
Unveiling Primary Cutaneous B-Cell Lymphomas: New Insights into Diagnosis and Treatment Strategies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Indolent/Low-Grade Primary Cutaneous B-Cell Lymphomas/Lymphoproliferative Disorders
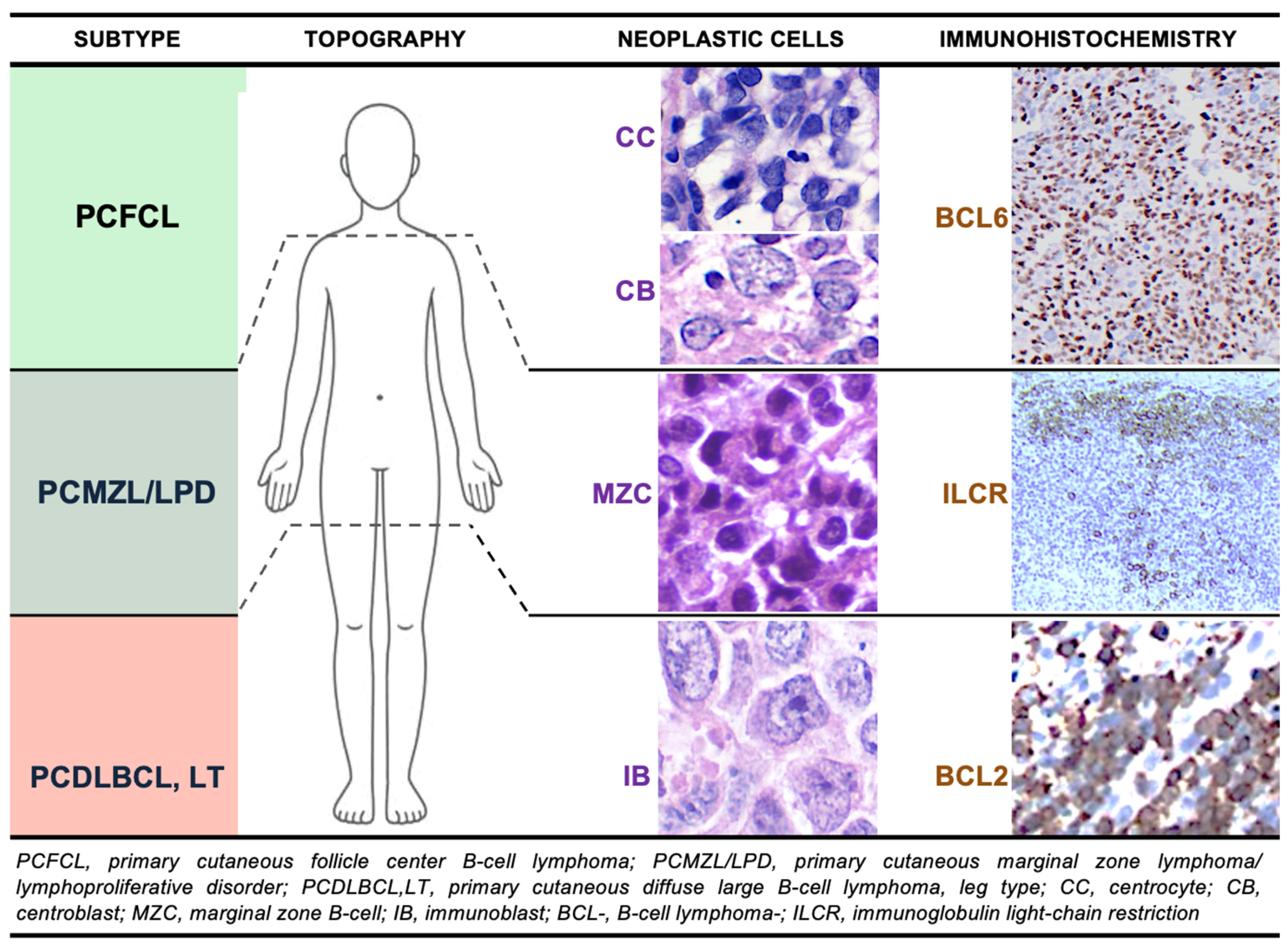
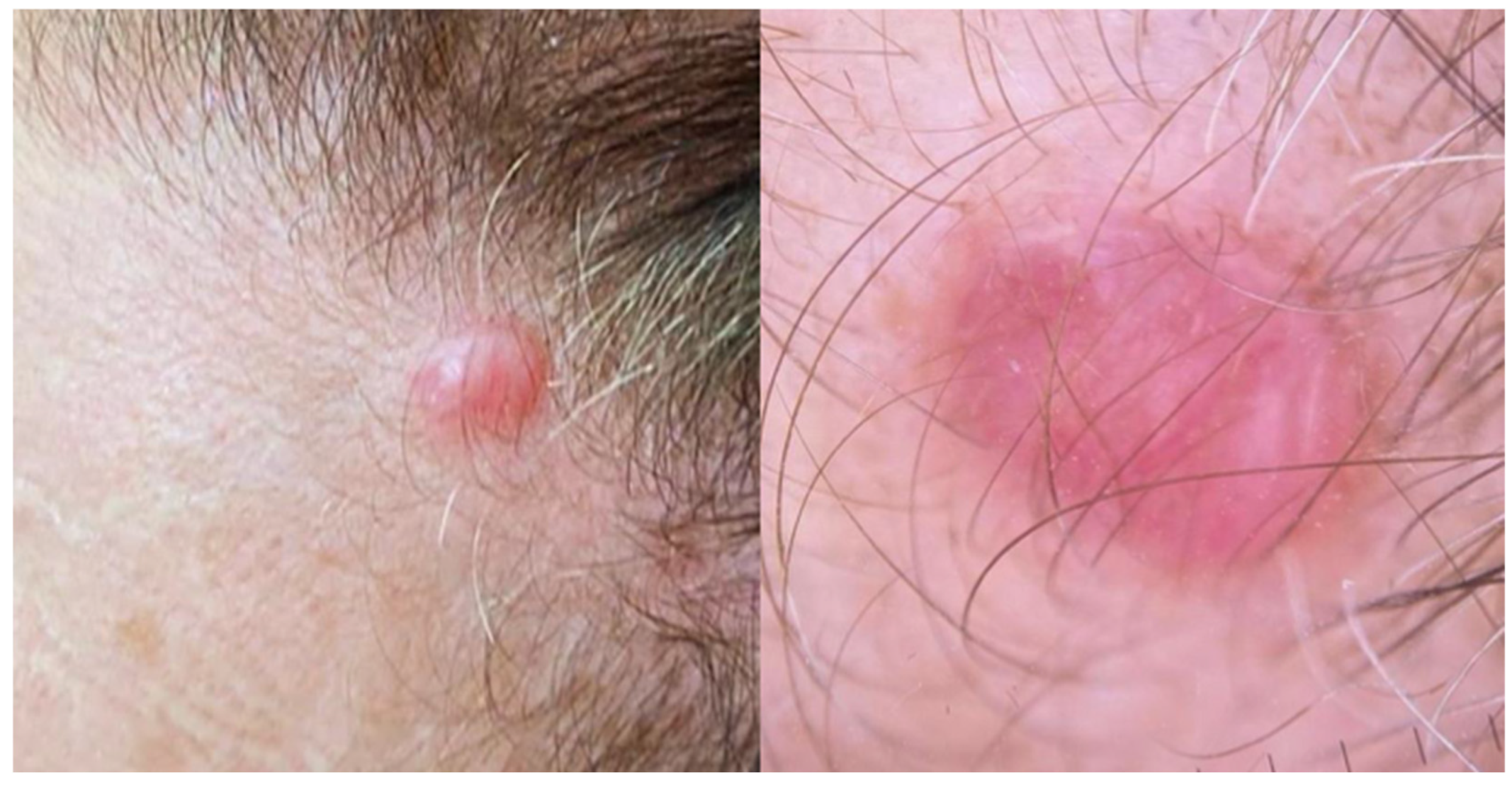
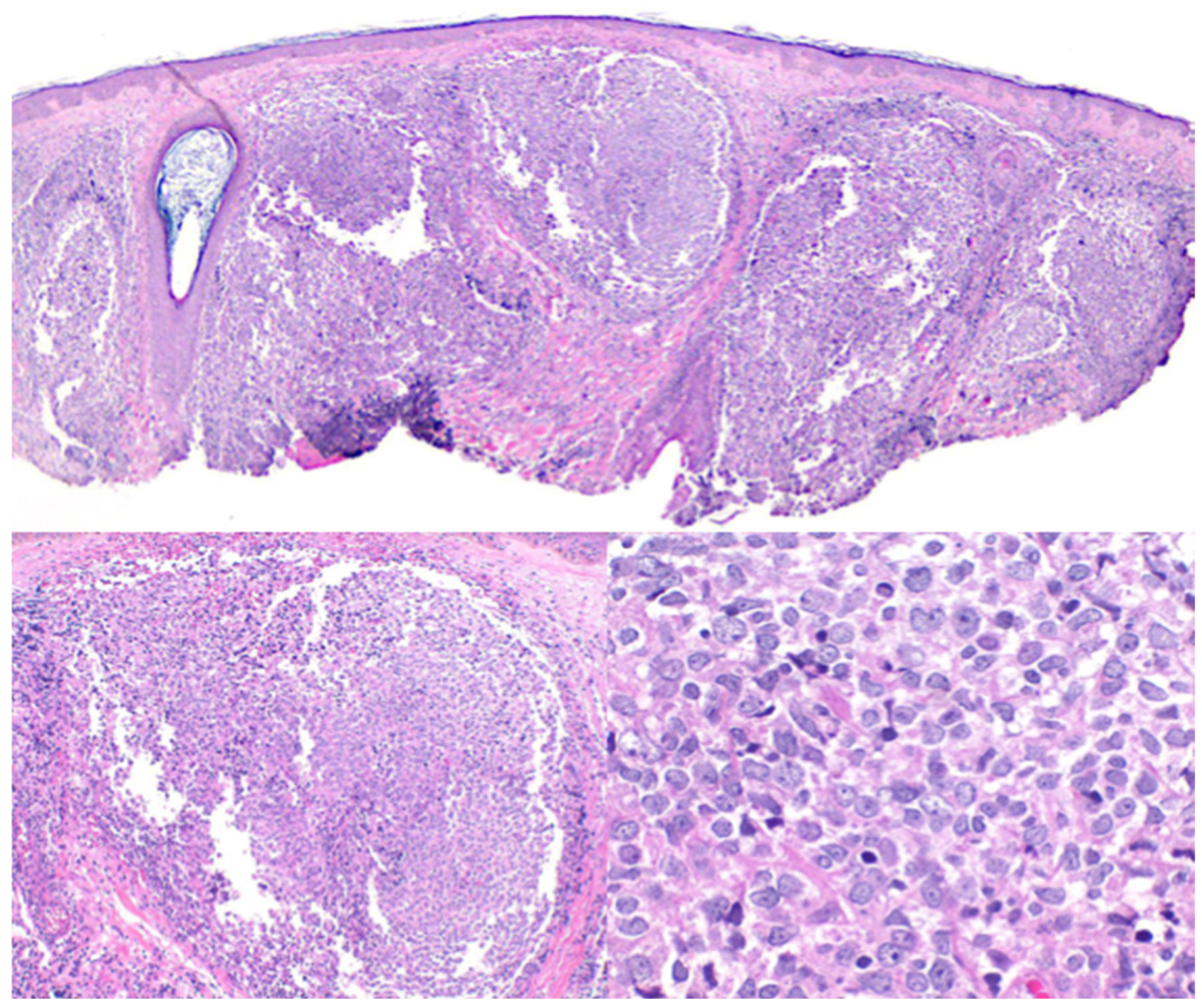
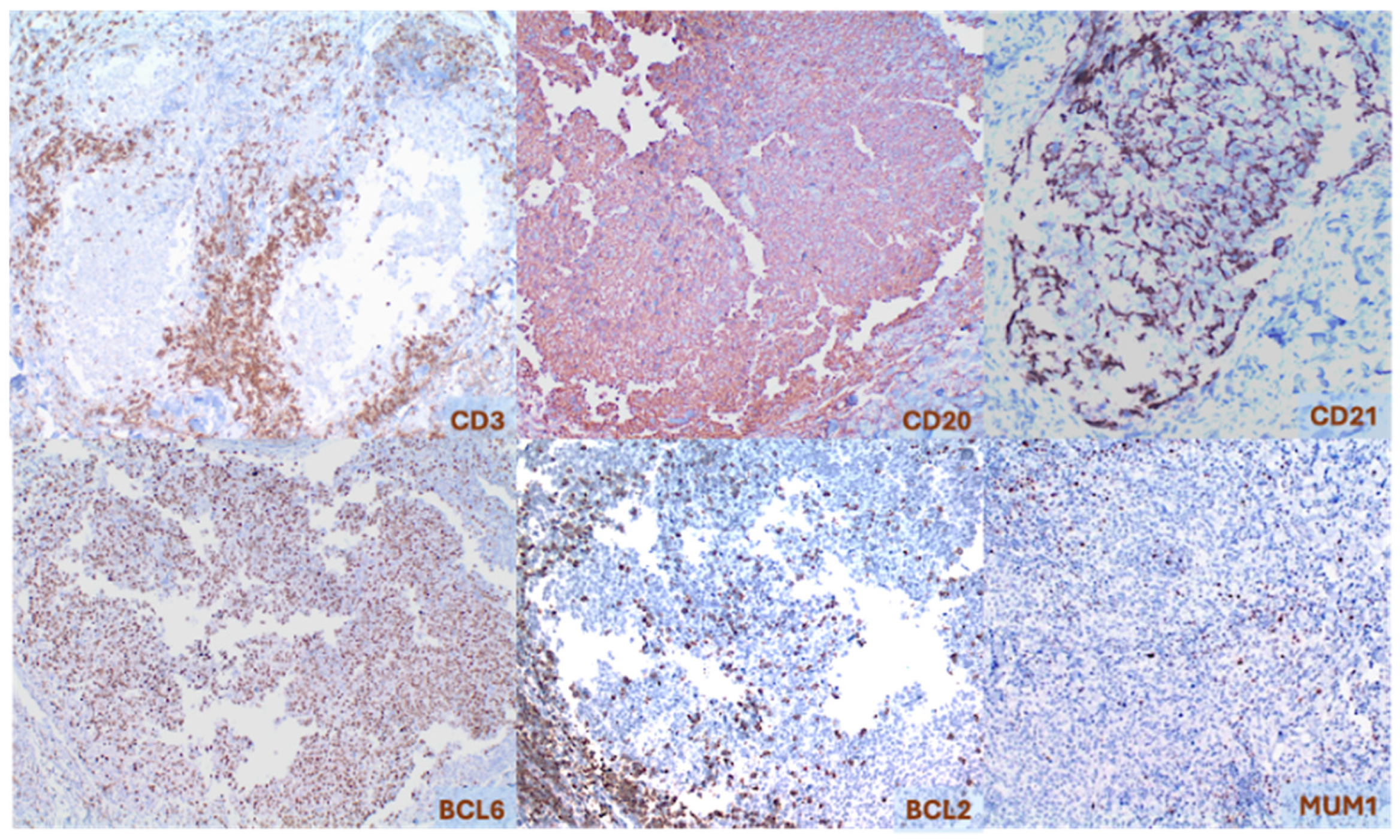
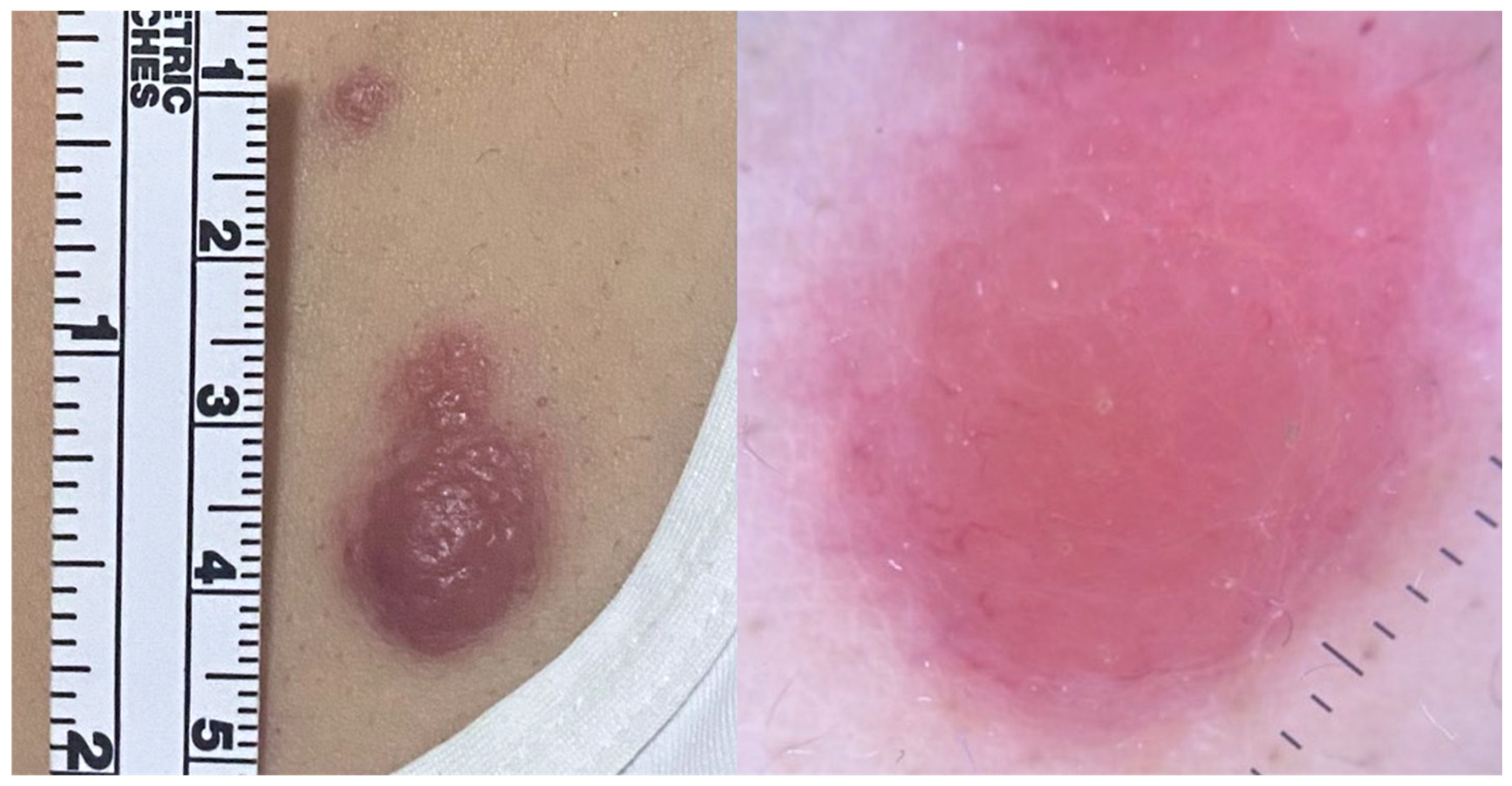
2.1. Primary Cutaneous Follicle Center Lymphoma (PCFCL)
2.2. Primary Cutaneous Marginal Zone Lymphoma/Lymphoproliferative Disorder (PCMZL/LPD)
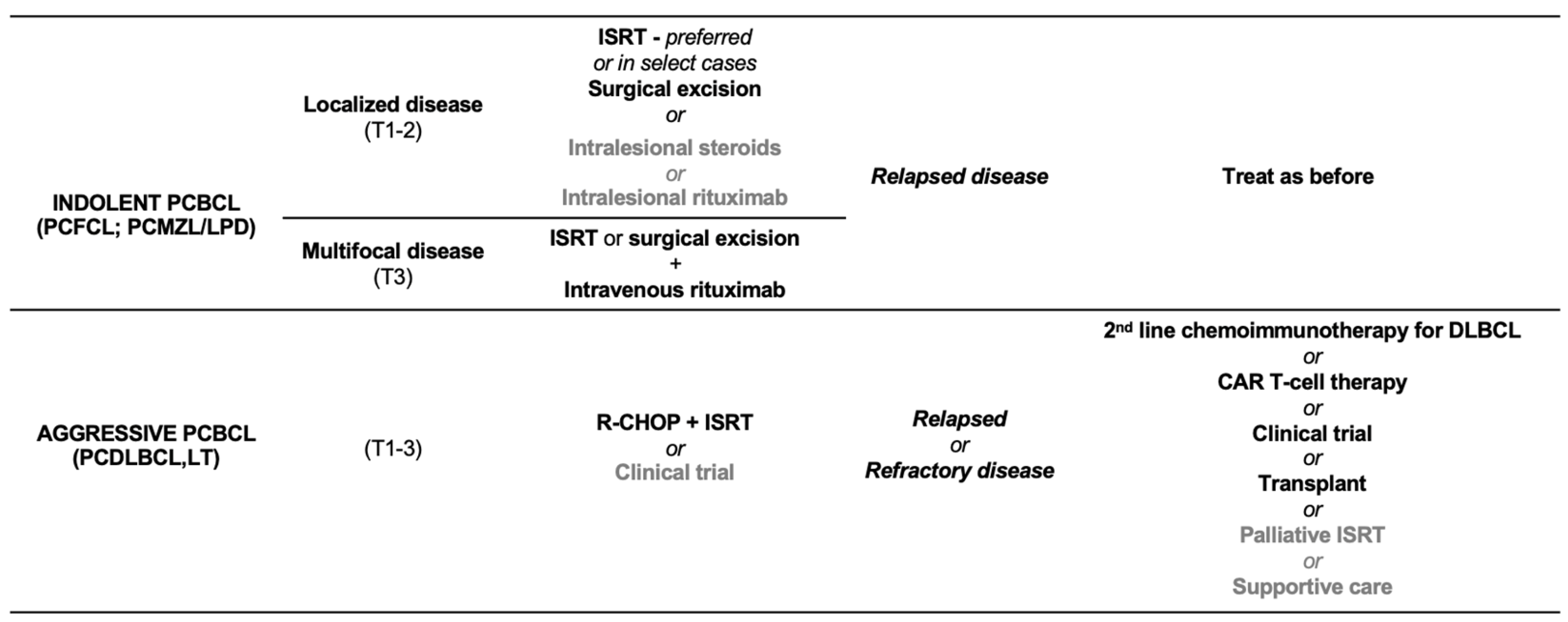
2.3. Treatment of Indolent/Low-Grade Primary Cutaneous B-Cell Lymphomas/Lymphoproliferative Disorders
2.4. Differential Diagnosis for Indolent/Low-Grade Primary Cutaneous B-Cell Lymphomas/Lymphoproliferative Disorders
3. Intermediate to Aggressive/High-Grade Primary Cutaneous B-Cell Lymphomas
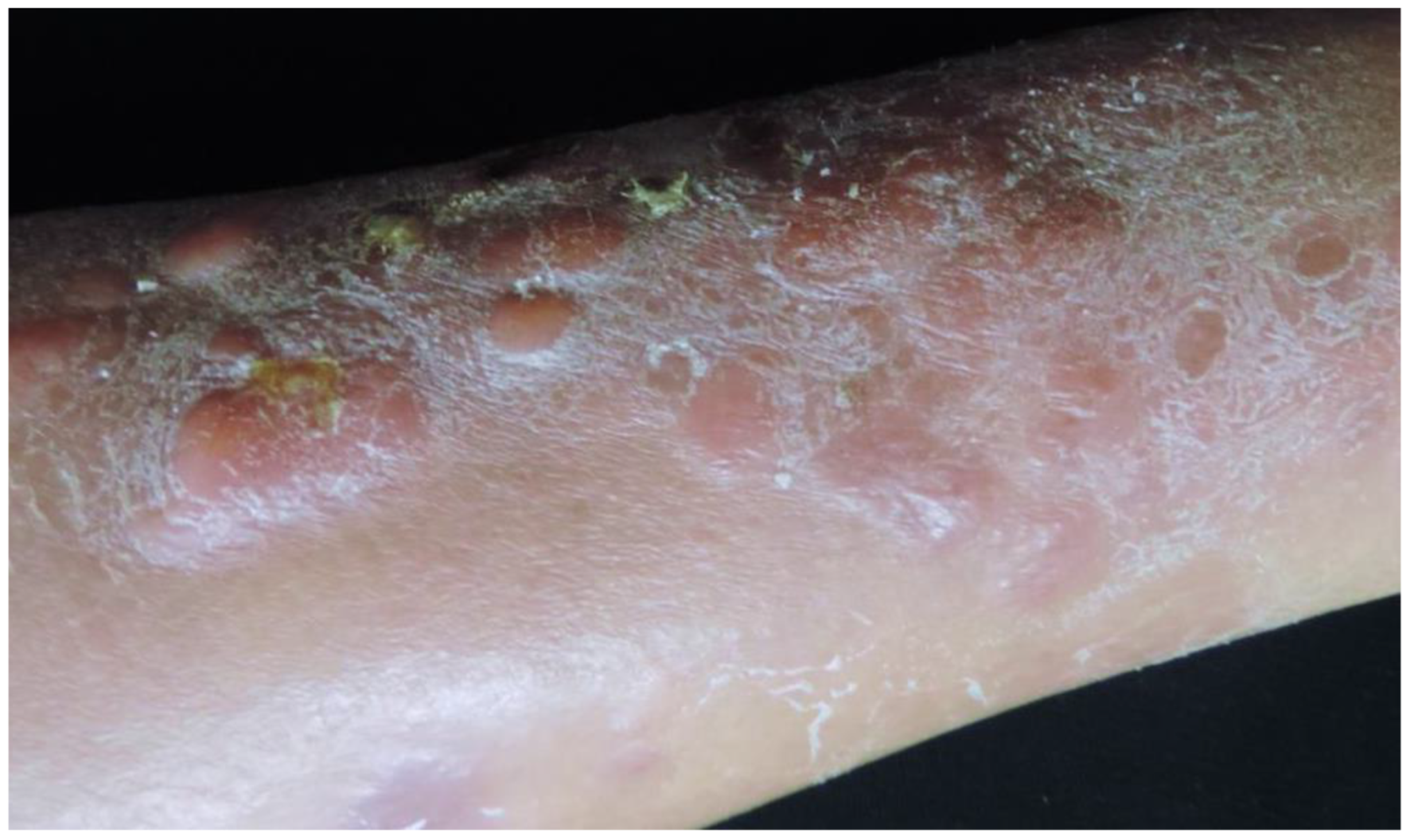
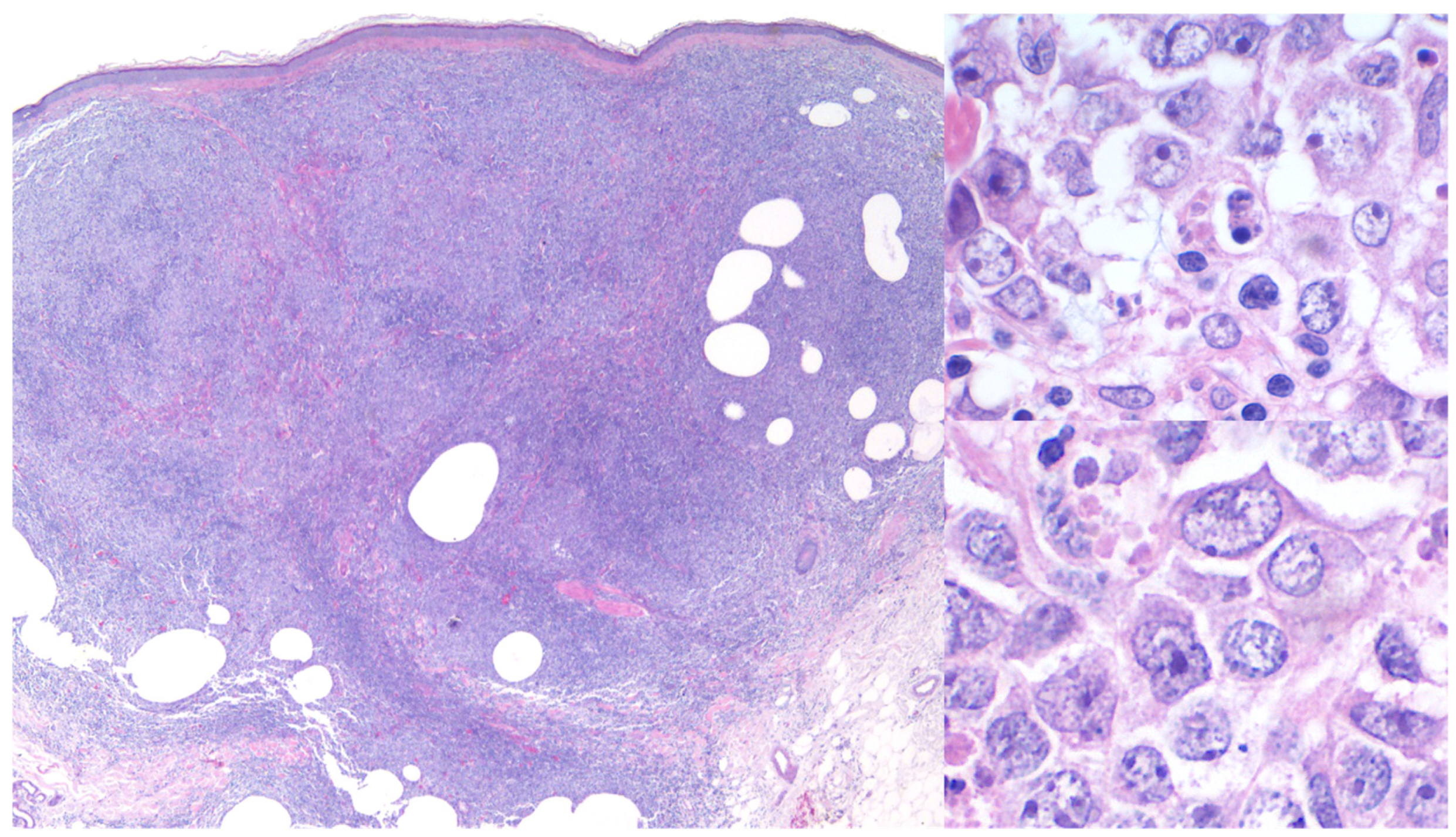
3.1. Primary Cutaneous Diffuse Large B-Cell Lymphoma, Leg Type (PCDLBCL,LT)
3.2. Treatment of PCDLBCL,LT
3.3. Differential Diagnosis for PCDLBCL,LT
4. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Goyal, N.; O’Leary, D.; Carter, J.B.; Comfere, N.; Sokumbi, O.; Goyal, A. A Practical Review of the Presentation, Diagnosis, and Management of Cutaneous B-Cell Lymphomas. Dermatol. Clin. 2023, 41, 187–208. [Google Scholar] [CrossRef] [PubMed]
- Bradford, P.T.; Devesa, S.S.; Anderson, W.F.; Toro, J.R. Cutaneous lymphoma incidence patterns in the United States: A population-based study of 3884 cases. Blood 2009, 113, 5064–5073. [Google Scholar] [CrossRef] [PubMed]
- Charli-Joseph, Y.V.; Gatica-Torres, M.; Pincus, L.B. Approach to Cutaneous Lymphoid Infiltrates: When to Consider Lymphoma? Indian J. Dermatol. 2016, 61, 351–374. [Google Scholar] [CrossRef] [PubMed]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef]
- Villasenor-Park, J.; Chung, J.; Kim, E.J. Cutaneous B-Cell Lymphomas. Hematol. Oncol. Clin. N. Am. 2024, 38, 1111–1131. [Google Scholar] [CrossRef]
- Suárez, A.L.; Pulitzer, M.; Horwitz, S.; Moskowitz, A.; Querfeld, C.; Myskowski, P.L. Primary cutaneous B-cell lymphomas: Part I. Clinical features, diagnosis, and classification. J. Am. Acad. Dermatol. 2013, 69, 329.e1–329.e13. [Google Scholar] [CrossRef]
- Campo, E.; Jaffe, E.S.; Cook, J.R.; Quintanilla-Martinez, L.; Swerdlow, S.H.; Anderson, K.C.; Brousset, P.; Cerroni, L.; de Leval, L.; Dirnhofer, S.; et al. The International Consensus Classification of Mature Lymphoid Neoplasms: A report from the Clinical Advisory Committee. Blood 2022, 140, 1229–1253. [Google Scholar] [CrossRef]
- Kempf, W.; Mitteldorf, C.; Cerroni, L.; Willemze, R.; Berti, E.; Guenova, E.; Scarisbrick, J.J.; Battistella, M. Classifications of cutaneous lymphomas and lymphoproliferative disorders: An update from the EORTC cutaneous lymphoma histopathology group. J. Eur. Acad. Dermatol. Venereol. 2024, 38, 1491–1503. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network. Clinical Practice Guidelines in Oncology. Primary Cutaneous Lymphomas (Version 1.25). Available online: https://www.nccn.org/professionals/physician_gls/pdf/primary_cutaneous.pdf (accessed on 16 March 2025).
- Charli-Joseph, Y.; Toussaint-Claire, S.; Lome-Maldonado, C.; Montante-Montes de Oca, D.; Ortiz-Hidalgo, C. Approach to dermal-based lymphoid infiltrates and proliferations. Semin. Cutan. Med. Surg. 2018, 37, 61–74. [Google Scholar] [CrossRef]
- Leary, D.O.; Goyal, N.; Rubin, N.; Goyal, A. Characterization of Primary and Secondary Cutaneous B-Cell Lymphomas: A Population-Based Study of 4758 Patients. Clin. Lymphoma Myeloma Leuk. 2022, 22, e269–e278. [Google Scholar] [CrossRef]
- Kim, Y.H.; Willemze, R.; Pimpinelli, N.; Whittaker, S.; Olsen, E.A.; Ranki, A.; Dummer, R.; Hoppe, R.T.; ISCL and the EORTC. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sezary syndrome: A proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood 2007, 110, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Olsen, E.A.; Whittaker, S.; Willemze, R.; Pinter-Brown, L.; Foss, F.; Geskin, L.; Schwartz, L.; Horwitz, S.; Guitart, J.; Zic, J.; et al. Primary cutaneous lymphoma: Recommendations for clinical trial design and staging update from the ISCL, USCLC, and EORTC. Blood 2022, 140, 419–437. [Google Scholar] [CrossRef] [PubMed]
- Senff, N.J.; Noordijk, E.M.; Kim, Y.H.; Bagot, M.; Berti, E.; Cerroni, L.; Dummer, R.; Duvic, M.; Hoppe, R.T.; Pimpinelli, N.; et al. European Organization for Research and Treatment of Cancer and International Society for Cutaneous Lymphoma consensus recommendations for the management of cutaneous B-cell lymphomas. Blood 2008, 112, 1600–1609. [Google Scholar] [CrossRef] [PubMed]
- Fujii, K.; Hamada, T.; Shimauchi, T.; Asai, J.; Fujisawa, Y.; Ihn, H.; Katoh, N. Cutaneous lymphoma in Japan, 2012-2017: A nationwide study. J. Dermatol. Sci. 2020, 97, 187–193. [Google Scholar] [CrossRef]
- Moon, I.J.; Won, C.H.; Chang, S.E.; Park, C.S.; Yoon, D.H.; Song, S.Y.; Lee, M.W.; Lee, W.J. Prevalence, clinical features, and survival outcome trends of 627 patients with primary cutaneous lymphoma over 29 years: A retrospective review from single tertiary center in Korea. Sci. Rep. 2024, 14, 20118. [Google Scholar] [CrossRef]
- Skala, S.L.; Hristov, B.; Hristov, A.C. Primary Cutaneous Follicle Center Lymphoma. Arch. Pathol. Lab. Med. 2018, 142, 1313–1321. [Google Scholar] [CrossRef]
- Zinzani, P.L.; Quaglino, P.; Pimpinelli, N.; Berti, E.; Baliva, G.; Rupoli, S.; Martelli, M.; Alaibac, M.; Borroni, G.; Chimenti, S.; et al. Italian Study Group for Cutaneous Lymphomas. Prognostic factors in primary cutaneous B-cell lymphoma: The Italian Study Group for Cutaneous Lymphomas. J. Clin. Oncol. 2006, 24, 1376–1382. [Google Scholar] [CrossRef]
- Bessell, E.M.; Humber, C.E.; O’Connor, S.; English, J.S.; Perkins, W.; Dickinson, P.D.; Patel, A.N. Primary cutaneous B-cell lymphoma in Nottinghamshire U.K.: Prognosis of subtypes defined in the WHO-EORTC classification. Br. J. Dermatol. 2012, 167, 1118–1123. [Google Scholar] [CrossRef]
- Grange, F.; Bekkenk, M.W.; Wechsler, J.; Meijer, C.J.; Cerroni, L.; Bernengo, M.; Bosq, J.; Hedelin, G.; Fink Puches, R.; van Vloten, W.A.; et al. Prognostic factors in primary cutaneous large B-cell lymphomas: A European multicenter study. J. Clin. Oncol. 2001, 19, 3602–3610. [Google Scholar] [CrossRef]
- Bergman, R.; Kurtin, P.J.; Gibson, L.E.; Hull, P.R.; Kimlinger, T.K.; Schroeter, A.L. Clinicopathologic, immunophenotypic, and molecular characterization of primary cutaneous follicular B-cell lymphoma. Arch. Dermatol. 2001, 137, 432–439. [Google Scholar]
- Massone, C.; Fink-Puches, R.; Laimer, M.; Rütten, A.; Vale, E.; Cerroni, L. Miliary and agminated-type primary cutaneous follicle center lymphoma: Report of 18 cases. J. Am. Acad. Dermatol. 2011, 65, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Gulia, A.; Saggini, A.; Wiesner, T.; Fink-Puches, R.; Argenyi, Z.; Ferrara, G.; Müller, C.S.; Vale, E.; Cerroni, L. Clinicopathologic features of early lesions of primary cutaneous follicle center lymphoma, diffuse type: Implications for early diagnosis and treatment. J. Am. Acad. Dermatol. 2011, 65, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Geller, S.; Pulitzer, M.; Myskowski, P.; Kheterpal, M. Low-Grade Cutaneous B-cell Lymphoma in African American Patients. J. Drugs Dermatol. 2018, 17, 1334–1337. [Google Scholar] [PubMed] [PubMed Central]
- Senff, N.J.; Hoefnagel, J.J.; Jansen, P.M.; Vermeer, M.H.; van Baarlen, J.; Blokx, W.A.; Canninga-van Dijk, M.R.; Geerts, M.L.; Hebeda, K.M.; Kluin, P.M.; et al. Reclassification of 300 primary cutaneous B-Cell lymphomas according to the new WHO-EORTC classification for cutaneous lymphomas: Comparison with previous classifications and identification of prognostic markers. J. Clin. Oncol. 2007, 25, 1581–1587. [Google Scholar] [CrossRef]
- Charli-Joseph, Y.; Cerroni, L.; LeBoit, P.E. Cutaneous Spindle-Cell B-Cell Lymphomas: Most are Neoplasms of Follicular Center Cell Origin. Am. J. Surg. Pathol. 2015, 39, 737–743. [Google Scholar] [CrossRef]
- Cerroni, L. Skin Lymphoma: The Illustrated Guide, 5th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2020; ISBN 978-1-119-48590-2. [Google Scholar]
- Gru, A.; Schaffer, A. Hematopathology of the Skin: A Clinical and Practical Approach, 2nd ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2023; ISBN 978-1-97-515855-2. [Google Scholar]
- Hoefnagel, J.J.; Dijkman, R.; Basso, K.; Jansen, P.M.; Hallermann, C.; Willemze, R.; Tensen, C.P.; Vermeer, M.H. Distinct types of primary cutaneous large B-cell lymphoma identified by gene expression profiling. Blood 2005, 105, 3671–3678. [Google Scholar] [CrossRef]
- Dijkman, R.; Tensen, C.P.; Jordanova, E.S.; Knijnenburg, J.; Hoefnagel, J.J.; Mulder, A.A.; Rosenberg, C.; Raap, A.K.; Willemze, R.; Szuhai, K.; et al. Array-based comparative genomic hybridization analysis reveals recurrent chromosomal alterations and prognostic parameters in primary cutaneous large B-cell lymphoma. J. Clin. Oncol. 2006, 24, 296–305. [Google Scholar] [CrossRef]
- Zhou, X.A.; Yang, J.; Ringbloom, K.G.; Martinez-Escala, M.E.; Stevenson, K.E.; Wenzel, A.T.; Fantini, D.; Martin, H.K.; Moy, A.P.; Morgan, E.A.; et al. Genomic landscape of cutaneous follicular lymphomas reveals 2 subgroups with clinically predictive molecular features. Blood Adv. 2021, 5, 649–661. [Google Scholar] [CrossRef]
- Goodlad, J.R.; Krajewski, A.S.; Batstone, P.J.; McKay, P.; White, J.M.; Benton, E.C.; Kavanagh, G.M.; Lucraft, H.H.; Scotland and Newcastle Lymphoma Group. Primary cutaneous follicular lymphoma: A clinicopathologic and molecular study of 16 cases in support of a distinct entity. Am. J. Surg. Pathol. 2002, 26, 733–741. [Google Scholar] [CrossRef]
- Child, F.J.; Russell-Jones, R.; Woolford, A.J.; Calonje, E.; Photiou, A.; Orchard, G.; Whittaker, S.J. Absence of the t(14;18) chromosomal translocation in primary cutaneous B-cell lymphoma. Br. J. Dermatol. 2001, 144, 735–744. [Google Scholar] [CrossRef]
- Barasch, N.J.K.; Liu, Y.C.; Ho, J.; Bailey, N.; Aggarwal, N.; Cook, J.R.; Swerdlow, S.H. The molecular landscape and other distinctive features of primary cutaneous follicle center lymphoma. Hum. Pathol. 2020, 106, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Wahab, A.; Tang, S.Y.; Robson, A.; Morris, S.; Agar, N.; Wain, E.M.; Child, F.; Scarisbrick, J.; Neat, M.; Whittaker, S. Chromosomal anomalies in primary cutaneous follicle center cell lymphoma do not portend a poor prognosis. J. Am. Acad. Dermatol. 2014, 70, 1010–1020. [Google Scholar] [CrossRef] [PubMed]
- Servitje, O.; Muniesa, C.; Benavente, Y.; Monsálvez, V.; Garcia-Muret, M.P.; Gallardo, F.; Domingo-Domenech, E.; Lucas, A.; Climent, F.; Rodriguez-Peralto, J.L.; et al. Primary cutaneous marginal zone B-cell lymphoma: Response to treatment and disease-free survival in a series of 137 patients. J. Am. Acad. Dermatol. 2013, 69, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Hoefnagel, J.J.; Vermeer, M.H.; Jansen, P.M.; Heule, F.; van Voorst Vader, P.C.; Sanders, C.J.; Gerritsen, M.J.; Geerts, M.L.; Meijer, C.J.; Noordijk, E.M.; et al. Primary cutaneous marginal zone B-cell lymphoma: Clinical and therapeutic features in 50 cases. Arch. Dermatol. 2005, 141, 1139–1145. [Google Scholar] [CrossRef]
- Kempf, W.; Kazakov, D.V.; Mitteldorf, C. Cutaneous lymphomas: An update. Part 2: B-cell lymphomas and related conditions. Am. J. Dermatopathol. 2014, 36, 197–208, quiz 209-10. [Google Scholar] [CrossRef]
- Hodak, E.; Feuerman, H.; Barzilai, A.; David, M.; Cerroni, L.; Feinmesser, M. Anetodermic primary cutaneous B-cell lymphoma: A unique clinicopathological presentation of lymphoma possibly associated with antiphospholipid antibodies. Arch. Dermatol. 2010, 146, 175–182. [Google Scholar] [CrossRef]
- Magro, C.M.; Davis, T.L.; Kurtzman, D.J.B. Epidermotropic marginal zone lymphoma: An uncommon cutaneous B-cell lymphoma responsive to rituximab. JAAD Case Rep. 2017, 3, 474–476. [Google Scholar] [CrossRef]
- Schafernak, K.T.; Variakojis, D.; Goolsby, C.L.; Tucker, R.M.; Martínez-Escala, M.E.; Smith, F.A.; Dittman, D.; Chenn, A.; Guitart, J. Clonality assessment of cutaneous B-cell lymphoid proliferations: A comparison of flow cytometry immunophenotyping, molecular studies, and immunohistochemistry/in situ hybridization and review of the literature. Am. J. Dermatopathol. 2014, 36, 781–795. [Google Scholar] [CrossRef]
- Edinger, J.T.; Kant, J.A.; Swerdlow, S.H. Cutaneous marginal zone lymphomas have distinctive features and include 2 subsets. Am. J. Surg. Pathol. 2010, 34, 1830–1841. [Google Scholar] [CrossRef]
- van Maldegem, F.; van Dijk, R.; Wormhoudt, T.A.; Kluin, P.M.; Willemze, R.; Cerroni, L.; van Noesel, C.J.; Bende, R.J. The majority of cutaneous marginal zone B-cell lymphomas expresses class-switched immunoglobulins and develops in a T-helper type 2 inflammatory environment. Blood 2008, 112, 3355–3361. [Google Scholar] [CrossRef]
- Carlsen, E.D.; Swerdlow, S.H.; Cook, J.R.; Gibson, S.E. Class-switched Primary Cutaneous Marginal Zone Lymphomas Are Frequently IgG4-positive and Have Features Distinct from IgM-positive Cases. Am. J. Surg. Pathol. 2019, 43, 1403–1412. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, A.J.; Frühwirth, M.; Aigelsreiter, A.; Cerroni, L.; Neumeister, P. Primary cutaneous marginal zone B-cell lymphomas are targeted by aberrant somatic hypermutation. J. Investig. Dermatol. 2009, 129, 476–479. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.; Pacchiarotti, A.; Frontani, M.; Pescarmona, E.; Caprini, E.; Lombardo, G.A.; Russo, G.; Faraggiana, T. Primary cutaneous B-cell lymphoma is associated with somatically hypermutated immunoglobulin variable genes and frequent use of VH1-69 and VH4-59 segments. Br. J. Dermatol. 2010, 162, 611–618. [Google Scholar] [CrossRef]
- Takino, H.; Li, C.; Hu, S.; Kuo, T.T.; Geissinger, E.; Muller-Hermelink, H.K.; Kim, B.; Swerdlow, S.H.; Inagaki, H. Primary cutaneous marginal zone B-cell lymphoma: A molecular and clinicopathological study of cases from Asia, Germany, and the United States. Mod. Pathol. 2008, 21, 1517–1526. [Google Scholar] [CrossRef]
- Golling, P.; Cozzio, A.; Dummer, R.; French, L.; Kempf, W. Primary cutaneous B-cell lymphomas—Clinicopathological, prognostic and therapeutic characterization of 54 cases according to the WHO-EORTC classification and the ISCL/EORTC TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sezary syndrome. Leuk. Lymphoma 2008, 49, 1094–1103. [Google Scholar] [CrossRef]
- Haverkos, B.; Tyler, K.; Gru, A.A.; Winardi, F.K.; Frederickson, J.; Hastings, J.; Elkins, C.; Zhang, X.; Xu-Welliver, M.; Wong, H.K.; et al. Primary Cutaneous B-Cell Lymphoma: Management and Patterns of Recurrence at the Multimodality Cutaneous Lymphoma Clinic of The Ohio State University. Oncologist 2015, 20, 1161–1166. [Google Scholar] [CrossRef]
- Goyal, A.; Carter, J.B.; Pashtan, I.; Gallotto, S.; Wang, I.; Isom, S.; Ng, A.; Winkfield, K.M. Very low-dose versus standard dose radiation therapy for indolent primary cutaneous B-cell lymphomas: A retrospective study. J. Am. Acad. Dermatol. 2018, 78, 408–410. [Google Scholar] [CrossRef]
- Cozzio, A.; Kempf, W.; Schmid-Meyer, R.; Gilliet, M.; Michaelis, S.; Schärer, L.; Burg, G.; Dummer, R. Intra-lesional low-dose interferon alpha2a therapy for primary cutaneous marginal zone B-cell lymphoma. Leuk. Lymphoma 2006, 47, 865–869. [Google Scholar] [CrossRef]
- Specht, L.; Dabaja, B.; Illidge, T.; Wilson, L.D.; Hoppe, R.T.; International Lymphoma Radiation Oncology Group. Modern radiation therapy for primary cutaneous lymphomas: Field and dose guidelines from the International Lymphoma Radiation Oncology Group. Int. J. Radiat. Oncol. Biol. Phys. 2015, 92, 32–39. [Google Scholar] [CrossRef]
- Neelis, K.J.; Schimmel, E.C.; Vermeer, M.H.; Senff, N.J.; Willemze, R.; Noordijk, E.M. Low-dose palliative radiotherapy for cutaneous B- and T-cell lymphomas. Int. J. Radiat. Oncol. Biol. Phys. 2009, 74, 154–158. [Google Scholar] [CrossRef]
- Perry, A.; Vincent, B.J.; Parker, S.R. Intralesional corticosteroid therapy for primary cutaneous B-cell lymphoma. Br. J. Dermatol. 2010, 163, 223–225. [Google Scholar] [CrossRef] [PubMed]
- Kollipara, R.; Hans, A.; Hall, J.; Lisle, A. A case report of primary cutaneous marginal zone lymphoma treated with intralesional steroids. Dermatol. Online J. 2015, 21, 13030/qt9s15929m. [Google Scholar] [CrossRef]
- Ponzoni, M.; Ferreri, A.J.; Mappa, S.; Pasini, E.; Govi, S.; Facchetti, F.; Fanoni, D.; Tucci, A.; Vino, A.; Doglioni, C.; et al. Prevalence of Borrelia burgdorferi infection in a series of 98 primary cutaneous lymphomas. Oncologist 2011, 16, 1582–1588. [Google Scholar] [CrossRef] [PubMed]
- Wood, G.S.; Kamath, N.V.; Guitart, J.; Heald, P.; Kohler, S.; Smoller, B.R.; Cerroni, L. Absence of Borrelia burgdorferi DNA in cutaneous B-cell lymphomas from the United States. J. Cutan. Pathol. 2001, 28, 502–507. [Google Scholar] [CrossRef]
- Trent, J.T.; Romanelli, P.; Kerdel, F.A. Topical targretin and intralesional interferon alfa for cutaneous lymphoma of the scalp. Arch. Dermatol. 2002, 138, 1421–1423. [Google Scholar] [CrossRef]
- Kerl, K.; Prins, C.; Saurat, J.H.; French, L.E. Intralesional and intravenous treatment of cutaneous B-cell lymphomas with the monoclonal anti-CD20 antibody rituximab: Report and follow-up of eight cases. Br. J. Dermatol. 2006, 155, 1197–1200. [Google Scholar] [CrossRef]
- Heinzerling, L.; Dummer, R.; Kempf, W.; Schmid, M.H.; Burg, G. Intralesional therapy with anti-CD20 monoclonal antibody rituximab in primary cutaneous B-cell lymphoma. Arch. Dermatol. 2000, 136, 374–378. [Google Scholar] [CrossRef]
- Kyrtsonis, M.C.; Siakantaris, M.P.; Kalpadakis, C.; Dimopoulou, M.N.; Vassilakopoulos, T.P.; Kontopidou, F.N.; Antoniou, C.; Korkolopoulou, P.; Panayiotidis, P.; Pangalis, G.A. Favorable outcome of primary cutaneous marginal zone lymphoma treated with intralesional rituximab. Eur. J. Haematol. 2006, 77, 300–303. [Google Scholar] [CrossRef]
- Heinzerling, L.M.; Urbanek, M.; Funk, J.O.; Peker, S.; Bleck, O.; Neuber, K.; Burg, G.; von Den Driesch, P.; Dummer, R. Reduction of tumor burden and stabilization of disease by systemic therapy with anti-CD20 antibody (rituximab) in patients with primary cutaneous B-cell lymphoma. Cancer 2000, 89, 1835–1844. [Google Scholar] [CrossRef] [PubMed]
- Gellrich, S.; Muche, J.M.; Wilks, A.; Jasch, K.C.; Voit, C.; Fischer, T.; Audring, H.; Sterry, W. Systemic eight-cycle anti-CD20 monoclonal antibody (rituximab) therapy in primary cutaneous B-cell lymphomas--an applicational observation. Br. J. Dermatol. 2005, 153, 167–173. [Google Scholar] [CrossRef]
- Morales, A.V.; Advani, R.; Horwitz, S.M.; Riaz, N.; Reddy, S.; Hoppe, R.T.; Kim, Y.H. Indolent primary cutaneous B-cell lymphoma: Experience using systemic rituximab. J. Am. Acad. Dermatol. 2008, 59, 953–957. [Google Scholar] [CrossRef] [PubMed]
- Valencak, J.; Weihsengruber, F.; Rappersberger, K.; Trautinger, F.; Chott, A.; Streubel, B.; Muellauer, L.; Der-Petrossian, M.; Jonak, C.; Binder, M.; et al. Rituximab monotherapy for primary cutaneous B-cell lymphoma: Response and follow-up in 16 patients. Ann. Oncol. 2009, 20, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, A.; Humme, D.; Terhorst, D.; Gellrich, S.; Sterry, W.; Beyer, M. Long-term outcome of intravenous therapy with rituximab in patients with primary cutaneous B-cell lymphomas. Br. J. Dermatol. 2013, 169, 1126–1132. [Google Scholar] [CrossRef]
- Vitiello, P.; Sica, A.; Ronchi, A.; Caccavale, S.; Franco, R.; Argenziano, G. Primary Cutaneous B-Cell Lymphomas: An Update. Front. Oncol. 2020, 10, 651. [Google Scholar] [CrossRef]
- Grogg, K.L.; Jung, S.; Erickson, L.A.; McClure, R.F.; Dogan, A. Primary cutaneous CD4-positive small/medium-sized pleomorphic T-cell lymphoma: A clonal T-cell lymphoproliferative disorder with indolent behavior. Mod. Pathol. 2008, 21, 708–715. [Google Scholar] [CrossRef]
- Rodríguez Pinilla, S.M.; Roncador, G.; Rodríguez-Peralto, J.L.; Mollejo, M.; García, J.F.; Montes-Moreno, S.; Camacho, F.I.; Ortiz, P.; Limeres-González, M.A.; Torres, A.; et al. Primary cutaneous CD4+ small/medium-sized pleomorphic T-cell lymphoma expresses follicular T-cell markers. Am. J. Surg. Pathol. 2009, 33, 81–90. [Google Scholar] [CrossRef]
- Beltraminelli, H.; Leinweber, B.; Kerl, H.; Cerroni, L. Primary cutaneous CD4+ small-/medium-sized pleomorphic T-cell lymphoma: A cutaneous nodular proliferation of pleomorphic T lymphocytes of undetermined significance? A study of 136 cases. Am. J. Dermatopathol. 2009, 31, 317–322. [Google Scholar] [CrossRef]
- Baum, C.L.; Link, B.K.; Neppalli, V.T.; Swick, B.L.; Liu, V. Reappraisal of the provisional entity primary cutaneous CD4+ small/medium pleomorphic T-cell lymphoma: A series of 10 adult and pediatric patients and review of the literature. J. Am. Acad. Dermatol. 2011, 65, 739–748. [Google Scholar] [CrossRef]
- Cetinözman, F.; Jansen, P.M.; Willemze, R. Expression of programmed death-1 in primary cutaneous CD4-positive small/medium-sized pleomorphic T-cell lymphoma, cutaneous pseudo-T-cell lymphoma, and other types of cutaneous T-cell lymphoma. Am. J. Surg. Pathol. 2012, 36, 109–116. [Google Scholar] [CrossRef]
- Alberti-Violetti, S.; Torres-Cabala, C.A.; Talpur, R.; Corti, L.; Fanoni, D.; Venegoni, L.; Berti, E.; Duvic, M. Clinicopathological and molecular study of primary cutaneous CD4+ small/medium-sized pleomorphic T-cell lymphoma. J. Cutan. Pathol. 2016, 43, 1121–1130. [Google Scholar] [CrossRef]
- Pileri, A.; Grandi, V.; Agostinelli, C.; Santucci, M.; Lastrucci, I.; Guglielmo, A.; Pipitò, C.; Pimpinelli, N. BCL-2 expression in primary cutaneous follicle center lymphoma is associated with a higher risk of cutaneous relapses: A study of 126 cases. J. Eur. Acad. Dermatol. Venereol. 2022, 36, e811–e813. [Google Scholar] [CrossRef] [PubMed]
- Ronchi, A.; Vitiello, P.; D’Abbronzo, G.; Caccavale, S.; Argenziano, G.; Sica, A.; Alfan, R.; Savarese, G.; Berretta, M.; Cozzolino, I.; et al. Primary Cutaneous B-Cell Lymphomas with Large Cell Morphology: A Practical Review. Int. J. Mol. Sci. 2023, 24, 6204. [Google Scholar] [CrossRef] [PubMed]
- Grange, F.; Beylot-Barry, M.; Courville, P.; Maubec, E.; Bagot, M.; Vergier, B.; Souteyrand, P.; Machet, L.; Dalac, S.; Esteve, E.; et al. Primary cutaneous diffuse large B-cell lymphoma, leg type: Clinicopathologic features and prognostic analysis in 60 cases. Arch. Dermatol. 2007, 143, 1144–1150. [Google Scholar] [CrossRef] [PubMed]
- Vermeer, M.H.; Geelen, F.A.; van Haselen, C.W.; van Voorst Vader, P.C.; Geerts, M.L.; van Vloten, W.A.; Willemze, R. Primary cutaneous large B-cell lymphomas of the legs. A distinct type of cutaneous B-cell lymphoma with an intermediate prognosis. Dutch Cutaneous Lymphoma Working Group. Arch. Dermatol. 1996, 132, 1304–1308. [Google Scholar] [PubMed]
- Grange, F.; Joly, P.; Barbe, C.; Bagot, M.; Dalle, S.; Ingen-Housz-Oro, S.; Maubec, E.; D’Incan, M.; Ram-Wolff, C.; Dalac, S.; et al. Improvement of survival in patients with primary cutaneous diffuse large B-cell lymphoma, leg type, in France. JAMA Dermatol. 2014, 150, 535–541. [Google Scholar] [CrossRef]
- Kraft, R.M.; Ansell, S.M.; Villasboas, J.C.; Bennani, N.N.; Wang, Y.; Habermann, T.M.; Thanarajasingam, G.; Lester, S.C.; Macon, W.; Inwards, D.J.; et al. Outcomes in primary cutaneous diffuse large B-cell lymphoma, leg type. Hematol. Oncol. 2021, 39, 658–663. [Google Scholar] [CrossRef]
- Willemze, R.; Meijer, C.J.; Sentis, H.J.; Scheffer, E.; van Vloten, W.A.; Toonstra, J.; van der Putte, S.C. Primary cutaneous large cell lymphomas of follicular center cell origin. A clinical follow-up study of nineteen patients. J. Am. Acad. Dermatol. 1987, 16 Pt 1, 518–526. [Google Scholar] [CrossRef]
- Schrader, A.M.R.; de Groen, R.A.L.; Willemze, R.; Jansen, P.M.; Quint, K.D.; van Wezel, T.; van Eijk, R.; Ruano, D.; Tensen, C.P.; Hauben, E.; et al. Cell-of-origin classification using the Hans and Lymph2Cx algorithms in primary cutaneous large B-cell lymphomas. Virchows Arch. 2022, 480, 667–675. [Google Scholar] [CrossRef]
- Prieto-Torres, L.; Rodríguez-Pinilla, S.M. New insights and advances in defining primary cutaneous B-cell lymphomas and cutaneous B-cell-rich lymphoid proliferations. Diagn. Histopathol. 2024, 30, 419–429. [Google Scholar] [CrossRef]
- Pham-Ledard, A.; Prochazkova-Carlotti, M.; Deveza, M.; Laforet, M.P.; Beylot-Barry, M.; Vergier, B.; Parrens, M.; Feuillard, J.; Merlio, J.P.; Gachard, N. Molecular analysis of immunoglobulin variable genes supports a germinal center experienced normal counterpart in primary cutaneous diffuse large B-cell lymphoma, leg-type. J. Dermatol. Sci. 2017, 88, 238–246. [Google Scholar] [CrossRef]
- Schrader, A.M.R.; de Groen, R.A.L.; Willemze, R.; Jansen, P.M.; Quint, K.D.; Cleven, A.H.G.; van Wezel, T.; van Eijk, R.; Ruano, D.; Veelken, J.H.H.; et al. Genetic Stability of Driver Alterations in Primary Cutaneous Diffuse Large B-Cell Lymphoma, Leg Type and Their Relapses: A Rationale for the Use of Molecular-Based Methods for More Effective Disease Monitoring. Cancers 2022, 14, 5152. [Google Scholar] [CrossRef] [PubMed]
- Senff, N.J.; Zoutman, W.H.; Vermeer, M.H.; Assaf, C.; Berti, E.; Cerroni, L.; Espinet, B.; de Misa Cabrera, R.F.; Geerts, M.L.; Kempf, W.; et al. Fine-mapping chromosomal loss at 9p21: Correlation with prognosis in primary cutaneous diffuse large B-cell lymphoma, leg type. J. Investig. Dermatol. 2009, 129, 1149–1155. [Google Scholar] [CrossRef] [PubMed]
- Schrader, A.M.R.; Jansen, P.M.; Vermeer, M.H.; Kleiverda, J.K.; Vermaat, J.S.P.; Willemze, R. High Incidence and Clinical Significance of MYC Rearrangements in Primary Cutaneous Diffuse Large B-Cell Lymphoma, Leg Type. Am. J. Surg. Pathol. 2018, 42, 1488–1494. [Google Scholar] [CrossRef]
- Pham-Ledard, A.; Prochazkova-Carlotti, M.; Andrique, L.; Cappellen, D.; Vergier, B.; Martinez, F.; Grange, F.; Petrella, T.; Beylot-Barry, M.; Merlio, J.P. Multiple genetic alterations in primary cutaneous large B-cell lymphoma, leg type support a common lymphomagenesis with activated B-cell-like diffuse large B-cell lymphoma. Mod. Pathol. 2014, 27, 402–411. [Google Scholar] [CrossRef]
- Menguy, S.; Frison, E.; Prochazkova-Carlotti, M.; Dalle, S.; Dereure, O.; Boulinguez, S.; Dalac, S.; Machet, L.; Ram-Wolff, C.; Verneuil, L.; et al. Double-hit or dual expression of MYC and BCL2 in primary cutaneous large B-cell lymphomas. Mod. Pathol. 2018, 31, 1332–1342. [Google Scholar] [CrossRef]
- Lucioni, M.; Pescia, C.; Bonometti, A.; Fraticelli, S.; Moltrasio, C.; Ramponi, A.; Riboni, R.; Roccio, S.; Ferrario, G.; Arcaini, L.; et al. Double expressor and double/triple hit status among primary cutaneous diffuse large B-cell lymphoma: A comparison between leg type and not otherwise specified subtypes. Hum. Pathol. 2021, 111, 1–9. [Google Scholar] [CrossRef]
- Elsayad, K.; Guenova, E.; Assaf, C.; Nicolay, J.P.; Trautinger, F.; Stadler, R.; Waldstein, C.; Boterberg, T.; Meijnders, P.; Kirova, Y.; et al. Radiotherapy in cutaneous lymphomas: Recommendations from the EORTC cutaneous lymphoma tumour group. Eur. J. Cancer 2024, 212, 115064. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network. Clinical Practice Guidelines in Oncology. B-Cell Lymphomas (Version 2.25). Available online: https://www.nccn.org/professionals/physician_gls/pdf/b-cell.pdf (accessed on 18 February 2025).
- Zoli, S.; Pellegrini, C.; Casadei, B.; Broccoli, A.; Argnani, L.; Nanni, L.; Stefoni, V.; Zinzani, P.L. Prolonged Complete Response with Lenalidomide in a Relapsed Diffuse Large B-Cell Lymphoma, Leg-Type: A Case Report. Chemotherapy 2024, 69, 23–26. [Google Scholar] [CrossRef]
- Deng, A.L.; Kim, Y.R.; Lichtenstein, E.A.; O’Connor, O.A.; Deng, C. Combination of ibrutinib and chemotherapy produced a durable remission in multiply relapsed diffuse large B-cell lymphoma leg type with mutant MYD88 and wildtype CD79. Haematologica 2017, 102, e275–e277. [Google Scholar] [CrossRef]
- Gupta, E.; Accurso, J.; Sluzevich, J.; Menke, D.M.; Tun, H.W. Excellent Outcome of Immunomodulation or Bruton’s Tyrosine Kinase Inhibition in Highly Refractory Primary Cutaneous Diffuse Large B-Cell Lymphoma, Leg Type. Rare Tumors 2015, 7, 6067. [Google Scholar] [CrossRef]
- Oka, S.; Ono, K.; Nohgawa, M. Effective treatment with polatuzumab vedotin of relapsed and refractory primary cutaneous diffuse large B-cell lymphoma leg type. Ann. Hematol. 2022, 101, 2353–2354. [Google Scholar] [CrossRef] [PubMed]
- Bandali, T. Treatment of primary cutaneous diffuse large B-cell lymphoma, leg-type (PCDLBCL-LT) in the modern era, Abstract. In Proceedings of the 5th World Congress of Cutaneous Lymphomas, Pasadena, CA, USA, 11–13 April 2024. [Google Scholar]
- Beylot-Barry, M.; Mermin, D.; Maillard, A.; Bouabdallah, R.; Bonnet, N.; Duval-Modeste, A.B.; Mortier, L.; Ingen-Housz-Oro, S.; Ram-Wolff, C.; Barete, S.; et al. A Single-Arm Phase II Trial of Lenalidomide in Relapsing or Refractory Primary Cutaneous Large B-Cell Lymphoma, Leg Type. J. Investig. Dermatol. 2018, 138, 1982–1989. [Google Scholar] [CrossRef] [PubMed]
- Westin, J.; Sehn, L.H. CAR T cells as a second-line therapy for large B-cell lymphoma: A paradigm shift? Blood 2022, 139, 2737–2746, Erratum in Blood 2023, 141, 683. https://doi.org/10.1182/blood.2022018793. [Google Scholar] [CrossRef] [PubMed]
- Locke, F.L.; Miklos, D.B.; Jacobson, C.A.; Perales, M.A.; Kersten, M.J.; Oluwole, O.O.; Ghobadi, A.; Rapoport, A.P.; McGuirk, J.; Pagel, J.M.; et al. All ZUMA-7 Investigators and Contributing Kite Members. Axicabtagene Ciloleucel as Second-Line Therapy for Large B-Cell Lymphoma. N. Engl. J. Med. 2022, 386, 640–654. [Google Scholar] [CrossRef]
- Bishop, M.R.; Dickinson, M.; Purtill, D.; Barba, P.; Santoro, A.; Hamad, N.; Kato, K.; Sureda, A.; Greil, R.; Thieblemont, C.; et al. Second-Line Tisagenlecleucel or Standard Care in Aggressive B-Cell Lymphoma. N. Engl. J. Med. 2022, 386, 629–639. [Google Scholar] [CrossRef]
- Datta, D.; Pandey, R.R.; Kumar, R.; Sharma, R.; Vedant, D. F-18 FDG PET/CT in staging and response assessment of primary cutaneous diffuse large B-cell lymphoma (leg type). Eur. J. Hybrid Imaging 2023, 7, 15. [Google Scholar] [CrossRef]
- Schrader, A.M.R.; van Engeland, J.; Willemze, R.; Vermaat, J.S.P.; Ottevanger, R.; Kersten, J.M.; Zoutman, W.H.; Jansen, P.M.; van Eijk, R.; van Egmond, D.; et al. Detection of Circulating Tumor DNA for Disease Monitoring in Patients with Primary Cutaneous Diffuse Large B-Cell Lymphoma, Leg Type. J. Investig. Dermatol. 2024, 145, 440–444. [Google Scholar] [CrossRef]
- Winkler, M.; Albrecht, J.D.; Sauer, C.; Kordaß, T.; Guenova, E.; Livingstone, E.; Wobser, M.; Mitteldorf, C.; Géraud, C.; Nicolay, J.P. Spontaneous regression of primary cutaneous diffuse large B-cell lymphoma, leg type: A case series and review of the literature. J. Dermatol. 2024, 51, 1233–1239. [Google Scholar] [CrossRef]
- Shi, Y.; Han, Y.; Yang, J.; Liu, P.; He, X.; Zhang, C.; Zhou, S.; Zhou, L.; Qin, Y.; Song, Y.; et al. Clinical features and outcomes of diffuse large B-cell lymphoma based on nodal or extranodal primary sites of origin: Analysis of 1,085 WHO classified cases in a single institution in China. Chin. J. Cancer Res. 2019, 31, 152–161. [Google Scholar] [CrossRef]
- Choi, W.W.; Weisenburger, D.D.; Greiner, T.C.; Piris, M.A.; Banham, A.H.; Delabie, J.; Braziel, R.M.; Geng, H.; Iqbal, J.; Lenz, G.; et al. A new immunostain algorithm classifies diffuse large B-cell lymphoma into molecular subtypes with high accuracy. Clin. Cancer Res. 2009, 15, 5494–5502. [Google Scholar] [CrossRef]
- Ducharme, O.; Beylot-Barry, M.; Pham-Ledard, A.; Bohers, E.; Viailly, P.J.; Bandres, T.; Faur, N.; Frison, E.; Vergier, B.; Jardin, F.; et al. Mutations of the B-Cell Receptor Pathway Confer Chemoresistance in Primary Cutaneous Diffuse Large B-Cell Lymphoma Leg Type. J. Investig. Dermatol. 2019, 139, 2334–2342.e8. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.A.; Louissaint AJr Wenzel, A.; Yang, J.; Martinez-Escala, M.E.; Moy, A.P.; Morgan, E.A.; Paxton, C.N.; Hong, B.; Andersen, E.F.; Guitart, J.; et al. Genomic Analyses Identify Recurrent Alterations in Immune Evasion Genes in Diffuse Large B-Cell Lymphoma, Leg Type. J. Investig. Dermatol. 2018, 138, 2365–2376. [Google Scholar] [CrossRef] [PubMed]
- Egbuniwe, I.U.; Karagiannis, S.N.; Nestle, F.O.; Lacy, K.E. Revisiting the role of B cells in skin immune surveillance. Trends Immunol. 2015, 36, 102–111. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbati, Z.R.; Charli-Joseph, Y. Unveiling Primary Cutaneous B-Cell Lymphomas: New Insights into Diagnosis and Treatment Strategies. Cancers 2025, 17, 1202. https://doi.org/10.3390/cancers17071202
Barbati ZR, Charli-Joseph Y. Unveiling Primary Cutaneous B-Cell Lymphomas: New Insights into Diagnosis and Treatment Strategies. Cancers. 2025; 17(7):1202. https://doi.org/10.3390/cancers17071202
Chicago/Turabian StyleBarbati, Zachary R., and Yann Charli-Joseph. 2025. "Unveiling Primary Cutaneous B-Cell Lymphomas: New Insights into Diagnosis and Treatment Strategies" Cancers 17, no. 7: 1202. https://doi.org/10.3390/cancers17071202
APA StyleBarbati, Z. R., & Charli-Joseph, Y. (2025). Unveiling Primary Cutaneous B-Cell Lymphomas: New Insights into Diagnosis and Treatment Strategies. Cancers, 17(7), 1202. https://doi.org/10.3390/cancers17071202