Simple Summary
Fanconi anemia (FA) is the most common inherited bone marrow failure syndrome, characterized by chromosomal instability and a high risk of cancer. Hematologic malignancy in FA, mainly myelodysplastic neoplasm (MDS) and acute myeloid leukemia (AML), associates with characteristic clonal chromosomal abnormalities in the bone marrow. Although clonal chromosome abnormalities (CCAs) associated with malignant progression in FA involve chromosomes 1, 3, and 7, information on the broader preleukemic bone marrow cytogenetic landscape is scarce. In this study, we report the type and frequency of every kind of non-clonal chromosomal abnormality (NCCA) appearing in the bone marrow of a group of patients with FA, spanning from cancer-free patients to hematologic malignancy, where CCA appears. This study unveils the dynamism of emerging karyotypes in the FA bone marrow and its potential association with the patient’s hematological state.
Abstract
Background/objectives: Fanconi anemia (FA) is an inherited bone marrow failure syndrome characterized by chromosome instability and predisposition to develop myelodysplastic neoplasm (MDS) and acute myeloid leukemia (AML). Clonal chromosome aberrations (CCAs) in chromosomes 1, 3, and 7 frequently appear in the bone marrow (BM) of patients with FA and are associated with MDS/AML progression. Given the underlying DNA repair defect that characterizes FA, non-clonal chromosomal abnormalities (NCCAs) are expected to be common events in the FA BM; in this study, we investigated the presence and significance of NCCA and CCA in the bone marrow (BM) of patients with FA. Methods: Here, we transversally examined the BM karyotypes of 43 non-transplanted patients with FA, 41 with non-clinically detectable hematologic neoplasia and two with diagnosed MDS. We searched for the presence of NCCAs, complex karyotypes (CKs), and CCAs as well as their association with the natural history of the disease, including age, degree of BM failure, and neoplastic transformation. Results: NCCAs were observed in the metaphase spreads of 41/43 FA patients; CKs were observed in 25/43 patients; CCAs were found in 15/43 patients; CCAs involving chromosomes 1, 3 and/or 7 were found in four patients; and other autosomes were found in the remaining 11 patients. Overall, we observed a baseline large karyotypic heterogeneity in the BM of FA patients, demonstrated by the ubiquitous presence of NCCA; such karyotypic heterogeneity precedes the eventual emergence of CKs and selection of cells carrying fitness-improving CCAs. Finally, CCAs involving chromosomes 1, 3 and 7, well-known drivers of hematological malignancy in FA, become established. Overall, we observed that the frequency of NCCAs and CCAs increased with age, even though a significant correlation was not found. Conclusions: These observations fit the model of evolution towards cancer that comprises a first phase of macroevolution represented by NCCAs and karyotypic heterogeneity, followed by the establishment of clones with CCAs, leading to microevolution and cancer. NCCAs are the most frequent chromosomal alterations in the bone marrow of patients with AF and constitute a genome with extensive karyotypic heterogeneity that evolves into clones with more complex genomes and can eventually progress to cancer.
1. Introduction
Fanconi anemia (FA) is a chromosome instability syndrome affecting 1 to 5 individuals per 1,000,000 individuals. FA is also the most frequently inherited bone marrow failure syndrome and originates from the inheritance of pathogenic variants (PVs) in one of any 23 genes (FANCA-FANCX) that operate in the FA/BRCA pathway, responsible for recognizing and repairing DNA interstrand cross-links (ICL), through homologous recombination, an error-free pathway of DNA repair [1,2,3]. In the absence of a functional FA/BRCA pathway, the ICLs can be repaired by low-fidelity DNA repair pathways, such as the non-homologous end joining (NHEJ) or the microhomology-mediated end joining (MMEJ) pathways [4,5], which promote cell viability at the expense of increasing chromosome instability (CIN). The latter is a pathognomonic feature of FA, which allows its diagnosis due to the sensitivity of FA cells to ICL-inducing agents, like mitomycin C (MMC) and Diepoxybutane (DEB) [2,6,7,8,9].
The FA clinical phenotype is highly heterogeneous, affecting multiple organs and systems. The phenotype includes developmental abnormalities that can be detected from birth and that are present in 80–96% of patients [10,11]. Bone marrow (BM) failure, which appears in over 86% of patients with FA, consists of aplastic BM accompanied by peripheral blood cytopenia of different degrees: mild, moderate, or severe; patients with FA will develop BM failure around 7 years of age [11,12]. Also, FA patients are prone to develop cancer, both hematologic and solid tumors, with a cumulative incidence by 40 years of age of 30–33% and 20–28%, respectively [13,14]. The relative risk for a patient with FA to develop cancer with respect to the general population is 19X and rises post-HSC transplant to 55X; specifically, 527X for head and neck squamous cell carcinomas (HNSCC), and after transplant, 933X; 582X for vulvar squamous cell carcinomas, and after transplant, 6298X; 213X for acute myeloid leukemia (AML); and 5669X for myelodysplastic neoplasia (MDS). The risk for hematological cancer, MDS and AML, increases by the age of 10 years old, plateauing at 20–30 years old [14,15].
BM failure in FA has been proposed to be the consequence of damage accumulation in the DNA of hematopoietic stem and progenitor cells (HSPCs) and hyperactivation of growth suppressing pathways, such as the TGFβ (Transforming growth factor β) and the p53/p21 pathways, thus resulting in critical reductions in the number of HSPCs [16,17]. Despite this harsh BM microenvironment, certain HSPCs thrive, probably due to the inherent CIN in FA and the acquisition of chromosome abnormalities that allow temporary survival, although not in the long term.
CIN, one of the hallmarks of cancer, is characterized by aneuploidy and structural chromosomal abnormalities (CAs) and can be observed in the early stages of cancer in the general population [18,19,20]. Importantly, CIN is a constant in FA patients; from zygote formation and embryonic development, it produces single cell genomic variation in the form of heterogeneous karyotypes and has the potential to produce, across the body of patients with FA, diverse altered genomes with short-term survival and elevated level of cell death, leading to the loss of somatic and HSPS cells [21,22]. This karyotypic heterogeneity and its inherent proapoptotic tendency might also be the foundation for the well-known appearance of clonal hematopoiesis and eventual progression to cancer in FA [22,23]. The latter might be the reason why up to 40% of children and young patients with FA show signs of clonal evolution in their BM, and 15 to 60% will develop MDS or leukemia, mainly AML, at an early age [2,24].
Multiple studies have described, and confirmed, the involvement of clonal chromosome abnormalities (CCAs) in the emergence of MDS or AML in patients with FA [23,24,25,26,27,28]. Most of these studies screened for common CCAs using microarrays or fluorescent in situ hybridization (FISH) directed to regions of interest in chromosomes 1q, 3q, and 7q, since these are the most informative cytogenetic markers of clonal evolution towards MDS/AML [24,29,30]. On the other hand, non-Clonal Chromosome Aberrations (NCCAs) are often overlooked in cytogenetic analysis because they appear in only a single cell and are dismissed as “background noise” [31], especially in patients with CIN, as is the case for FA [24].
NCCAs involve large-scale genomic changes, including numerical and structural chromosomal abnormalities resulting in the gain and loss of genomic material; they also include balanced structural alterations such as translocations or inversions, which modify the topology of chromosome segments and change the gene interaction at the scale of an entire genome [32], leading to a karyotypically diverse cell population, with each cell potentially having unique evolutionary properties. NCCAs have gained importance because they are known to alter the genomic information system by changing the karyotype coding, either through a loss or gain of genetic material or by changes in the topology of genes and their regulatory elements, or both. Thus, each cell with a specific NCCA has unique evolutionary potential, and this karyotypic heterogeneity of a cell population is essential for macroevolution toward disease, and specifically, toward cancer [33].
Cancer evolution is known to be a dynamic process, directed by large-scale chromosomal copy number variations that create karyotypic heterogeneity between the cells that compose a tissue, privileging the selection of the genomes with the greatest fitness [18] and eventually leading to the selection of clones with CCAs that will orchestrate cancer development and evolution [18,34]. FA-inherent CIN is expected to create large levels of NCCA, generating a BM with high karyotypic heterogeneity, years before full-blown MDS or AML. In this sense, NCCA could precede the formation of complex genomes and stable CCAs, eventually transitioning to cancer.
The aim of this study was to cytogenetically analyze the BM of a group of non-transplanted patients with FA, to investigate whether the multiple steps of karyotypic evolution towards cancer can be considered in relation to the age of the patients and their hematological condition. We did not find an association between the complexity of the BM karyotype and the age of the patients or their hematological condition, suggesting that the age and stage of bone marrow failure are not the only drivers of leukemogenesis in FA. However, inspecting the group of FA patients as a whole revealed transitions from a karyotypically diverse population of BM cells with NCCA to the presence of complex karyotypes and the emergence of CCAs involving CA of indeterminate potential in patients with FA [30] and CCAs with the classic high-risk chromosome alterations found in chromosomes 1, 3, and 7; the only two patients in the group who were identified with MDS presented with the classic alterations in chromosomes 3 and 7.
2. Materials and Methods
2.1. Patients
We performed a transversal study including non-transplanted patients with a confirmed diagnosis of FA with the DEB chromosome breakage test. The samples analyzed corresponded to the first cytogenetic BM evaluation of each patient within the framework of the recommended annual follow-up in the search for MDS/AML progression markers. All samples were processed at the Cytogenetics Laboratory of the National Institute of Pediatrics (Mexico) between 2015 and 2024. In accordance with the Declaration of Helsinki, all patients or their relatives gave written informed consent for genetic analyses under project numbers INP 2014/041 and 2020/012; of the 43 patients studied, 33 had available genotyping data. The distribution of PVs among FANC genes in this cohort is similar to that reported in other studied populations. As expected, the most frequently mutated gene was FANCA. Variants in genes other than FANCA showed slight differences in frequency compared to those reported in previous studies [11,35]. Among the 33 genotyped patients, 22 (67%) had a PV in FANCA. The remaining 33% were distributed as follows: 3/33 in FANCE, 2/33 in FANCG, 2/33 in FANCL, and 1/33 each in FANCF, FANCD2, FANCJ, and FANCN. The available genetic information for FA is presented in Supplementary Table S2. Clinical and demographic data were obtained from the patient’s medical records and interviews conducted under the framework of the Fanconi Anemia Registry from Mexico (RAFMex), project INP 2020/053. These projects were approved by the institutional research and ethics committees at Instituto Nacional de Pediatría (México).
Age at diagnosis was calculated considering the date of a positive DEB test. BMF severity at the time of cytogenetic study was classified according to Fanconi anemia guidelines for diagnosis and management [35], using the hemogram data (free of transfusion) nearest to the cytogenetic study. The use of androgens was recorded irrespective of the length of treatment or if being taken at the time of sample collection. Transfusion dependency was considered as the time when the patient could not maintain adequate blood counts without sequential transfusions at any timepoint.
2.2. Cytogenetics
Conventional karyotypes were performed on BM cells using the standard direct method. Briefly, 1–2 mL of heparinized BM samples were incubated in a 5% CO2 incubator, cultured during 48 h in MarrowMax medium (Gibco, Life technologies, Grand Island, NY, USA) without cell division stimulant at 37 °C, after which colcemid [10 μg/mL] (Gibco, Life technologies, NY, USA) was added for the final 2 h of culture. Harvesting, slide preparation, and GTG banding were conducted according to the classic methodology [36]. On average, 20 metaphase spreads were analyzed per patient and reported according to the International System for Human Cytogenetic Nomenclature (ISCN) [37]; fewer than 18 metaphases were analyzed in four patients with severe aplastic anemia due to hypocellular BM.
Chromosome abnormalities were classified as (a) NCCA, non-clonal chromosomal abnormality, both numerical and structural; (b) CK, complex karyotype, defined in this study as a cell with ≥3 independent cytogenetic abnormalities [38]; (c) CCA, clonal chromosomal abnormalities, when at least two cells bear the same chromosome gains or structural rearrangements (including deletions and duplications) or when at least three cells present with the same whole chromosome loss [37]. CCA 1,3,7 refers to clonal abnormalities involving at least one of the known recurrent alterations, i.e., duplication of the long arm of chromosome 1 (commonly known as 1q+), duplication of the long arm of chromosome 3 (commonly known as 3q+), complete monosomy of chromosome 7 or deletion of the long arm of chromosome 7 (commonly known as –7/7q–), while CCA refers to clonal chromosome alterations in chromosomes other than 1q+,3q+ and –7/7q–. When aneuploidies of a whole chromosome were detected at a given metaphase, and to reduce potential technical errors, the periphery of such metaphases was analyzed with a 10Xobjective to increase the visibility margin by 10 fields and to detect whether the lost chromosome was located at the periphery of the metaphase or whether the gained chromosome belonged to a nearby metaphase. Chromosome breaks and radial exchange figures were recorded during the screening but are not part of the analysis in this report.
2.3. Statistics
All statistical analyses were performed in Prism (version 10.4.0). Normal distribution of the data was assessed with the Shapiro-Wilk test. Data with normal distribution were compared using one-way ANOVA with Tukey’s post-test for multiple comparisons. Comparisons between two groups were performed with the unpaired t test. The Fisher’s exact test was used to compare proportions between two groups. The Pearson correlation test was used to probe correlation between age and frequency of chromosome abnormalities. The log-rank (Mantel-Cox) test was used to compare the survival of the patients, according to the presence of different types of CA.
3. Results
3.1. Patients
Samples from 50 non-transplanted patients with a positive diagnosis of FA were sent to the cytogenetics laboratory at Instituto Nacional de Pediatría (Mexico) for routine annual follow-up between 2015 and 2024; six samples had insufficient material to perform cytogenetic analysis, and one patient was excluded since the provided sample was obtained once antineoplastic treatment for AML was started. Appropriate cells for cytogenetic analysis, in quality and number, were obtained from 43 patients (Table S1); in two of these (FANC031 and RAFMex015), the concurrence of cytopenia and a monosomy of chromosome 7 clone allowed the diagnosis of MDS in accordance with International Consensus Classification criteria [39]. Over the course of this study, some of the patients received more than one cytogenetic analysis on more than one sample; only the first cytogenetic study was considered for the purposes of this report.
Median age at diagnosis was 8.1 (1.4–15.6) years old, and median age at the time of the cytogenetic study was 9.14 (4.7–28.6) years old, with a female-male relationship of 1.4:1. A summary of the population demographics is shown in Table 1, and the main clinical data are shown in Table 2. Eleven patients died during the time that elapsed between the cytogenetic study and this report, due to BM failure or HSCT-related complications in eight patients, MDS in two patients, and acute lymphoblastic leukemia (ALL) in the remaining one.

Table 1.
Clinical characteristics of study population.
Table 2.
Population and clinical data.
3.2. BM Cytogenetics of Patients with FA
As expected for patients with FA, their BM showed enormous karyotypic heterogeneity, evidencing the underlying CIN. The cytogenetic analysis in the 43 included patients showed that all but one patient displayed chromosomal abnormalities, which were classified in three types: NCCA, non-recurrent CK, and CCA. Representative karyotypes per CA type are shown in Figure 1.
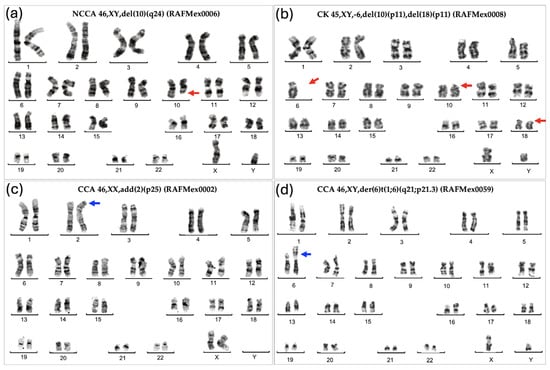
Figure 1.
Representative karyotypes showing the various types of chromosomal abnormalities found in the BM from patients with FA. (a) NCCA: Non-Clonal Chromosome Aberration; (b) CK: Complex Karyotype showing monosomy of chromosome 6 and deletion of 10p and 18p; (c) CCA = Clonal Chromosome Aberration of indeterminate potential; (d) Clonal Chromosome Aberration involving the duplication of the region 1q, a high risk chromosomal abnormality. Red arrows indicate deletions; blue arrows indicate duplications.
NCCAs were observed in 41/43 patients; karyotypes where NCCAs were the only findings occurred in 26/43 patients; the presence of only CCA was observed in a single patient; and the combination of both NCCA and CCA was observed in 15/43 patients. In addition, 25 patients presented with non-recurrent CK. Among the 15 patients with CCA, eight had clones with aneuploidy for one complete chromosome, six had clones with structural chromosomal aberrations, and one patient had clones with both types of chromosomal aberration (Figure 2). Chromosome losses were more commonly observed in comparison to chromosome gains. The chromosomes more frequently involved in any kind of CA were chromosomes 3, 7, 8, and 18, followed by chromosomes 6, 16 12, 17, 20, and 22; also, we found additional material (add), marker chromosomes (mar), and neutral aberrations like inversions and balanced translocations (Figure 3 and Table 3).
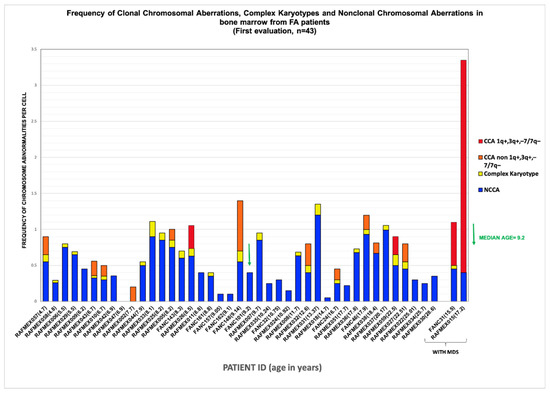
Figure 2.
Frequency of chromosomal aberrations found in BM of FA patients ordered by age and MDS diagnosis at time of cytogenetic study. Distribution of the frequencies of the different type of chromosomal aberrations, per patient, in ascendent order of age. Age is indicated in parentheses.
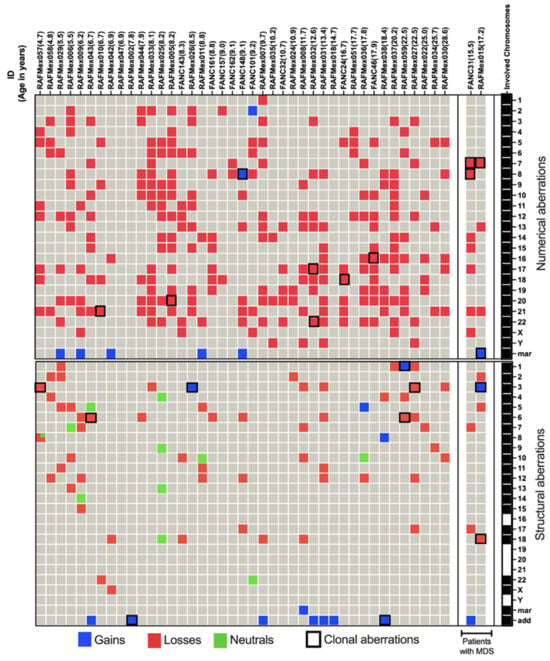
Figure 3.
NCCA and CCA abnormalities in 43 FA patients. In the upper panel, patients are ordered by age and MDS diagnosis. The rightmost column signals which chromosomes are involved in CA. Complete aneuploidy, including loss and gain, was observed for all 24 chromosomes. Structural aberrations, depicted in the lower section, involved all chromosomes except 16, 19, 20, 21, and Y. Marker chromosomes refer to non-identified abnormal chromosomes and add refers to additional chromosomal material of unknown origin. Neutral aberrations signal inversions and balanced translocations. Patient RAFMex057 showed two structural alterations on chromosome 8, one deletion and one inversion, as indicated by  .
.
.
Table 3.
Cytogenetic findings in the BM of 43 patients with FA.
The complexity and heterogeneity of the karyotypes present in the bone marrow (BM) of patients with Fanconi anemia (FA) are evident in Figure 2 and Figure 3 and Table 3. As shown, all but one patient (RAFMex047) presented with composite karyotypes. Unlike a classic karyotype, in which no CAs are found or in which the same alteration is found in all cells, we refer to a composite karyotype as a karyotype in which multiple chromosomal changes, both numerical or structural, are identified in different cells, meaning there is cell-to-cell variation in CA within the same cell population [40], forming a “composite” genetic profile. Specifically in neoplasias, the composite karyotype is useful to track how chromosomal alterations change in a cell population over time by evidencing the karyotypic heterogeneity of the clones, where the metaphases are different, although they share CCA due to the accompanying alterations, which can subsequently form subclones such that clonal evolution is evident [37]. We found composite karyotypes in patients with and without clones, reflecting a highly complex cell population harboring distinct chromosomal alterations, primarily copy-number variations, which underscore the karyotypic diversity resulting from chromosomal instability in patients with FA [23,37].
3.2.1. Non-Clonal Chromosomal Abnormalities
Patients with inherent CIN, such as in FA, are expected to have enormous karyotypic heterogeneity, as was evidenced in their BM, where stochastic chromosomal alterations were found (Table 3 and Figure 2). All 24 human chromosomes were involved in these CAs, and numerical aberrations were the most common (Figure 3).
NCCAs were observed in the vast majority of FA patients, 95% (41/43), including non-recurrent CK. NCCAs without evidence of any additional chromosomal clones were found in 26 patients. NCCAs were the most prevalent type of CA, accounting for 74.6% of the total abnormalities observed, and included CK in 25/43 patients, with most being non-recurrent. Patient RAFMex015 was exceptional, since clonal and complex karyotypes were found in 100% of the analyzed cells, concurrent with an MDS diagnosis (Table 3 and Figure 2).
In most patients, NCCAs were the most frequent CAs; these were highly diverse and included numerical and structural alterations. Among the structural CAs, 11/78 were neutral in that they did not appear to condition a loss or gain of chromosome information; most were inversions, but there was also one translocation (Figure 3, green cells).
3.2.2. Clonal Chromosomal Abnormalities (CCAs)
CCAs accounted for 25.4% of the total CAs found in the 43 patients with FA (Table 3). They were found in 35% (15/43); among these, the presence of CCAs involving monosomy of chromosome 7 in two of these patients led to a MDS diagnosis, and both presented with a composite karyotype, RAFMex015: 45,XY,–7,–18,+der?(18)t(3;18)(q13.3;q?12)[15]/45,idem,del(2)(p23)[1]/45,idem,del(5)(p14)[1]/45,idem,–13,+mar2[1]/45,idem,–21,+mar1[2][cp20]. In this karyotype, a main stem line with a clone with monosomy 7 and a subclone with monosomy 21 were found; both alterations were recognized as high risk in patients with FA, with an evident clonal evolution. The patient FANC31: 45,XX,–7[8]/44,sl1,add(1)(p36),–17[1]/45,XX,–8[2]/44,sl2,–15[1]/45,X,–X[1]/44,XX,del(7p),–17,–21[1]/45,XX,–14[1]/46,XX,del(17)(p11.2)[1][cp16]/46,XX[4], presented with two unrelated stem lines, one of them -sl1- with monosomy 7, also a high-risk CA. Within the 15 patients that presented with clonal chromosome abnormalities (WC), seven presented clones consisting of whole-chromosome aneuploidy, losses involving chromosomes 7, 8, 16, 17, 18, 20, 21, and 22, gains involving chromosome 8, and/or a marker chromosome. Seven patients had clones with structural CA in chromosomes 1, 3, 6, and 18, and one patient presented a clone with both numerical (chromosomes 7 and +mar) and structural (chromosomes 3 and 18) CAs. Among 15 patients WC, 11 presented CCAs involving autosomes other than 1, 3, and 7, and in 4/15 patients WC, the clone involved the regions on chromosomes 1, 3, and 7 associated with the evolution towards hematological malignancy; one of them had a duplication 1q21-qter, due to a translocation t(1;6)(q21;p21.3). Patients WC, in addition to having cytogenetic clones, revealed a variety of NCCAs: 3/15 had whole-chromosome aneuploidies, 11 had numerical and structural NCCAs, and only one patient did not present NCCAs (Figure 3 and Table 3). Within the chromosomes or chromosomal regions involved in CCAs, in addition to the genes commonly associated with MDS and AML in patients with FA, such as MDM4, EVI1, and RUNX1 [23,26], we identified other genes contained in CCAs that may also play a role in the progression to these malignancies, as detailed in Supplementary Table S3.
3.3. Clonicity and Chromosomal Damage
In this group of patients, we find that the average frequency of CCAs (0.16) is only ~20% of the observed average frequency of NCCAs (0.47) (Table 3). In addition, it is evident that when the total frequency of ab/cell is higher, CCA appears in a patient (Figure 4). In patients WC who have MDS, a large number of CAs are observed in their BM, because the fittest clone is present in a large proportion (Figure 1 and Figure 4). In patients WC, CCA alterations coexist with NCCAs, although the latter are generally the most common alteration. However, it should be noted that in the two patients who already presented MDS, CCAs prevailed over NCCAs (Figure 2).
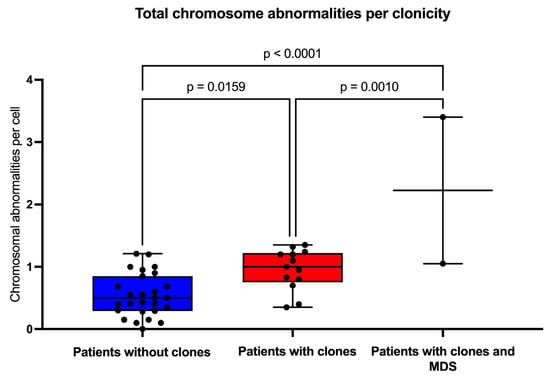
Figure 4.
Frequency of chromosomal aberrations per cell. The population was divided into patients without clones (blue box), with clones (red box), and with –7/MDS (myelodysplastic neoplasm). Statistical analyses were performed using one-way ANOVA with Tukey’s post-test for multiple comparisons.
3.4. Complex Karyotypes and Clonicity
Several analyzed metaphases had more than one CA; we classified the patients that had at least one metaphase that also had three or more CAs as having a CK. Twenty-five out of 43 patients had metaphases with these CKs, which represent a greater complexity within the population of cells with NCCAs. When comparing the frequency of CKs in patients with and without clones, there is a higher proportion of patients with CKs among those who have already developed clones, showcasing an increased complexity of the diverse karyotypes found in their BM (Figure 5).
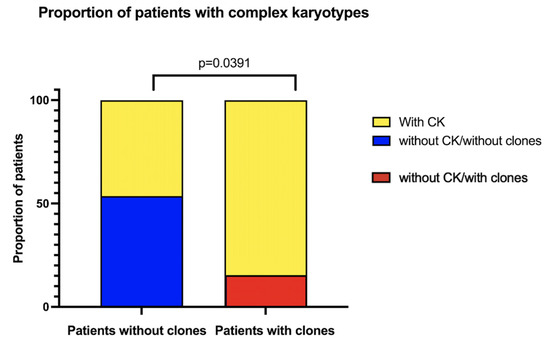
Figure 5.
Patients with complex karyotypes (CKs), according to clonicity. In yellow, the proportion of patients who present at least one metaphase with CKs (≥3 CA). Patients with MDS were excluded.
3.5. Chromosomal Damage and Patient Demographic and Clinical Characteristics
Chromosomal Abnormalities and the Age and Gender of Patients
Pearson correlation analysis revealed that as the patients grow older, the total frequency of CAs does not increase (R = 0.056, p = 0.73) (Figure 6a). However, when patients were sub-divided into three groups of age, 4–9 years old, 10–15 years old, and >16 years old, a significantly higher frequency of CAs was observed between the older patients WC (excluding MDS patients) and the younger patients without clones (one-way ANOVA with Tukey’s post-test) (Figure 6b). Finally, there were no significant differences in AC frequency when comparing males and females (Figure 7).
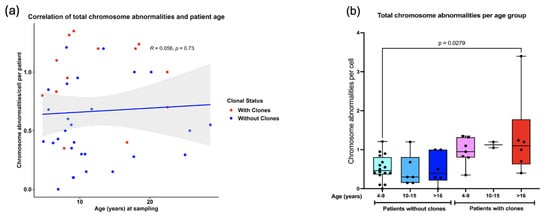
Figure 6.
Correlation between the total frequency of CAs and patient age. (a) No correlation was found between the amount of CAs and increasing age of patients WC and without clones (Pearson R = 0.056 p = 0.73). (b) A significant increase in total CAs was found only in patients 4–9 years old without clones and patients > 16 years with clones.
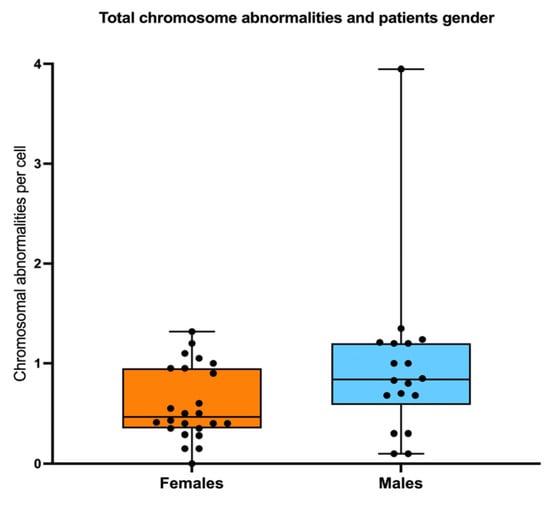
Figure 7.
Sex does not influence the frequency of CAs. No differences in the total frequency of CAs were found when comparing females vs. males.
We searched for an association between the frequency of chromosomal aberrations and bone marrow failure severity: 5/43 did not have BMF, 28/43 had moderate or severe BMF. As a group, the patients WC show slightly more CA than WOC, and although no statistical differences were found, it would appear that in individuals WC, CAs would increase as the BMF becomes more severe (Figure 8a). Other hematological status parameters, including transfusion dependency and androgen treatment, did not influence the total frequency of CAs observed in patients with FA (Figure 8b,c).
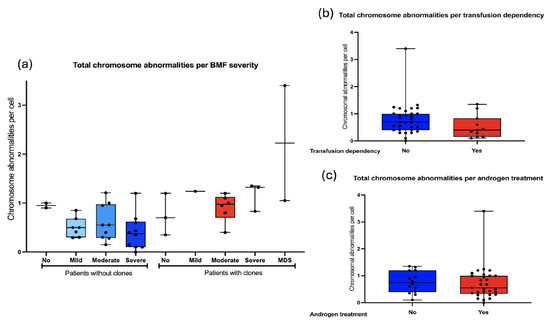
Figure 8.
Frequency of CA with respect to hematologic status. (a) The frequency of CA in FA does not correlate with the severity of bone marrow failure, determined in accordance with the FA clinical care guidelines [35]. (b) CA per cell with respect to transfusion dependency. (c) CA per cell in reference to androgen therapy. No differences in the frequency of CA were observed with respect to hematological conditions or patients’ treatment.
Finally, we performed a survival analysis, according to the presence of the diverse types of cytogenetic abnormalities in general and contrasting the presence of the well-known adverse abnormalities in FA (affecting chromosomes 1, 3, and 7) and other clonal chromosome abnormalities of indeterminate potential. In addition, we examined the impact of complex karyotypes upon survival (Figure 9). We did not observe significant differences in patient survival based on the type of CA. However, the presence of clonal cytogenetic abnormalities (CCAs) involving the known high-risk cytogenetic alterations (such as duplication of 1q, duplication of 3q, monosomy 7) complex karyotypes, or composite karyotypes with high-risk alterations plus monosomy 17 and monosomy 21 appears to negatively impact survival in patients with FA. Although these trends were evident, statistical significance may not have been reached, likely due to the small sample size.
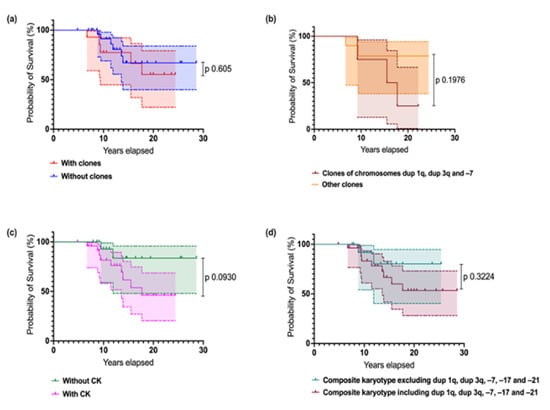
Figure 9.
Survival estimates in Mexican individuals with FA according to the presence of the diverse types of cytogenetic abnormalities. (a) Survival according to the presence of clonal cytogenetic abnormalities. Patients without clones are in blue, and patients with clones are in red. (b) Survival in individuals with cytogenetic clones: aberrations affecting chromosome 1 (1q duplication), 3 (3q duplication), and 7 (7q deletion or –7) in pink, and all other clonal aberrations of indeterminate potential in orange. (c) Survival according to the presence of complex karyotypes (CKs): absence of CKs in blue, presence of CKs in red. (d) Survival according with the presence of composite karyotypes, including or excluding recognized high-risk CA plus –17 and –21 in FA patients. Log-rank (Mantel-Cox) Test.
4. Discussion
In this study, we conducted a cytogenetic cross-sectional analysis of the BM of 43 patients with the chromosome instability and cancer predisposition syndrome, FA. As anticipated, a high number of CAs were observed in virtually all participants. NCCAs were the most prevalent, reflecting an underlying karyotype heterogeneity caused by faulty DNA repair mechanisms in individuals with FA. CCAs, mostly abnormalities of indeterminate potential, were also observed, with high-risk CAs being detected in four patients, two of whom had MDS defining CAs. This comprehensive analysis of the type and frequency of CAs in the BM of individuals with FA stresses the natural history of hematological cancer in FA, from heterogeneous NCCA towards CK, and finally CCA and cancer. This study complements the information on the genomic changes involved in the evolution towards hematological cancer, which has been extensively studied by other authors [22,23,30], given that we analyzed the chromosomes of pre-leukemic BM and at the individual cell level, revealing the presence of composite karyotypes in the BM of practically all patients.
As shown in Figure 3, the observed NCCAs included both structural alterations and aneuploidies (gains and losses of whole chromosomes). Structural alterations included gains of chromosome material and chromosome deletions. Specifically, chromosome gains may arise from the joining of non-homologous chromosome segments through the non-homologous end-joining (NHEJ) pathway, causing chromosome translocations. Additionally, the segregation of radial exchange figures during mitosis could produce daughter cells harboring single derivative chromosomes (with the corresponding gain and/or loss of chromosome material). On the other hand, chromosome deletions were six times more frequent than gains. Their origin could be chromosomal rearrangements or non-repaired double-strand breaks (DSB) that reached mitosis and whose broken segment was not retained in the same cell after exiting mitosis, leaving a deleted chromosome.
We saw an overrepresentation of loss-of-chromosome aneuploidies, raising the possibility of technical errors during the preparation of metaphase spreads; nonetheless, given our strict scoring criteria during chromosome analysis (see methodology), we consider that most of these alterations are genuine chromosome numerical losses. In this context of genomic instability, we hypothesize that chromosome losses are related to the mis-segregation of structurally altered chromosomes that became incorporated into micronuclei and further shattered, as others have shown to occur with lagged chromosomes [41].
Interestingly, we also identified neutral CA, i.e., balanced CA such as chromosome inversions and translocations. Traditionally, these alterations were thought to be less detrimental than deletions and duplications, but it is now recognized that they can have a significant impact in cancer evolution, since the change in the 3D distribution pattern of genes and regulatory elements is responsible for emerging network dynamics [42]. Inversions are not commonly reported in previous cytogenetic studies of patients with FA, most likely because cytogenomic analysis using microarrays is usually preferred over GTG banded chromosomes [23]. These inversions could result from the presence of two DSBs in the same chromosome that were rejoined after a 180-degree rotation. Other mechanisms may also explain the presence of inversions, including chromothripsis-like rearrangements, but with fewer breakpoints than traditional chromothripsis, which usually involves extensive fragmentation and rearrangement of a single chromosome. It is known that an intact FA/BRCA pathway plays a crucial role in the micronucleus-related chromosome fragmentation that leads to chromothripsis, while a deficient FA/BRCA pathway generates significantly less chromosome fragmentation under similar conditions [41]. This type of alteration, evidenced here by G banding, may represent just the tip of the iceberg of more complex ACs; these can be studied at higher resolution, with methodologies such as SKY or M-FISH, with which one can identify CA in greater number and complexity. This undoubtedly deserves further study.
Of note, NCCAs in FA are quite variable from cell to cell, and this is probably due to multiple factors, including the following: first, a continuous generation of DSBs, which are incorrectly and not homogeneously rejoined due to the reliance of the FA cells on error-prone DNA repair pathways [4]; second, dividing cells harboring NCCAs will not parent chromosomally identical daughter cells, particularly when dicentric chromosomes and radial figures are present, as mitotic segregation will not equally distribute the CAs into the daughter cells [2]; third, NCCAs might confer differential surviving capacities, and therefore alterations incompatible with life will not thrive, while fitness-providing CAs will persist, in a constant cycle of cell replacement and cell attrition. During this cycle of depletion and emergence of cells with NCCAs, new genome compositions in the tissue will arise, producing a unique combination of CAs that will benefit the survival of cells harboring stable karyotypes with CCAs.
Cells carrying a specific CCA can be considered evidence of a successful genome arrangement. However, this specific clone could also be transient, as the continuous production of CAs does not cease, leading to the emergence of CKs, most of which are NCCAs. Some CKs may appear in cells that already contain a specific CCA, generating genomes that may either disappear or adapt, eventually evolving towards a clone with higher fitness. In a cell population exhibiting CIN, NCCAs will create dynamic karyotypic diversity. This heterogeneity of cells, with stochastic CAs, eventually gives rise to CCAs, which may be transient, creating a dynamic cycle of NCCA/CCA. The NCCA/CCA cycle establishes a macroevolutive phase that may continue until the emergence of stable clones that encompasses a phase transition. One surviving genome with CCAs carrying high-risk CAs, such as clones with dup(1q), dup(3q), del(7q), and –7, with a composite karyotype showing clonal evolution and a decrease in NCCAs (as in patients with MDS), could be indicative of evolution towards leukemia. Clonal expansion is characteristic for the microevolutionary phase [43]. These specific karyotype alterations are known to facilitate the progression to cancer. The cells with these CAs will proliferate until they become a clone, thus completing the transition from NCCA to CCA. In patients with FA, the transition to cancer also involves specific genetic alterations such as RUNX1 and TP53, among others [23].
Importantly, progression towards cancer, which typically takes a long time in DNA repair-proficient individuals, is accelerated in patients with FA. In this work, through an in-depth cytogenetic analysis of the BM of patients with FA, we recapitulate macroevolution patterns previously described [23,30,31], including an initial phase of high karyotypic heterogeneity leading to more stable “end products of evolution”, i.e., CCA abnormalities that combine large-scale chromosomal changes with genetic mutations and copy number alterations, which will ultimately drive micro-evolution steps towards cancer.
5. Conclusions
In this study, we observed that the frequency of ACs was not associated with gender, severity of bone marrow failure, or androgen treatment. The frequency of NCCAs and CCAs increased with age; although no significant correlation was found, a significant difference was observed between older patients with CCAs and younger patients without CCAs.
In general, the preleukemic bone marrow of patients with FA exhibits significant basal karyotypic heterogeneity, evidenced by the widespread presence of NCCAs. This karyotypic heterogeneity precedes the eventual appearance of CKs and the selection of CCA-bearing cells that enhance adaptation, which may be transient until the appearance of a stable CCA. The NCCA/CCA cycle establishes a macroevolutionary phase that could continue until the emergence of stable clones, leading to a phase transition. A surviving CCA genome harboring high-risk ACs emerges, such as clones with dup(1q), dup(3q), del(7q), and –7, and the associated NCCAs decline. These observations fit the model of evolution towards cancer that comprises a first phase of macroevolution represented by NCCA and karyotypic heterogeneity, followed by the establishment of clones with CCAs, which leads to microevolution and cancer.
These observations warrant a longitudinal follow-up study of patients with FA to determine the macro- and microevolutionary phases and to detect potential cytogenetic biomarkers that precede clonal hematopoiesis.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cancers17111805/s1, Supplementary Table S1. Database containing the cytogenetics of 43 patients with FA. Complete data. Supplementary Table S2. Table containing data on the genes and variants of patients with FA. Supplementary Table S3. Genes located in CCA, involved in MDS and AML [16,17,23,26,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69].
Author Contributions
Conceptualization, S.F., S.S. and B.G.-d.-T.; methodology, S.F., S.S. and M.A.M.-B.; formal analysis, S.F., S.S., B.G.-d.-T., M.Ó.F.-R. and A.R.; investigation, S.F., S.S., B.G.-d.-T., P.V.R.-J., A.P.-M., M.A.M. and B.M.; resources, B.G.-d.-T., A.M.-O., M.Ó.F.-R., B.M., M.A.M.-B. and L.T.; data curation, S.S. and B.G.-d.-T. writing—original draft preparation, S.F., S.S. and B.G.-d.-T.; writing—review and editing, S.F., S.S., B.G.-d.-T., and A.R.; visualization, S.S., B.G.-d.-T., B.M., P.V.R.-J. and A.R.; supervision, S.F., S.S., B.M., L.T., B.G.-d.-T. and A.M.-O.; project administration, S.F. and B.M.; funding acquisition, S.F. and B.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Dirección General de Asuntos del Personal Académico, Universidad Nacional Autónoma de México, UNAM-PAPIIT, grant number IN205120; Consejo Nacional de Humanidades, Ciencias y Tecnologías, grant number CF-2023-G-800; Instituto Nacional de Pediatría, Recursos Fiscales, Projects 2020/012 and 2020/053. The APC was funded by Instituto Nacional de Pediatría.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Ethics Committee of Instituto Nacional de Pediatría (protocol code 2014/041 approved on 14 July 2014, 2020/053 approved on 15 October 2020, and 2020/012 approved on 23 March 2020).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The original contributions presented in this study are included in the article/Supplementary Materials. Further inquiries can be directed to the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Rodríguez, A.; D’Andrea, A. Fanconi anemia pathway. Curr. Biol. 2017, 27, R986–R988. [Google Scholar] [CrossRef] [PubMed]
- García-De-teresa, B.; Rodríguez, A.; Frias, S. Chromosome instability in fanconi anemia: From breaks to phenotypic consequences. Genes 2020, 11, 1528. [Google Scholar] [CrossRef] [PubMed]
- Kuehl, J.; Zhang, Y.; Schindler, D. Genetic inactivation of FAAP100 causes Fanconi anemia due to disruption of the monoubiquitin ligase core complex. J. Clin. Investig. 2025. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Zambrano, C.; Yuste, M.; Frías, S.; García de Teresa, B.; Mendoza, L.; Azpeitia, E.; Rodríguez, A.; Torres, L. A Boolean network model of the double-strand break repair pathway choice. J. Theor. Biol. 2023, 573, 111608. [Google Scholar] [CrossRef]
- Rogers, C.B.; Kram, R.E.; Lin, K.; Myers, C.L.; Sobeck, A.; Hendrickson, E.A.; Bielinsky, A.-K. Fanconi anemia-associated chromosomal radial formation is dependent on POL θ -mediated alternative end joining. Cell Rep. 2023, 42, 112428. [Google Scholar] [CrossRef]
- Auerbach, A.D. Fanconi anemia and its diagnosis. Mutat. Res.—Fundam. Mol. Mech. Mutagen. 2009, 668, 4–10. [Google Scholar] [CrossRef]
- Moreno, O.M.; Paredes, A.C.; Suarez-Obando, F.; Rojas, A. An update on Fanconi anemia: Clinical, cytogenetic and molecular approaches (review). Biomed. Rep. 2021, 15, 74. [Google Scholar] [CrossRef]
- Esmer, C.; Sánchez, S.; Ramos, S.; Molina, B.; Frias, S.; Carnevale, A. DEB Test for Fanconi Anemia Detection in Patients with Atypical Phenotypes. Am. J. Med. Genet. 2004, 124, 35–39. [Google Scholar] [CrossRef]
- Merfort, L.W.; Lisboa, M.D.O.; Cavalli, L.R.; Bonfim, C.M.S. Cytogenetics in Fanconi Anemia: The Importance of Follow-Up and the Search for New Biomarkers of Genomic Instability. Int. J. Mol. Sci. 2022, 23, 14119. [Google Scholar] [CrossRef]
- Fiesco-Roa, M.O.; Giri, N.; McReynolds, L.J.; Best, A.F.; Alter, B.P. Genotype-phenotype associations in Fanconi anemia: A literature review. Blood Rev. 2019, 37, 100589. [Google Scholar] [CrossRef]
- Altintas, B.; Giri, N.; McReynolds, L.J.; Best, A.; Alter, B.P. Genotype-phenotype and outcome associations in patients with Fanconi anemia: The National Cancer Institute cohort. Haematologica 2023, 108, 69–82. [Google Scholar] [CrossRef]
- Risitano, A.M.; Marotta, S.; Calzone, R.; Grimaldi, F.; Zatterale, A. Twenty years of the Italian Fanconi Anemia Registry: Where we stand and what remains to be learned. Haematologica 2016, 101, 319–327. [Google Scholar] [CrossRef]
- Kutler, D.I.; Singh, B.; Satagopan, J.; Batish, S.D.; Berwick, M.; Giampietro, P.F.; Hanenberg, H.; Auerbach, A.D. A 20-year perspective on the International Fanconi Anemia Registry (IFAR). Blood 2003, 101, 1249–1256. [Google Scholar] [CrossRef]
- Alter, B.P. Fanconi anemia and the development of leukemia. Best Pract. Res. Clin. Haematol. 2014, 27, 214–221. [Google Scholar] [CrossRef]
- Alter, B.P.; Giri, N.; Savage, S.A.; Rosenberg, P.S. Cancer in the national cancer institute inherited bone marrow failure syndrome cohort after fifteen years of follow-up. Haematologica 2018, 103, 30–39. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Parmar, K.; Mouly, E.; Delord, M.; Kim, J.M.; Regairaz, M.; Pla, M.; Vasquez, N.; Zhang, Q.S.; Pondarre, C.; et al. Bone marrow failure in fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell 2012, 11, 36–49. [Google Scholar] [CrossRef]
- Rodríguez, A.; Zhang, K.; Färkkilä, A.; Filiatrault, J.; Yang, C.; Velázquez, M.; Furutani, E.; Goldman, D.C.; García de Teresa, B.; Garza-Mayén, G.; et al. MYC Promotes Bone Marrow Stem Cell Dysfunction in Fanconi Anemia. Cell Stem Cell 2021, 28, 33–47. [Google Scholar] [CrossRef]
- Tijhuis, A.E.; Foijer, F. Characterizing chromosomal instability-driven cancer evolution and cell fitness at a glance. J. Cell Sci. 2024, 137, jcs260199. [Google Scholar] [CrossRef]
- Abdallah, B.Y.; Horne, S.D.; Stevens, J.B.; Liu, G.; Ying, A.Y.; Vanderhyden, B.; Krawetz, S.A.; Gorelick, R.; Heng, H.H.Q. Single cell heterogeneity: Why unstable genomes are incompatible with average profiles. Cell Cycle 2013, 12, 3640–3649. [Google Scholar] [CrossRef]
- Davoli, T.; Xu, A.W.; Mengwasser, K.E.; Sack, L.M.; Yoon, J.C.; Park, P.J.; Elledge, S.J. XCumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 2013, 155, 948. [Google Scholar] [CrossRef]
- Liu, G.; Stevens, J.B.; Horne, S.D.; Abdallah, B.Y.; Ye, K.J.; Bremer, S.W.; Ye, C.J.; Chen, D.J.; Heng, H.H. Genome chaos: Survival strategy during crisis. Cell Cycle 2014, 13, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Cioc, A.M.; Wagner, J.E.; MacMillan, M.L.; DeFor, T.; Hirsch, B. Diagnosis of myelodysplastic syndrome among a cohort of 119 patients with fanconi anemia: Morphologic and cytogenetic characteristics. Am. J. Clin. Pathol. 2010, 133, 92–100. [Google Scholar] [CrossRef]
- Sebert, M.; Gachet, S.; Leblanc, T.; Rousseau, A.; Bluteau, O.; Kim, R.; Ben Abdelali, R.; Sicre de Fontbrune, F.; Maillard, L.; Fedronie, C.; et al. Clonal hematopoiesis driven by chromosome 1q/MDM4 trisomy defines a canonical route toward leukemia in Fanconi anemia. Cell Stem Cell 2023, 30, 153–170.e9. [Google Scholar] [CrossRef]
- Butturini, A.; Gale, R.P.; Verlander, P.C.; Adler-Brecher, B.; Gillio, A.P.; Auerbach, A.D. Hematologic abnormalities in Fanconi anemia: An International Fanconi Anemia Registry study. Blood 1994, 84, 1650–1655. [Google Scholar] [CrossRef]
- Berger, R.; Jonveaux, P. Clonal chromosome abnormalities in Fanconi anemia. Hematol. Cell Ther. 1996, 38, 291–296. [Google Scholar] [CrossRef]
- Quentin, S.; Cuccuini, W.; Ceccaldi, R.; Nibourel, O.; Pondarre, C.; Pagès, M.P.; Vasquez, N.; D’Enghien, C.D.; Larghero, J.; De Latour, R.P.; et al. Myelodysplasia and leukemia of fanconi anemia are associated with a specific pattern of genomic abnormalities that includes cryptic RUNX1/AML1 lesions. Blood 2011, 117, 161–170. [Google Scholar] [CrossRef]
- Mehta, P.A.; Harris, R.E.; Davies, S.M.; Kim, M.O.; Mueller, R.; Lampkin, B.; Mo, J.; Myers, K.; Smolarek, T.A. Numerical chromosomal changes and risk of development of myelodysplastic syndrome-acute myeloid leukemia in patients with Fanconi anemia. Cancer Genet. Cytogenet. 2010, 203, 180–186. [Google Scholar] [CrossRef]
- Lisker, R.; Gutiérrez, A.C. de Cytogenetic studies in Fanconi’s anemia. Description of a case with bone marrow clonal evolution. Clin. Genet. 1974, 5, 72–76. [Google Scholar] [CrossRef]
- Tönnies, H.; Huber, S.; Kühl, J.S.; Gerlach, A.; Ebell, W.; Neitzel, H. Clonal chromosomal aberrations in bone marrow cells of Fanconi anemia patients: Gains of the chromosomal segment 3q26q29 as an adverse risk factor. Blood 2003, 101, 3872–3874. [Google Scholar] [CrossRef]
- Behrens, Y.L.; Göhring, G.; Bawadi, R.; Cöktü, S.; Reimer, C.; Hoffmann, B.; Sänger, B.; Käfer, S.; Thol, F.; Erlacher, M.; et al. A novel classification of hematologic conditions in patients with fanconi anemia. Haematologica 2021, 106, 3000–3003. [Google Scholar] [CrossRef]
- Heng, H.H.Q.; Regan, S.M.; Liu, G.; Ye, C.J. Why it is crucial to analyze non clonal chromosome aberrations or NCCAs? Mol. Cytogenet. 2016, 9, 15. [Google Scholar] [CrossRef]
- Ye, C.J.; Stilgenbauer, L.; Moy, A.; Liu, G.; Heng, H.H. What Is Karyotype Coding and Why Is Genomic Topology Important for Cancer and Evolution? Front. Genet. 2019, 10, 1082. [Google Scholar] [CrossRef]
- Heng, J.; Heng, H.H. Two-phased evolution: Genome chaos-mediated information creation and maintenance. Prog. Biophys. Mol. Biol. 2021, 165, 29–42. [Google Scholar] [CrossRef]
- Heng, J.; Heng, H.H. Genome Chaos, Information Creation, and Cancer Emergence: Searching for New Frameworks on the 50th Anniversary of the “War on Cancer”. Genes 2022, 13, 101. [Google Scholar] [CrossRef]
- Fanconi Anemia Research Fund. Fanconi Anemia Clinical Care Guidelines, 5th ed.; Sroka, I., Frohnmayer, L., Van Ravenhorst, S., Wirkkula, L., Eds.; Fanconi Anemia Research Fund: Eugene, OR, USA, 2020. [Google Scholar]
- Sánchez, S.; Reyes, P.; Barrera, M.A.M.; Mar-Tínez, A.P.; Frias, S. Fanconi anemia, Part 3. Cytogenetic monitoring in the bone marrow of patients with Fanconi anemia. Acta Pediatr. Mex. 2024, 45, 343–368. [Google Scholar] [CrossRef]
- Hastings, R.J.; Moore, S.; Chia, N. ISCN 2024—An International System for Human Cytogenomic Nomenclature (2024). Cytogenet. Genome Res. 2024, 164, 1–224. [Google Scholar]
- Nguyen-Khac, F.; Bidet, A.; Daudignon, A.; Lafage-Pochitaloff, M.; Ameye, G.; Bilhou-Nabéra, C.; Chapiro, E.; Collonge-Rame, M.A.; Cuccuini, W.; Douet-Guilbert, N.; et al. The complex karyotype in hematological malignancies: A comprehensive overview by the Francophone Group of Hematological Cytogenetics (GFCH). Leukemia 2022, 36, 1451–1466. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef]
- Zneimer, S.M. Composite karyotypes and other complex rearrangements. In Cytogenetic Abnormalities: Chromosomal, FISH and Microarray-Based Clinical Reporting; Wiley: Hoboken, NJ, USA, 2014; pp. 107–114. [Google Scholar]
- Engel, J.L.; Zhang, X.; Wu, M.; Wang, Y.; Espejo Valle-Inclán, J.; Hu, Q.; Woldehawariat, K.S.; Sanders, M.A.; Smogorzewska, A.; Chen, J.; et al. The Fanconi anemia pathway induces chromothripsis and ecDNA-driven cancer drug resistance. Cell 2024, 187, 6055–6070.e22. [Google Scholar] [CrossRef]
- Heng, J.; Heng, H.H. Genome chaos: Creating new genomic information essential for cancer macroevolution. Semin. Cancer Biol. 2022, 81, 160–175. [Google Scholar] [CrossRef]
- Ye, J.C.; Editors, H.H.H. Cancer Cytogenetics and Cytogenomics; Springer: Berlin/Heidelberg, Germany, 2024; ISBN 9781071639450. [Google Scholar]
- Schratz, K.E.; Gaysinskaya, V.; Cosner, Z.L.; DeBoy, E.A.; Xiang, Z.; Kasch-Semenza, L.; Florea, L.; Shah, P.D.; Armanios, M. Somatic reversion impacts myelodysplastic syndromes and acute myeloid leukemia evolution in the short telomere disorders. J. Clin. Investig. 2021, 131, 1–9. [Google Scholar] [CrossRef]
- He, X.; Zou, H.; Wang, F. SOX4-induced upregulation of ARHGAP9 promotes the progression of acute myeloid leukemia. Drug Dev. Res. 2021, 82, 1227–1234. [Google Scholar] [CrossRef]
- Gou, J.; Bi, J.; Wang, K.; Lei, L.; Feng, Y.; Tan, Z.; Gao, J.; Song, Y.; Kang, E.; Guan, F.; et al. O-GlcNAcylated FTO promotes m6A modification of SOX4 to enhance MDS/AML cell proliferation. Cell Commun. Signal. 2025, 23, 43. [Google Scholar] [CrossRef]
- Kim, H.M.; Liu, Z. LSD2 Is an Epigenetic Player in Multiple Types of Cancer and Beyond. Biomolecules 2024, 14, 553. [Google Scholar] [CrossRef]
- Rudelius, M.; Weinberg, O.K.; Niemeyer, C.M.; Shimamura, A.; Calvo, K.R. The International Consensus Classification (ICC) of hematologic neoplasms with germline predisposition, pediatric myelodysplastic syndrome, and juvenile myelomonocytic leukemia. Virchows Arch. 2023, 482, 113–130. [Google Scholar] [CrossRef]
- Marion, W.; Koppe, T.; Chen, C.C.; Wang, D.; Frenis, K.; Fierstein, S.; Sensharma, P.; Aumais, O.; Peters, M.; Ruiz-Torres, S.; et al. RUNX1 mutations mitigate quiescence to promote transformation of hematopoietic progenitors in Fanconi anemia. Leukemia 2023, 37, 1698–1708. [Google Scholar] [CrossRef]
- Chang, L.; Cui, Z.; Shi, D.; Chu, Y.; Wang, B.; Wan, Y.; Ma, Q.; Zhang, R.; Li, H.; Cheng, X.; et al. Polyclonal evolution of Fanconi anemia to MDS and AML revealed at single cell resolution. Exp. Hematol. Oncol. 2022, 11, 1–14. [Google Scholar] [CrossRef]
- Ottema, S.; Mulet-Lazaro, R.; Beverloo, H.B.; Erpelinck, C.; van Herk, S.; van der Helm, R.; Havermans, M.; Grob, T.; Valk, P.J.M.; Bindels, E.; et al. Atypical 3q26/MECOM rearrangements genocopy inv(3)/t(3;3) in acute myeloid leukemia. Blood 2020, 136, 224–234. [Google Scholar] [CrossRef]
- Gajzer, D.; Logothetis, C.N.; Sallman, D.A.; Calon, G.; Babu, A.; Chan, O.; Vincelette, N.D.; Volpe, V.O.; Al Ali, N.H.; Basra, P.; et al. MYC overexpression is associated with an early disease progression from MDS to AML. Leuk. Res. 2021, 111, 1–17. [Google Scholar] [CrossRef]
- Huh, Y.O.; Tang, G.; Talwalkar, S.S.; Khoury, J.D.; Ohanian, M.; Bueso-Ramos, C.E.; Abruzzo, L.V. Double minute chromosomes in acute myeloid leukemia, myelodysplastic syndromes, and chronic myelomonocytic leukemia are associated with micronuclei, MYC or MLL amplification, and complex karyotype. Cancer Genet. 2016, 209, 313–320. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, W.; Yu, J.; Pei, S.; Zhang, Q.; Shi, J.; Huang, H.; Zhao, Y. TP53 in MDS and AML: Biological and clinical advances. Cancer Lett. 2024, 588, 216767. [Google Scholar] [CrossRef] [PubMed]
- Philpott, C.; Tovell, H.; Frayling, I.M.; Cooper, D.N.; Upadhyaya, M. The NF1 somatic mutational landscape in sporadic human cancers. Hum. Genomics 2017, 11, 1–19. [Google Scholar] [CrossRef]
- Miller, P.G.; Sperling, A.S.; Mayerhofer, C.; McConkey, M.E.; Ellegast, J.M.; Da Silva, C.; Cohen, D.N.; Wang, C.; Sharda, A.; Yan, N.; et al. PPM1D modulates hematopoietic cell fitness and response to DNA damage and is a therapeutic target in myeloid malignancy. Blood 2023, 142, 2079–2091. [Google Scholar] [CrossRef]
- Kam, A.Y.F.; Piryani, S.O.; Lee, C.L.; Rizzieri, D.A.; Spector, N.L.; Sarantopoulos, S.; Doan, P.L. Selective ERBB2 and BCL2 inhibition is synergistic for mitochondrial-mediated apoptosis in MDS and AML cells. Mol. Cancer Res. 2021, 19, 886–899. [Google Scholar] [CrossRef]
- Douet-Guilbert, N.; Soubise, B.; Bernard, D.G.; Troadec, M.B. Cytogenetic and Genetic Abnormalities with Diagnostic Value in Myelodysplastic Syndromes (MDS): Focus on the Pre-Messenger RNA Splicing Process. Diagnostics 2022, 12, 1658. [Google Scholar] [CrossRef]
- Attar, N.; Kurdistani, S.K. Acetyltransferases by Cancer. Cold Spring Harb Perspect. Med. 2017, 7, a026534. [Google Scholar] [CrossRef]
- Tanaka, A.; Nishimura, K.; Saika, W.; Kon, A.; Koike, Y.; Tatsumi, H.; Takeda, J.; Nomura, M.; Zang, W.; Nakayama, M.; et al. SETBP1 is dispensable for normal and malignant hematopoiesis. Leukemia 2023, 37, 1802–1811. [Google Scholar] [CrossRef]
- Debaugny, R.E.; Skok, J.A. CTCF and CTCFL in cancer. Curr. Opin. Genet. Dev. 2020, 61, 44–52. [Google Scholar] [CrossRef]
- Levavasseur, F.; Oussous, S.; Zubaidan, T.; Kosmider, O.; Pendino, F.; Rombaut, D.; Bouscary, D.; Fontenay, M.; Lauret, E.; Dusanter-Fourt, I. FOXP1 regulates oxidative stress, SIRT1 expression, and resistance to chemotherapies in acute myeloid leukemia cells. Blood Adv. 2023, 7, 3265–3275. [Google Scholar] [CrossRef]
- Herzig, J.K.; Bullinger, L.; Tasdogan, A.; Zimmermann, P.; Schlegel, M.; Teleanu, V.; Weber, D.; Rücker, F.G.; Paschka, P.; Dolnik, A.; et al. Protein phosphatase 4 regulatory subunit 2 (PPP4R2) is recurrently deleted in acute myeloid leukemia and required for efficient DNA double strand break repair. Oncotarget 2017, 8, 95038–95053. [Google Scholar] [CrossRef]
- Pellagatti, A.; Boultwood, J. The molecular pathogenesis of the myelodysplastic syndromes. Eur. J. Haematol. 2015, 95, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Tanase-Nakao, K.; Shima, H.; Shirai, R.; Yoshida, K.; Osumi, T.; Deguchi, T.; Mori, M.; Arakawa, Y.; Takagi, M.; et al. Prevalence of germline GATA2 and SAMD9/9L variants in paediatric haematological disorders with monosomy 7. Br. J. Haematol. 2020, 191, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Liu, Y.; Rappaport, A.R.; Kitzing, T.; Schultz, N.; Zhao, Z.; Shroff, A.S.; Dickins, R.A.; Vakoc, C.R.; Bradner, J.E.; et al. MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer Cell 2014, 25, 652–665. [Google Scholar] [CrossRef]
- Zheng, Z.; Wang, W.; Feng, M.; Chen, X.; Ren, F.; Hou, Y. The mechanism of EZH2/H3K27me3 downregulating CXCL10 to affect CD8+ T cell exhaustion to participate in the transformation from myelodysplastic syndrome to acute myeloid leukaemia. Br. J. Haematol. 2025, 206, 1335–1349. [Google Scholar] [CrossRef]
- Lainey, E.; Thépot, S.; Bouteloup, C.; Sébert, M.; Ads, L.; Tailler, M.; Gardin, C.; De Botton, S.; Baruchel, A.; Fenaux, P.; et al. Tyrosine kinase inhibitors for the treatment of acute myeloid leukemia: Delineation of anti-leukemic mechanisms of action. Biochem. Pharmacol. 2011, 82, 1457–1466. [Google Scholar] [CrossRef]
- Hershberger, C.E.; Moyer, D.C.; Adema, V.; Kerr, C.M.; Hutter, S.; Meggendorfer, M.; Baer, C.; Kern, W.; Nadarajah, N.; Twardziok, S.; et al. Complex landscape of alternative splicing in myeloid neoplasms. Leukemia 2021, 35, 1108–1120. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).