, J.; Abe, K.T.; Coelho, P.P.; Schwartz, M.; Del Corpo, J.; Zou, D.; Morin, G.; et al. Evolution of chromosome-arm aberrations in breast cancer through genetic network rewiring. Cell Rep. 2024, 43, 113988. [Google Scholar] [CrossRef]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Zhou, X.; Zhou, M.; Zheng, M.; Tian, S.; Yang, X.; Ning, Y.; Li, Y.; Zhang, S. Polyploid giant cancer cells and cancer progression. Front. Cell Dev. Biol. 2022, 10, 1017588. [Google Scholar] [CrossRef]
- Niu, N.; Mercado-Uribe, I.; Liu, J. Dedifferentiation into blastomere-like cancer stem cells via formation of polyploid giant cancer cells. Oncogene 2017, 36, 4887–4900. [Google Scholar] [CrossRef]
- Mosieniak, G.; Sliwinska, M.A.; Alster, O.; Strzeszewska, A.; Sunderland, P.; Piechota, M.; Was, H.; Sikora, E. Polyploidy Formation in Doxorubicin-Treated Cancer Cells Can Favor Escape from Senescence. Neoplasia 2015, 17, 882–893. [Google Scholar] [CrossRef]
- Zhang, S.; Mercado-Uribe, I.; Sood, A.; Bast, R.C.; Liu, J. Coevolution of neoplastic epithelial cells and multilineage stroma via polyploid giant cells during immortalization and transformation of mullerian epithelial cells. Genes Cancer 2016, 7, 60–72. [Google Scholar] [CrossRef]
- Davoli, T.; de Lange, T. Telomere-Driven Tetraploidization Occurs in Human Cells Undergoing Crisis and Promotes Transformation of Mouse Cells. Cancer Cell 2012, 21, 765–776. [Google Scholar] [PubMed]
- Erenpreisa, J.; Cragg, M.S. Three steps to the immortality of cancer cells: Senescence, polyploidy and self-renewal. Cancer Cell Int. 2013, 13, 92. [Google Scholar]
- Saini, G.; Joshi, S.; Garlapati, C.; Li, H.; Kong, J.; Krishnamurthy, J.; Reid, M.D.; Aneja, R. Polyploid giant cancer cell characterization: New frontiers in predicting response to chemotherapy in breast cancer. Semin. Cancer Biol. 2022, 81, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Fei, F.; Zhang, D.; Yang, Z.; Wang, S.; Wang, X.; Wu, Z.; Wu, Q.; Zhang, S. The number of polyploid giant cancer cells and epithelial-mesenchymal transition-related proteins are associated with invasion and metastasis in human breast cancer. J. Exp. Clin. Cancer Res. 2015, 34, 158. [Google Scholar] [CrossRef]
- Bielski, C.M.; Zehir, A.; Penson, A.V.; Donoghue, M.T.A.; Chatila, W.; Armenia, J.; Chang, M.T.; Schram, A.M.; Jonsson, P.; Bandlamudi, C.; et al. Genome doubling shapes the evolution and prognosis of advanced cancers. Nat. Genet. 2018, 50, 1189–1195. [Google Scholar]
- Heng, H.H. Chapter 3—Genome Chaos and Macrocellular Evolution: How Evolutionary Cytogenetics Unravels the Mystery of Cancer. In Genome Chaos; Heng, H.H., Ed.; Academic Press: New York, NY, USA, 2019; pp. 95–168. Available online: https://www.sciencedirect.com/science/article/pii/B9780128136355000033 (accessed on 3 March 2025).
- Heng, J.; Heng, H.H. Two-phased evolution: Genome chaos-mediated information creation and maintenance. Prog. Biophys. Mol. Biol. 2021, 165, 29–42. [Google Scholar]
- Heng, H.H.Q.; Stevens, J.B.; Bremer, S.W.; Liu, G.; Abdallah, B.Y.; Ye, C.J. Evolutionary Mechanisms and Diversity in Cancer. In Advances in Cancer Research; Gisselsson, D., Ed.; Academic Press: New York, NY, USA, 2011; pp. 217–253. Available online: https://www.sciencedirect.com/science/article/pii/B9780123876881000089 (accessed on 3 March 2025).
- Heng, H.H.Q.; Stevens, J.B.; Lawrenson, L.; Liu, G.; Ye, K.J.; Bremer, S.W.; Ye, C.J. Patterns of Genome Dynamics and Cancer Evolution. Cell. Oncol. 2008, 30, 513–514. [Google Scholar]
- Heng, H.H.Q.; Liu, G.; Stevens, J.B.; Bremer, S.W.; Ye, K.J.; Abdallah, B.Y.; Horne, S.D.; Ye, C.J. Decoding the genome beyond sequencing: The new phase of genomic research. Genomics 2011, 98, 242–252. [Google Scholar]
- Ye, C.J.; Liu, G.; Bremer, S.W.; Heng, H.H.Q. The dynamics of cancer chromosomes and genomes. Cytogenet. Genome Res. 2007, 118, 237–246. [Google Scholar]
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour evolution inferred by single-cell sequencing. Nature 2011, 472, 90–94. [Google Scholar] [CrossRef]
- McClelland, S.E. Role of chromosomal instability in cancer progression. Endocr. Relat. Cancer 2017, 24, T23–T31. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yu, X.; Xu, Y. Breast cancer gene expression signatures: Development and clinical significance-a narrative review. Transl. Breast Cancer Res. 2023, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Van Loo, P.; Wedge, D.C.; Alexandrov, L.B.; Greenman, C.D.; Lau, K.W.; Raine, K.; Jones, D.; Marshall, J.; Ramakrishna, M.; et al. The life history of 21 breast cancers. Cell 2012, 149, 994–1007. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; Van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational Processes Molding the Genomes of 21 Breast Cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef]
- Wagenblast, E.; Soto, M.; Gutiérrez-Ángel, S.; Hartl, C.A.; Gable, A.L.; Maceli, A.R.; Erard, N.; Williams, A.M.; Kim, S.Y.; Dickopf, S.; et al. A model of breast cancer heterogeneity reveals vascular mimicry as a driver of metastasis. Nature 2015, 520, 358–362. [Google Scholar] [CrossRef]
- Merino, D.; Weber, T.S.; Serrano, A.; Vaillant, F.; Liu, K.; Pal, B.; Di Stefano, L.; Schreuder, J.; Lin, D.; Chen, Y.; et al. Barcoding reveals complex clonal behavior in patient-derived xenografts of metastatic triple negative breast cancer. Nat. Commun. 2019, 10, 766. [Google Scholar]
- McGranahan, N.; Burrell, R.A.; Endesfelder, D.; Novelli, M.R.; Swanton, C. Cancer chromosomal instability: Therapeutic and diagnostic challenges. EMBO Rep. 2012, 13, 528–538. [Google Scholar] [CrossRef]
- Swanton, C.; Nicke, B.; Schuett, M.; Eklund, A.C.; Ng, C.; Li, Q.; Hardcastle, T.; Lee, A.; Roy, R.; East, P.; et al. Chromosomal instability determines taxane response. Proc. Natl. Acad. Sci. USA 2009, 106, 8671–8676. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2019, 2, 141–160. [Google Scholar] [CrossRef]
- Carter, S.L.; Eklund, A.C.; Kohane, I.S.; Harris, L.N.; Szallasi, Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat. Genet. 2006, 38, 1043–1048. [Google Scholar] [CrossRef]
- Biermann, J.; Nemes, S.; Parris, T.Z.; Engqvist, H.; Werner Rönnerman, E.; Kovács, A.; Karlsson, P.; Helou, K. A 17-marker panel for global genomic instability in breast cancer. Genomics 2020, 112, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Spurr, L.F.; Weichselbaum, R.R.; Pitroda, S.P. Tumor aneuploidy predicts survival following immunotherapy across multiple cancers. Nat. Genet. 2022, 54, 1782–1785. [Google Scholar] [CrossRef] [PubMed]
- Kohlruss, M.; Krenauer, M.; Grosser, B.; Pfarr, N.; Jesinghaus, M.; Slotta-Huspenina, J.; Novotny, A.; Hapfelmeier, A.; Schmidt, T.; Steiger, K.; et al. Diverse “just-right” levels of chromosomal instability and their clinical implications in neoadjuvant treated gastric cancer. Br. J. Cancer 2021, 125, 1621–1631. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.C.; Sheltzer, J.M. Genome-wide identification and analysis of prognostic features in human cancers. Cell Rep. 2022, 38, 110569. [Google Scholar] [CrossRef]
- Steele, C.D.; Abbasi, A.; Islam, S.M.A.; Bowes, A.L.; Khandekar, A.; Haase, K.; Hames-Fathi, S.; Ajayi, D.; Verfaillie, A.; Dhami, P.; et al. Signatures of copy number alterations in human cancer. Nature 2022, 606, 984–991. [Google Scholar] [CrossRef]
- Greene, S.B.; Dago, A.E.; Leitz, L.J.; Wang, Y.; Lee, J.; Werner, S.L.; Gendreau, S.; Patel, P.; Jia, S.; Zhang, L.; et al. Chromosomal Instability Estimation Based on Next Generation Sequencing and Single Cell Genome Wide Copy Number Variation Analysis. PLoS ONE 2016, 11, e0165089. [Google Scholar] [CrossRef]
- Lynch, A.R.; Bradford, S.; Zhou, S.; Oxendine, K.; Henderson, L.; Horner, V.L.; Weaver, B.A.; Burkard, M.E. A survey of chromosomal instability measures across mechanistic models. Proc. Natl. Acad. Sci. USA 2024, 121, e2309621121. [Google Scholar] [CrossRef]
- Bakhoum, S.F.; Cantley, L.C. The Multifaceted Role of Chromosomal Instability in Cancer and Its Microenvironment. Cell 2018, 174, 1347–1360. [Google Scholar] [CrossRef]
- Santaguida, S.; Amon, A. Short- and long-term effects of chromosome mis-segregation and aneuploidy. Nat. Rev. Mol. Cell Biol. 2015, 16, 473–485. [Google Scholar] [CrossRef]
- Sheltzer, J.M. A transcriptional and metabolic signature of primary aneuploidy is present in chromosomally unstable cancer cells and informs clinical prognosis. Cancer Res. 2013, 73, 6401–6412. [Google Scholar] [CrossRef]
- Acharya, C.R.; Hsu, D.S.; Anders, C.K.; Anguiano, A.; Salter, K.H.; Walters, K.S.; Redman, R.C.; Tuchman, S.A.; Moylan, C.A.; Mukherjee, S.; et al. Gene expression signatures, clinicopathological features, and individualized therapy in breast cancer. JAMA 2008, 299, 1574–1587. [Google Scholar]
- Vishwakarma, R.; McManus, K.J. Chromosome Instability; Implications in Cancer Development, Progression, and Clinical Outcomes. Cancers 2020, 12, 824. [Google Scholar] [CrossRef] [PubMed]
- Andor, N.; Maley, C.C.; Ji, H.P. Genomic Instability in Cancer: Teetering on the Limit of Tolerance. Cancer Res. 2017, 77, 2179–2185. [Google Scholar] [CrossRef] [PubMed]
- Jovic, D.; Liang, X.; Zeng, H.; Lin, L.; Xu, F.; Luo, Y. Single-cell RNA sequencing technologies and applications: A brief overview. Clin. Transl. Med. 2022, 12, e694. [Google Scholar]
- Wills, Q.F.; Mead, A.J. Application of single-cell genomics in cancer: Promise and challenges. Hum. Mol. Genet. 2015, 24, R74–R84. [Google Scholar] [CrossRef]
- Xu, L.; Saunders, K.; Huang, S.P.; Knutsdottir, H.; Martinez-Algarin, K.; Terrazas, I.; Chen, K.; McArthur, H.M.; Maués, J.; Hodgdon, C.; et al. A comprehensive single-cell breast tumor atlas defines epithelial and immune heterogeneity and interactions predicting anti-PD-1 therapy response. Cell Rep. Med. 2024, 5, 101511. [Google Scholar]
- Zhao, Y.; Carter, R.; Natarajan, S.; Varn, F.S.; Compton, D.A.; Gawad, C.; Cheng, C.; Godek, K.M. Single-cell RNA sequencing reveals the impact of chromosomal instability on glioblastoma cancer stem cells. BMC Med. Genom. 2019, 12, 79. [Google Scholar] [CrossRef]
- Laumont, C.M.; Nelson, B.H. B cells in the tumor microenvironment: Multi-faceted organizers, regulators, and effectors of anti-tumor immunity. Cancer Cell 2023, 41, 466–489. [Google Scholar]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar]
- Lim, S.Y.; Lin, Y.; Lee, J.H.; Pedersen, B.; Stewart, A.; Scolyer, R.A.; Long, G.V.; Yang, J.Y.H.; Rizos, H. Single-cell RNA sequencing reveals melanoma cell state-dependent heterogeneity of response to MAPK inhibitors. eBioMedicine 2024, 107, 105308. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Suva, M.L. Cancer cell states: Lessons from ten years of single-cell RNA-sequencing of human tumors. Cancer Cell 2024, 42, 1497–1506. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; Chu, H.; Jin, Z.; Long, H.; Zhu, B. High-throughput single-cell sequencing in cancer research. Sig. Transduct. Target. Ther. 2022, 7, 1–20. [Google Scholar] [CrossRef]
- Wu, S.Z.; Al-Eryani, G.; Roden, D.L.; Junankar, S.; Harvey, K.; Andersson, A.; Thennavan, A.; Wang, C.; Torpy, J.R.; Bartonicek, N.; et al. A single-cell and spatially resolved atlas of human breast cancers. Nat. Genet. 2021, 53, 1334–1347. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hubisz, M.J.; Earlie, E.M.; Duran, M.A.; Hong, C.; Varela, A.A.; Lettera, E.; Deyell, M.; Tavora, B.; Havel, J.J.; et al. Non-cell-autonomous cancer progression from chromosomal instability. Nature 2023, 620, 1080–1088. [Google Scholar] [CrossRef]
- Ansari, D.; Bellido, C.D.P.; Bauden, M.; Andersson, R. Centrosomal Abnormalities in Pancreatic Cancer: Molecular Mechanisms and Clinical Implications. Anticancer Res. 2018, 38, 1241–1245. [Google Scholar]
- Watkins, T.B.K.; Lim, E.L.; Petkovic, M.; Elizalde, S.; Birkbak, N.J.; Wilson, G.A.; Moore, D.A.; Grönroos, E.; Rowan, A.; Dewhurst, S.M.; et al. Pervasive chromosomal instability and karyotype order in tumour evolution. Nature 2020, 587, 126–132. [Google Scholar] [CrossRef]
- Morden, C.R.; Farrell, A.C.; Sliwowski, M.; Lichtensztejn, Z.; Altman, A.D.; Nachtigal, M.W.; McManus, K.J. Chromosome instability is prevalent and dynamic in high-grade serous ovarian cancer patient samples. Gynecol. Oncol. 2021, 161, 769–778. [Google Scholar] [CrossRef]
- Choma, D.; Daurès, J.P.; Quantin, X.; Pujol, J.L. Aneuploidy and prognosis of non-small-cell lung cancer: A meta-analysis of published data. Br. J. Cancer 2001, 85, 14–22. [Google Scholar] [CrossRef]
- Al-Rawi, D.H.; Lettera, E.; Li, J.; DiBona, M.; Bakhoum, S.F. Targeting chromosomal instability in patients with cancer. Nat. Rev. Clin. Oncol. 2024, 21, 645–659. [Google Scholar] [CrossRef]
- Bakhoum, S.F. Targeting the undruggable. Science 2023, 380, 47. [Google Scholar]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Mao, J.H.; Zhu, W.; Jain, A.K.; Liu, K.; Brown, J.B.; Karpen, G.H. Centromere and kinetochore gene misexpression predicts cancer patient survival and response to radiotherapy and chemotherapy. Nat. Commun. 2016, 7, 12619. [Google Scholar]
- Vitale, I.; Shema, E.; Loi, S.; Galluzzi, L. Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nat. Med. 2021, 27, 212–224. [Google Scholar]
- Bhatia, S.; Khanna, K.K.; Duijf, P.H.G. Targeting chromosomal instability and aneuploidy in cancer. Trends Pharmacol. Sci. 2024, 45, 210–224. [Google Scholar]
- Peuget, S.; Zhou, X.; Selivanova, G. Translating p53-based therapies for cancer into the clinic. Nat. Rev. Cancer 2024, 24, 192–215. [Google Scholar] [CrossRef]
- PYNNACLE Study|Rezatapopt|PYNNACLE Clinical Study. 2024. Available online: https://www.pynnaclestudy.com/ (accessed on 25 November 2024).
- Wang, W.; Albadari, N.; Du, Y.; Fowler, J.F.; Sang, H.T.; Xian, W.; McKeon, F.; Li, W.; Zhou, J.; Zhang, R. MDM2 Inhibitors for Cancer Therapy: The Past, Present, and Future. Pharmacol. Rev. 2024, 76, 414–453. [Google Scholar]
- Maniswami, R.R.; Prashanth, S.; Karanth, A.V.; Koushik, S.; Govindaraj, H.; Mullangi, R.; Rajagopal, S.; Jegatheesan, S.K. PLK4: A link between centriole biogenesis and cancer. Expert Opin. Ther. Targets 2018, 22, 59–73. [Google Scholar]
- Mason, J.M.; Lin, D.C.C.; Wei, X.; Che, Y.; Yao, Y.; Kiarash, R.; Cescon, D.W.; Fletcher, G.C.; Awrey, D.E.; Bray, M.R.; et al. Functional characterization of CFI-400945, a Polo-like kinase 4 inhibitor, as a potential anticancer agent. Cancer Cell 2014, 26, 163–176. [Google Scholar] [CrossRef]
- Veitch, Z.W.; Cescon, D.W.; Denny, T.; Yonemoto, L.M.; Fletcher, G.; Brokx, R.; Sampson, P.; Li, S.W.; Pugh, T.J.; Bruce, J.; et al. Safety and tolerability of CFI-400945, a first-in-class, selective PLK4 inhibitor in advanced solid tumours: A phase 1 dose-escalation trial. Br. J. Cancer 2019, 121, 318–324. [Google Scholar]
- Murphy, T.; Mason, J.M.; Leber, B.; Bray, M.R.; Chan, S.M.; Gupta, V.; Khalaf, D.; Maze, D.; McNamara, C.J.; Schimmer, A.D.; et al. Preclinical characterization and clinical trial of CFI-400945, a polo-like kinase 4 inhibitor, in patients with relapsed/refractory acute myeloid leukemia and higher-risk myelodysplastic neoplasms. Leukemia 2024, 38, 502–512. [Google Scholar] [PubMed]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [PubMed]
- Yeow, Z.Y.; Lambrus, B.G.; Marlow, R.; Zhan, K.H.; Durin, M.A.; Evans, L.T.; Scott, P.M.; Phan, T.; Park, E.; Ruiz, L.A.; et al. Targeting TRIM37-driven centrosome dysfunction in 17q23-amplified breast cancer. Nature 2020, 585, 447–452. [Google Scholar] [PubMed]
- Meitinger, F.; Ohta, M.; Lee, K.Y.; Watanabe, S.; Davis, R.L.; Anzola, J.V.; Kabeche, R.; Jenkins, D.A.; Shiau, A.K.; Desai, A.; et al. TRIM37 controls cancer-specific vulnerability to PLK4 inhibition. Nature 2020, 585, 440–446. [Google Scholar] [CrossRef]
- Bhatnagar, S.; Gazin, C.; Chamberlain, L.; Ou, J.; Zhu, X.; Tushir, J.S.; Virbasius, C.M.; Lin, L.; Zhu, L.J.; Wajapeyee, N.; et al. TRIM37 is a new histone H2A ubiquitin ligase and breast cancer oncoprotein. Nature 2014, 516, 116–120. [Google Scholar]
- Newman, D.L.; Thurgood, L.A.; Gregory, S.L. The impact of aneuploidy on cellular homeostasis. Free Radic. Res. 2019, 53, 705–713. [Google Scholar]
- Li, R.; Zhu, J. Effects of aneuploidy on cell behaviour and function. Nat. Rev. Mol. Cell Biol. 2022, 23, 250–265. [Google Scholar]
- Liu, T.; Yang, K.; Chen, J.; Qi, L.; Zhou, X.; Wang, P. Comprehensive Pan-Cancer Analysis of KIF18A as a Marker for Prognosis and Immunity. Biomolecules 2023, 13, 326. [Google Scholar] [CrossRef]
- Zhang, C.; Zhu, C.; Chen, H.; Li, L.; Guo, L.; Jiang, W.; Lu, S.H. Kif18A is involved in human breast carcinogenesis. Carcinogenesis 2010, 31, 1676–1684. [Google Scholar]
- Marquis, C.; Fonseca, C.L.; Queen, K.A.; Wood, L.; Vandal, S.E.; Malaby, H.L.H.; Clayton, J.E.; Stumpff, J. Chromosomally unstable tumor cells specifically require KIF18A for proliferation. Nat. Commun. 2021, 12, 1213. [Google Scholar]
- Payton, M.; Belmontes, B.; Hanestad, K.; Moriguchi, J.; Chen, K.; McCarter, J.D.; Chung, G.; Ninniri, M.S.; Sun, J.; Manoukian, R.; et al. Small-molecule inhibition of kinesin KIF18A reveals a mitotic vulnerability enriched in chromosomally unstable cancers. Nat. Cancer 2024, 5, 66–84. [Google Scholar] [PubMed]
- Gliech, C.R.; Yeow, Z.Y.; Tapias-Gomez, D.; Yang, Y.; Huang, Z.; Tijhuis, A.E.; Spierings, D.C.; Foijer, F.; Chung, G.; Tamayo, N.; et al. Weakened APC/C activity at mitotic exit drives cancer vulnerability to KIF18A inhibition. EMBO J. 2024, 43, 666–694. [Google Scholar] [CrossRef] [PubMed]
- Belmontes, B.; Moriguchi, J.; Chung, G.; Sun, J.; Ninniri, M.S.S.; Hanestad, K.; Chen, K.; McCarter, J.D.; Dahal, U.P.; Ghimire-Rijal, S.; et al. Abstract 516: Discovery and preclinical characterization of AMG 650, a first-in-class inhibitor of kinesin KIF18A motor protein with potent activity against chromosomally unstable cancers. Cancer Res. 2023, 83 (Suppl. S7), 516. [Google Scholar]
- Tu, P.; Zhang, C.; Lu, B.; Xia, Y.; Yang, F. Abstract 664: GSC000190, a highly potent inhibitor of KIF18A, for tumors with chromosome instability. Cancer Res. 2024, 84 (Suppl. S6), 664. [Google Scholar]
- Gaillard, S.; Starodub, A.; O’Neil, B.; Mathews, C.A.; Dumbrava, E.E.; Friedman, C.F.; Swiecicki, P.; Oh, C.; Andreu, C.; Lee, L.; et al. A phase I/II, first-in-human study of VLS-1488, an oral KIF18A inhibitor, in patients with advanced cancer. J. Clin. Oncol. 2024, 42 (Suppl. S16), TPS3181. [Google Scholar]
- Samson, N.; Ablasser, A. The cGAS-STING pathway and cancer. Nat. Cancer 2022, 3, 1452–1463. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar]
- Zimmerli, D.; Brambillasca, C.S.; Talens, F.; Bhin, J.; Linstra, R.; Romanens, L.; Bhattacharya, A.; Joosten, S.E.P.; Da Silva, A.M.; Padrao, N.; et al. MYC promotes immune-suppression in triple-negative breast cancer via inhibition of interferon signaling. Nat. Commun. 2022, 13, 6579. [Google Scholar]
- Heijink, A.M.; Talens, F.; Jae, L.T.; van Gijn, S.E.; Fehrmann, R.S.N.; Brummelkamp, T.R.; van Vugt, M.A.T.M. BRCA2 deficiency instigates cGAS-mediated inflammatory signaling and confers sensitivity to tumor necrosis factor-alpha-mediated cytotoxicity. Nat. Commun. 2019, 10, 100. [Google Scholar]
- Hong, C.; Schubert, M.; Tijhuis, A.E.; Requesens, M.; Roorda, M.; van den Brink, A.; Ruiz, L.A.; Bakker, P.L.; van der Sluis, T.; Pieters, W.; et al. cGAS-STING drives the IL-6-dependent survival of chromosomally instable cancers. Nature 2022, 607, 366–373. [Google Scholar] [CrossRef]
- Chen, M.; Yu, S.; van der Sluis, T.; Zwager, M.C.; Schröder, C.P.; van der Vegt, B.; van Vugt, M.A.T.M. cGAS-STING pathway expression correlates with genomic instability and immune cell infiltration in breast cancer. NPJ Breast Cancer 2024, 10, 1. [Google Scholar]
- Liu, H.; Zhang, H.; Wu, X.; Ma, D.; Wu, J.; Wang, L.; Jiang, Y.; Fei, Y.; Zhu, C.; Tan, R.; et al. Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature 2018, 563, 131–136. [Google Scholar] [PubMed]
- Hong, C.; Tijhuis, A.E.; Foijer, F. The cGAS Paradox: Contrasting Roles for cGAS-STING Pathway in Chromosomal Instability. Cells 2019, 8, 1228. [Google Scholar] [CrossRef]
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; Taunk, N.K.; et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018, 553, 467–472. [Google Scholar] [PubMed]
- Shih, J.; Sarmashghi, S.; Zhakula-Kostadinova, N.; Zhang, S.; Georgis, Y.; Hoyt, S.H.; Cuoco, M.S.; Gao, G.F.; Spurr, L.F.; Berger, A.C.; et al. Cancer aneuploidies are shaped primarily by effects on tumour fitness. Nature 2023, 619, 793–800. [Google Scholar] [CrossRef]
- Watson, E.V.; Lee, J.J.K.; Gulhan, D.C.; Melloni, G.E.M.; Venev, S.V.; Magesh, R.Y.; Frederick, A.; Chiba, K.; Wooten, E.C.; Naxerova, K.; et al. Chromosome evolution screens recapitulate tissue-specific tumor aneuploidy patterns. Nat. Genet. 2024, 56, 900–912. [Google Scholar] [CrossRef]
- Klockner, T.C.; Campbell, C.S. Selection forces underlying aneuploidy patterns in cancer. Mol. Cell. Oncol. 2024, 11, 2369388. [Google Scholar] [CrossRef]
- Cai, Y.; Crowther, J.; Pastor, T.; Abbasi Asbagh, L.; Baietti, M.F.; De Troyer, M.; Vazquez, I.; Talebi, A.; Renzi, F.; Dehairs, J.; et al. Loss of Chromosome 8p Governs Tumor Progression and Drug Response by Altering Lipid Metabolism. Cancer Cell 2016, 29, 751–766. [Google Scholar]
- Huth, T.; Dreher, E.C.; Lemke, S.; Fritzsche, S.; Sugiyanto, R.N.; Castven, D.; Ibberson, D.; Sticht, C.; Eiteneuer, E.; Jauch, A.; et al. Chromosome 8p engineering reveals increased metastatic potential targetable by patient-specific synthetic lethality in liver cancer. Sci. Adv. 2023, 9, eadh1442. [Google Scholar] [CrossRef]
- Lukow, D.A.; Sausville, E.L.; Suri, P.; Chunduri, N.K.; Wieland, A.; Leu, J.; Smith, J.C.; Girish, V.; Kumar, A.A.; Kendall, J.; et al. Chromosomal instability accelerates the evolution of resistance to anti-cancer therapies. Dev. Cell 2021, 56, 2427–2439.e4. [Google Scholar] [CrossRef]
- Andrade, J.R.; Gallagher, A.D.; Maharaj, J.; McClelland, S.E. Disentangling the roles of aneuploidy, chromosomal instability and tumour heterogeneity in developing resistance to cancer therapies. Chromosome Res. 2023, 31, 28. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Kabeche, L.; Wood, M.D.; Laucius, C.D.; Qu, D.; Laughney, A.M.; Reynolds, G.E.; Louie, R.J.; Phillips, J.; Chan, D.A.; et al. Numerical chromosomal instability mediates susceptibility to radiation treatment. Nat. Commun. 2015, 6, 5990. [Google Scholar] [CrossRef] [PubMed]
- Cosper, P.F.; Paracha, M.; Jones, K.M.; Hrycyniak, L.; Henderson, L.; Bryan, A.; Eyzaguirre, D.; McCunn, E.; Boulanger, E.; Wan, J.; et al. Chromosomal instability increases radiation sensitivity. bioRxiv 2024. [Google Scholar] [CrossRef]
- Pellizzari, S.; Bhat, V.; Athwal, H.; Cescon, D.W.; Allan, A.L.; Parsyan, A. PLK4 as a potential target to enhance radiosensitivity in triple-negative breast cancer. Radiat. Oncol. 2024, 19, 24. [Google Scholar] [CrossRef]
- Berton, S.; Cusan, M.; Segatto, I.; Citron, F.; D’Andrea, S.; Benevol, S.; Avanzo, M.; Dall’Acqua, A.; Schiappacassi, M.; Bristow, R.G.; et al. Loss of p27kip1 increases genomic instability and induces radio-resistance in luminal breast cancer cells. Sci. Rep. 2017, 7, 595. [Google Scholar] [CrossRef]
- Mosca, L.; Ilari, A.; Fazi, F.; Assaraf, Y.G.; Colotti, G. Taxanes in cancer treatment: Activity, chemoresistance and its overcoming. Drug Resist. Updat. 2021, 54, 100742. [Google Scholar] [CrossRef]
- Zasadil, L.M.; Andersen, K.A.; Yeum, D.; Rocque, G.B.; Wilke, L.G.; Tevaarwerk, A.J.; Raines, R.T.; Burkard, M.E.; Weaver, B.A. Cytotoxicity of paclitaxel in breast cancer is due to chromosome missegregation on multipolar spindles. Sci. Transl. Med. 2014, 6, 229ra43. [Google Scholar] [CrossRef]
- Scribano, C.M.; Wan, J.; Esbona, K.; Tucker, J.B.; Lasek, A.; Zhou, A.S.; Zasadil, L.M.; Molini, R.; Fitzgerald, J.; Lager, A.M.; et al. Chromosomal instability sensitizes patient breast tumors to multipolar divisions induced by paclitaxel. Sci. Transl. Med. 2021, 13, eabd4811. [Google Scholar] [CrossRef]
- Replogle, J.M.; Zhou, W.; Amaro, A.E.; McFarland, J.M.; Villalobos-Ortiz, M.; Ryan, J.; Letai, A.; Yilmaz, O.; Sheltzer, J.; Lippard, S.J.; et al. Aneuploidy increases resistance to chemotherapeutics by antagonizing cell division. Proc. Natl. Acad. Sci. USA 2020, 117, 30566–30576. [Google Scholar] [CrossRef]
- Jagetia, G.C.; Shrinath Baliga, M. Vincristine increases the genomic instability in irradiated cultured human peripheral blood lymphocytes. Toxicol. Lett. 2002, 126, 179–186. [Google Scholar] [CrossRef]
- Vargas-Rondón, N.; Pérez-Mora, E.; Villegas, V.E.; Rondón-Lagos, M. Role of chromosomal instability and clonal heterogeneity in the therapy response of breast cancer cell lines. Cancer Biol. Med. 2020, 17, 970–985. [Google Scholar] [CrossRef] [PubMed]
- Barton, S.; Swanton, C. Recent Developments in Treatment Stratification for Metastatic Breast Cancer. Drugs 2011, 71, 2099–2113. [Google Scholar] [CrossRef] [PubMed]
- Kumari, L.; Kumar, Y.; Bhatia, A. The Link Between Chromosomal Instability and Immunity in Cancer. In Handbook of Cancer and Immunology; Rezaei, N., Ed.; Springer International Publishing: Cham, Switzerland, 2022; pp. 1–20. [Google Scholar] [CrossRef]
- Yum, S.; Li, M.; Frankel, A.E.; Chen, Z.J. Roles of the cGAS-STING Pathway in Cancer Immunosurveillance and Immunotherapy. Annu. Rev. Cancer Biol. 2019, 3, 323–344. [Google Scholar] [CrossRef]
- Nishida, H.; Ohara, N.; Kato, A.; Kaimori, R.; Kondo, Y.; Kusaba, T.; Kadowaki, H.; Kawamura, K.; Daa, T. The relationship between tumor immunity and the cGAS-STING pathway in breast cancer: An immunohistochemical study. Exp. Mol. Pathol. 2024, 139, 104917. [Google Scholar]
- Hines, J.B.; Kacew, A.J.; Sweis, R.F. The Development of STING Agonists and Emerging Results as a Cancer Immunotherapy. Curr. Oncol. Rep. 2023, 25, 189–199. [Google Scholar]
- Kandala, S.; Ramos, M.; Voith von Voithenberg, L.; Diaz-Jimenez, A.; Chocarro, S.; Keding, J.; Brors, B.; Imbusch, C.D.; Sotillo, R. Chronic chromosome instability induced by Plk1 results in immune suppression in breast cancer. Cell Rep. 2023, 42, 113166. [Google Scholar]
- Dumont, M.; Gamba, R.; Gestraud, P.; Klaasen, S.; Worrall, J.T.; De Vries, S.G.; Boudreau, V.; Salinas-Luypaert, C.; Maddox, P.S.; Lens, S.M.; et al. Human chromosome-specific aneuploidy is influenced by DNA-dependent centromeric features. EMBO J. 2020, 39, e102924. [Google Scholar]
- Doig, K.D.; Fellowes, A.P.; Fox, S.B. Homologous Recombination Repair Deficiency: An Overview for Pathologists. Mod. Pathol. 2023, 36, 100049. [Google Scholar]
- Perez-Lopez, R.; Ghaffari Laleh, N.; Mahmood, F.; Kather, J.N. A guide to artificial intelligence for cancer researchers. Nat. Rev. Cancer 2024, 24, 427–441. [Google Scholar] [CrossRef]
- Luchini, C.; Pea, A.; Scarpa, A. Artificial intelligence in oncology: Current applications and future perspectives. Br. J. Cancer 2022, 126, 4–9. [Google Scholar] [CrossRef]
- Xu, Z.; Verma, A.; Naveed, U.; Bakhoum, S.F.; Khosravi, P.; Elemento, O. Deep learning predicts chromosomal instability from histopathology images. Science 2021, 24, 102394. [Google Scholar] [CrossRef] [PubMed]
- Bergstrom, E.N.; Abbasi, A.; Díaz-Gay, M.; Galland, L.; Ladoire, S.; Lippman, S.M.; Alexandrov, L.B. Deep Learning Artificial Intelligence Predicts Homologous Recombination Deficiency and Platinum Response from Histologic Slides. J. Clin. Oncol. 2024, 42, 3550–3560. [Google Scholar] [CrossRef] [PubMed]
- Di Cosimo, S.; Silvestri, M.; De Marco, C.; Calzoni, A.; De Santis, M.C.; Carnevale, M.G.; Reduzzi, C.; Cristofanilli, M.; Cappelletti, V. Low-pass whole genome sequencing of circulating tumor cells to evaluate chromosomal instability in triple-negative breast cancer. Sci. Rep. 2024, 14, 20479. [Google Scholar] [CrossRef]
- Cai, Z.; Poulos, R.C.; Liu, J.; Zhong, Q. Machine learning for multi-omics data integration in cancer. Science 2022, 25, 103798. [Google Scholar] [CrossRef] [PubMed]
- Erbe, R.; Gore, J.; Gemmill, K.; Gaykalova, D.A.; Fertig, E.J. The use of machine learning to discover regulatory networks controlling biological systems. Mol. Cell 2022, 82, 260–273. [Google Scholar] [CrossRef]
- Shapiro, E.; Biezuner, T.; Linnarsson, S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat. Rev. Genet. 2013, 14, 618–630. [Google Scholar] [CrossRef]
- Huang, D.; Ma, N.; Li, X.; Gou, Y.; Duan, Y.; Liu, B.; Xia, J.; Zhao, X.; Wang, X.; Li, Q.; et al. Advances in single-cell RNA sequencing and its applications in cancer research. J. Hematol. Oncol. 2023, 16, 98. [Google Scholar] [CrossRef]
- Bischoff, P.; Trinks, A.; Obermayer, B.; Pett, J.P.; Wiederspahn, J.; Uhlitz, F.; Liang, X.; Lehmann, A.; Jurmeister, P.; Elsner, A.; et al. Single-cell RNA sequencing reveals distinct tumor microenvironmental patterns in lung adenocarcinoma. Oncogene 2021, 40, 6748–6758. [Google Scholar] [CrossRef]
- Baysoy, A.; Bai, Z.; Satija, R.; Fan, R. The technological landscape and applications of single-cell multi-omics. Nat. Rev. Mol. Cell Biol. 2023, 24, 695–713. [Google Scholar] [CrossRef]
- Williams, C.G.; Lee, H.J.; Asatsuma, T.; Vento-Tormo, R.; Haque, A. An introduction to spatial transcriptomics for biomedical research. Genome Med. 2022, 14, 68. [Google Scholar] [CrossRef]
- Xu, Z.; Zhang, T.; Chen, H.; Zhu, Y.; Lv, Y.; Zhang, S.; Chen, J.; Chen, H.; Yang, L.; Jiang, W.; et al. High-throughput single nucleus total RNA sequencing of formalin-fixed paraffin-embedded tissues by snRandom-seq. Nat. Commun. 2023, 14, 2734. [Google Scholar]
- Tovini, L.; Johnson, S.C.; Guscott, M.A.; Andersen, A.M.; Spierings, D.C.J.; Wardenaar, R.; Foijer, F.; McClelland, S.E. Targeted assembly of ectopic kinetochores to induce chromosome-specific segmental aneuploidies. EMBO J. 2023, 42, e111587. [Google Scholar] [CrossRef]
- Truong, M.A.; Cané-Gasull, P.; de Vries, S.G.; Nijenhuis, W.; Wardenaar, R.; Kapitein, L.C.; Foijer, F.; Lens, S.M. A kinesin-based approach for inducing chromosome-specific mis-segregation in human cells. EMBO J. 2023, 42, e111559. [Google Scholar] [CrossRef] [PubMed]
- Iemura, K.; Anzawa, H.; Funayama, R.; Iwakami, R.; Nakayama, K.; Kinoshita, K.; Tanaka, K. High levels of chromosomal instability facilitate the tumor growth and sphere formation. Cancer Sci. 2022, 113, 2727–2737. [Google Scholar] [PubMed]
- Tucker, J.B.; Bonema, S.C.; García-Varela, R.; Denu, R.A.; Hu, Y.; McGregor, S.M.; Burkard, M.E.; Weaver, B.A. Misaligned Chromosomes are a Major Source of Chromosomal Instability in Breast Cancer. Cancer Res. Commun. 2023, 3, 54–65. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Techniques | Advantages | Limitations |
|---|---|---|
| G-Banding Karyotype | Cell-by-cell evaluation of all chromosomes. | Requires dividing cells. |
| Detection of complex rearrangements (operator dependent). | Does not detect small alterations (<5–10 Mbp). | |
| Low cost and availability. | Labor intensive. | |
| Fluorescence In Situ Hybridization (FISH) | Cell-by-cell evaluation in interphase or metaphase nuclei. | Requires probes designed to target specific alterations. |
| A large number of cells can be analyzed in a single assay. | Detection of individual alterations rather than whole-genome evaluation. | |
| Better resolution (~150 Kbp). | Labor intensive. | |
| Spectral Karyotyping (SKY) | Cell-by-cell evaluation of all chromosomes. | Requires dividing cells. |
| Requires specialized equipment. | ||
| Better resolution for detecting complex rearrangements. | Labor intensive. | |
| High costs. | ||
| Array Comparative Genomic Hybridization (aCGH) | Genome-wide assessment of DNA gains and losses. | Assessment of a cell population rather than a single-cell resolution. |
| Better resolution for detecting smaller alterations (>50–100 kb) (according to the resolution level). | Inability to detect chromosome alterations that do not result in copy number changes (e.g., balanced translocations). | |
| It does not require dividing cells. | Requires specialized equipment. | |
| Labor intensive. | ||
| Optical Genome Mapping (OGM) | Detection of balanced and unbalanced chromosome alterations, as well as submicroscopic alterations (>500 bp). | Assessment of a cell population rather than a single-cell resolution. |
| Requires specialized equipment. | ||
| Labor intensive. | ||
| High costs. | ||
| Single-cell DNA Sequencing (scDNA-seq) | Cell-by-cell evaluation in interphase or metaphase nuclei. | It may require specialized equipment. |
| A large number of cells can be analyzed in a single assay. | Complex data analysis. | |
| Genome-level resolution to detect chromosome alterations | High costs. | |
| Limited number of studies assessing its reliability. | ||
| Single-cell RNA Sequencing (scRNA-seq) | Cell-by-cell evaluation in interphase or metaphase nuclei. | It may require specialized equipment. |
| A large number of cells can be analyzed in a single assay. | Detection of chromosome alterations is dependent on expression levels. | |
| High resolution to detect chromosome alterations. | Complex data analysis. | |
| Assessment of CIN gene expression signatures. | High costs. | |
| Limited number of studies assessing its reliability. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meléndez-Flórez, M.P.; Ortega-Recalde, O.; Rangel, N.; Rondón-Lagos, M. Chromosomal Instability and Clonal Heterogeneity in Breast Cancer: From Mechanisms to Clinical Applications. Cancers 2025, 17, 1222. https://doi.org/10.3390/cancers17071222
Meléndez-Flórez MP, Ortega-Recalde O, Rangel N, Rondón-Lagos M. Chromosomal Instability and Clonal Heterogeneity in Breast Cancer: From Mechanisms to Clinical Applications. Cancers. 2025; 17(7):1222. https://doi.org/10.3390/cancers17071222
Chicago/Turabian StyleMeléndez-Flórez, María Paula, Oscar Ortega-Recalde, Nelson Rangel, and Milena Rondón-Lagos. 2025. "Chromosomal Instability and Clonal Heterogeneity in Breast Cancer: From Mechanisms to Clinical Applications" Cancers 17, no. 7: 1222. https://doi.org/10.3390/cancers17071222
APA StyleMeléndez-Flórez, M. P., Ortega-Recalde, O., Rangel, N., & Rondón-Lagos, M. (2025). Chromosomal Instability and Clonal Heterogeneity in Breast Cancer: From Mechanisms to Clinical Applications. Cancers, 17(7), 1222. https://doi.org/10.3390/cancers17071222