
Targeting the TLK1-MK5 Axis Suppresses Prostate Cancer Metastasis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Results and Discussion
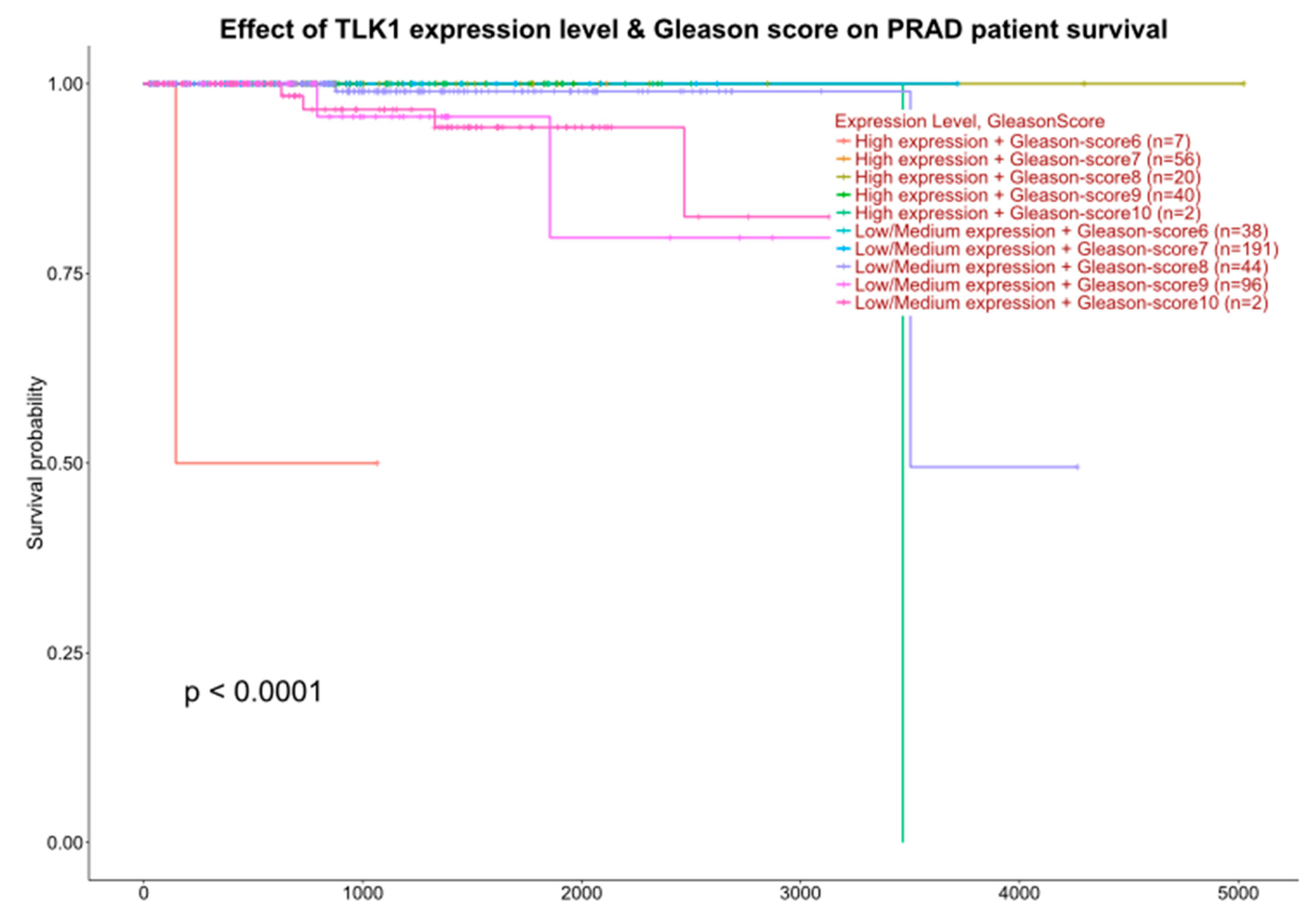
2.1. Interrogation of Expression Reports
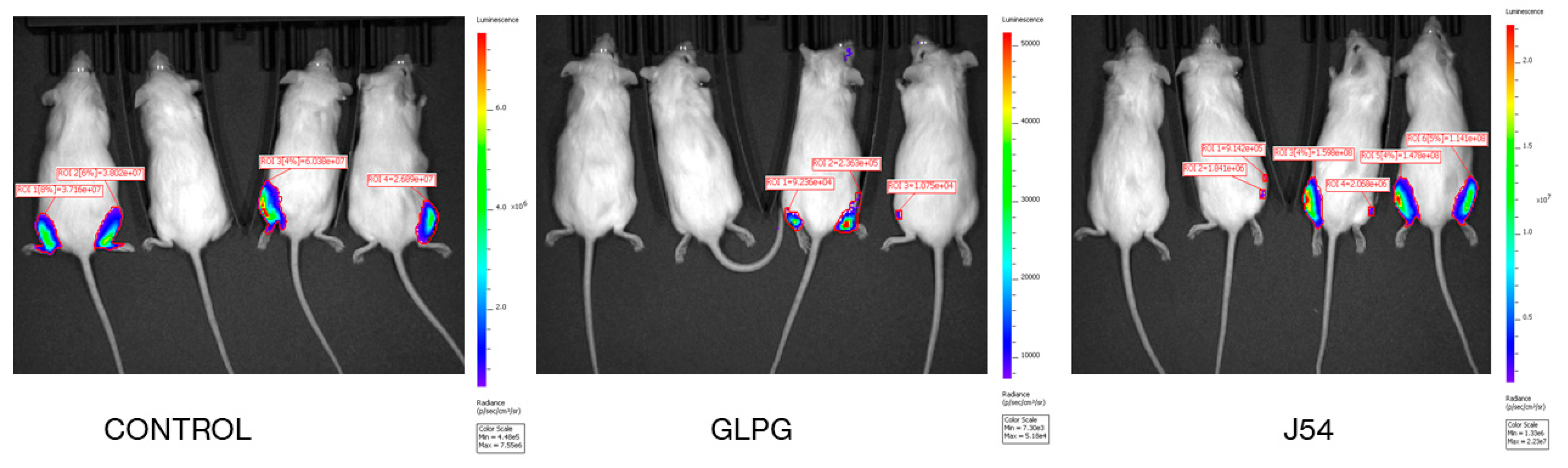
2.2. Reduction of Metastatic Spread with Inhibitors of TLK or MK5
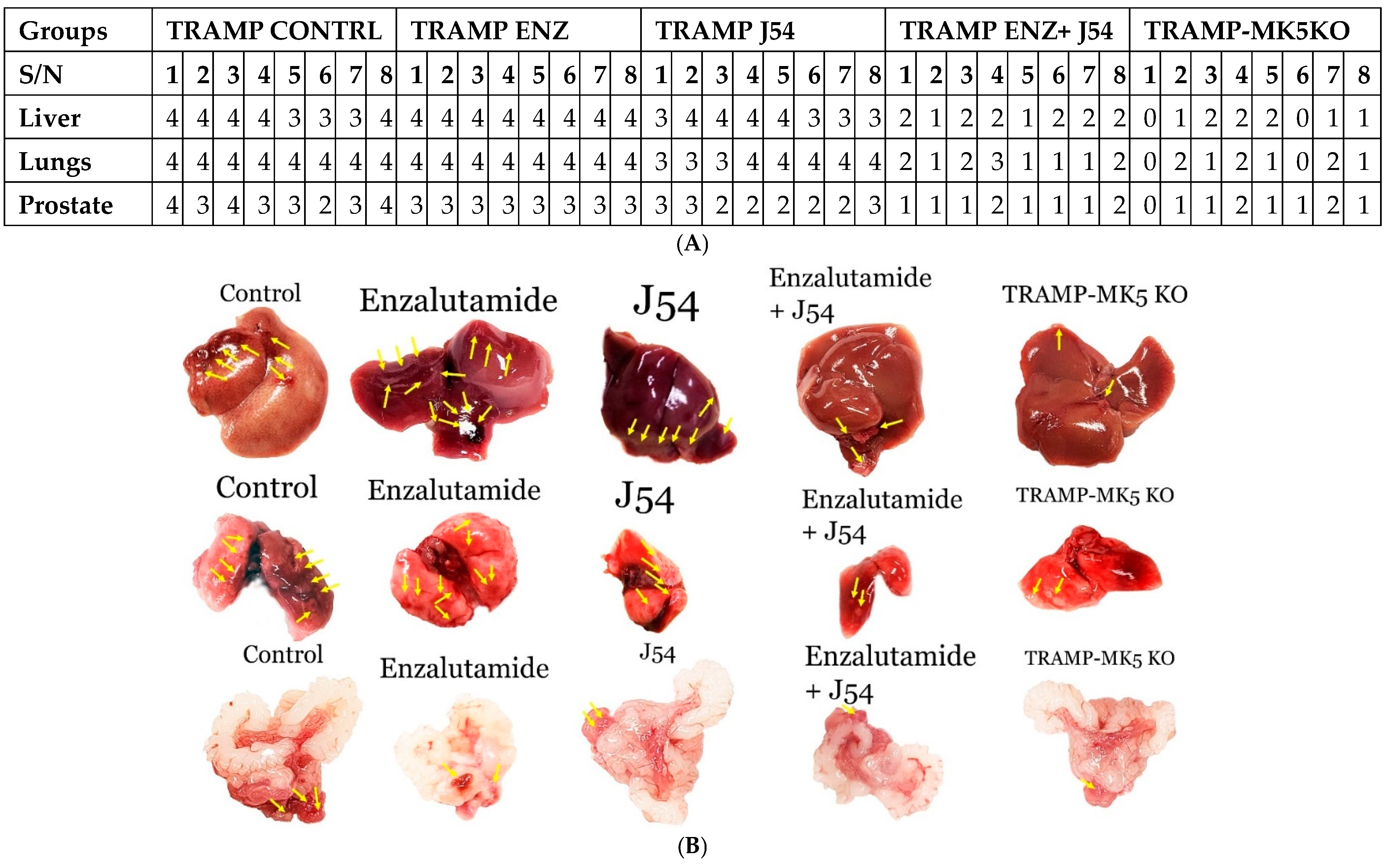
2.3. Role of TLK1>MK5 in Cancer Dissemination in a Mouse Model of Spontaneous PCa Progression
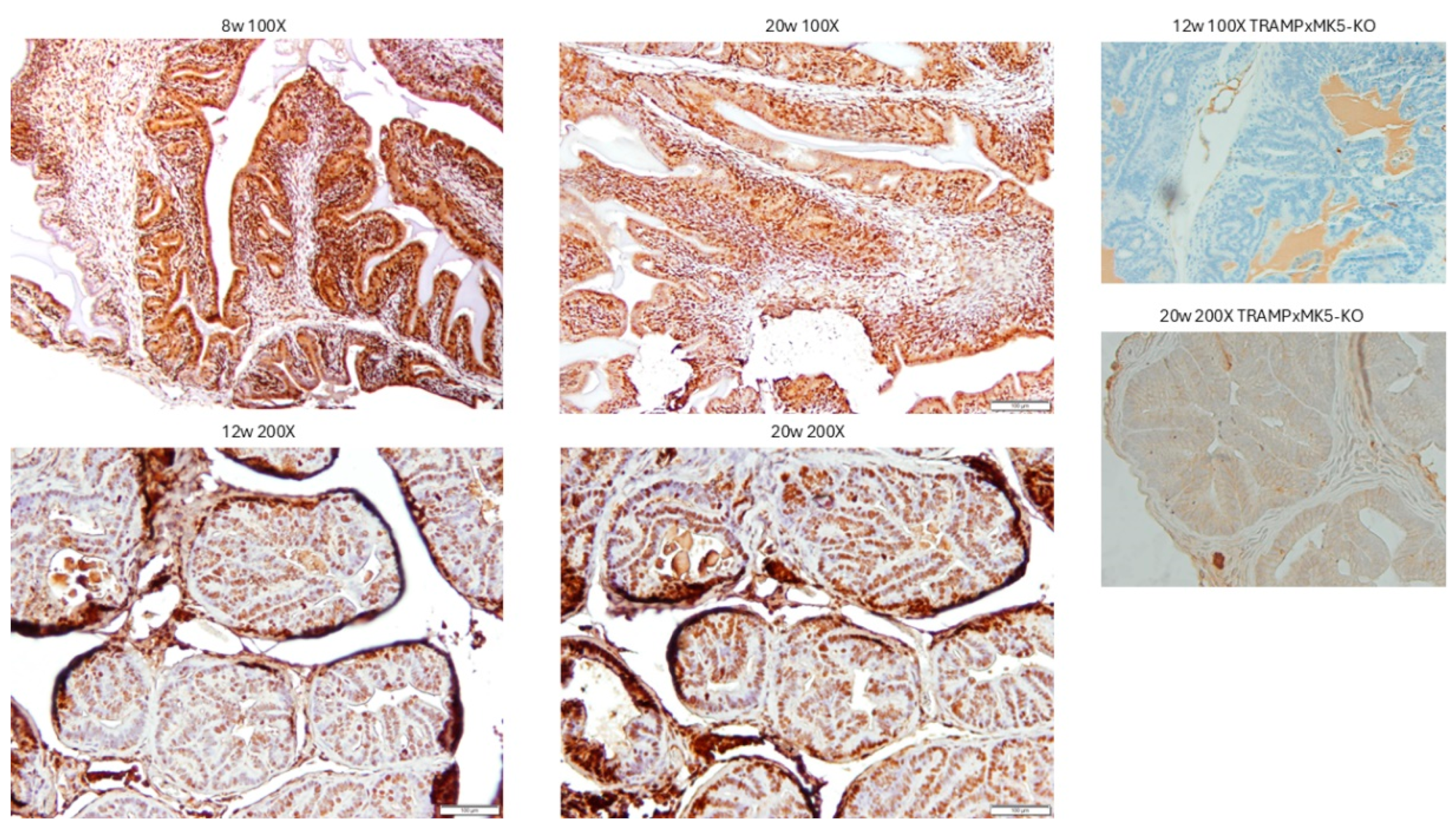
2.4. Evaluation of PCa Progression in TRAMP via IHC for pMK5 Ab
2.5. Evaluation of PCa TMA with pMK5 Ab
2.6. Direct Bone Engraftment
3. Discussion
3.1. Choice of GLPG vs. J54 in Clinical Translation
3.2. Specificity vs. General Toxicity Considerations
4. Conclusions
5. Materials and Methods
5.1. Cell Viability Assay and Treatments
5.2. Animal Studies
5.3. Immunohistochemistry and Fluorescence Imaging
5.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Halabi, S.; Kelly, W.K.; Ma, H.; Zhou, H.; Solomon, N.C.; Fizazi, K.; Tangen, C.M.; Rosenthal, M.; Petrylak, D.P.; Hussain, M.; et al. Meta-Analysis Evaluating the Impact of Site of Metastasis on Overall Survival in Men With Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2016, 34, 1652–1659. [Google Scholar] [CrossRef] [PubMed]
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef] [PubMed]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef]
- Hood, J.D.; Cheresh, D.A. Role of integrins in cell invasion and migration. Nat. Rev. Cancer 2002, 2, 91–100. [Google Scholar] [CrossRef]
- van Zijl, F.; Krupitza, G.; Mikulits, W. Initial steps of metastasis: Cell invasion and endothelial transmigration. Mutat. Res. 2011, 728, 23–34. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, J.; Ren, Y.; Liu, S.; Ba, Y.; Zuo, A.; Luo, P.; Cheng, Q.; Xu, H.; Han, X. Multi-stage mechanisms of tumor metastasis and therapeutic strategies. Signal Transduct. Target. Ther. 2024, 9, 270. [Google Scholar] [CrossRef]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef]
- Bhoir, S.; De Benedetti, A. Targeting Prostate Cancer, the ‘Tousled Way’. Int. J. Mol. Sci. 2023, 24, 11100. [Google Scholar] [CrossRef]
- Ghosh, I.; De Benedetti, A. Untousling the Role of Tousled-like Kinase 1 in DNA Damage Repair. Int. J. Mol. Sci. 2023, 24, 13369. [Google Scholar] [CrossRef]
- Segura-Bayona, S.; Stracker, T.H. The Tousled-like kinases regulate genome and epigenome stability: Implications in development and disease. Cell. Mol. Life Sci. 2019, 76, 3827–3841. [Google Scholar] [CrossRef]
- Kim, J.A.; Tan, Y.; Wang, X.; Cao, X.; Veeraraghavan, J.; Liang, Y.; Edwards, D.P.; Huang, S.; Pan, X.; Li, K.; et al. Comprehensive functional analysis of the tousled-like kinase 2 frequently amplified in aggressive luminal breast cancers. Nat. Commun. 2016, 7, 12991. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Yao, Z.; Zhao, N.; Zhang, C. TLK2 enhances aggressive phenotypes of glioblastoma cells through the activation of SRC signaling pathway. Cancer Biol. Ther. 2019, 20, 101–108. [Google Scholar] [CrossRef]
- Xiang, W.; Zhang, D.; Montell, D.J. Tousled-like kinase regulates cytokine-mediated communication between cooperating cell types during collective border cell migration. Mol. Biol. Cell. 2016, 27, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Bhoir, S.; Chikhale, R.V.; Hussain, J.; Dwyer, D.; Bryce, R.A.; Kirubakaran, S.; De Benedetti, A. Generation of Phenothiazine with Potent Anti-TLK1 Activity for Prostate Cancer Therapy. iScience 2020, 23, 101474. [Google Scholar] [CrossRef] [PubMed]
- Khalil, M.I.; De Benedetti, A. The TLK1-MK5 Axis Regulates Motility, Invasion, and Metastasis of Prostate Cancer Cells. Cancers 2022, 14, 5728. [Google Scholar] [CrossRef]
- Gelman, I.H. How the TRAMP Model Revolutionized the Study of Prostate Cancer Progression. Cancer Res. 2016, 76, 6137–6139. [Google Scholar] [CrossRef]
- Gingrich, J.R.; Barrios, R.J.; Kattan, M.W.; Nahm, H.S.; Finegold, M.J.; Greenberg, N.M. Androgen-independent prostate cancer progression in the TRAMP model. Cancer Res. 1997, 57, 4687–4691. [Google Scholar]
- Gingrich, J.R.; Barrios, R.J.; Morton, R.A.; Boyce, B.F.; DeMayo, F.J.; Finegold, M.J.; Angelopoulou, R.; Rosen, J.M.; Greenberg, N.M. Metastatic prostate cancer in a transgenic mouse. Cancer Res. 1996, 56, 4096–4102. [Google Scholar]
- Cerasuolo, M.; Maccarinelli, F.; Coltrini, D.; Mahmoud, A.M.; Marolda, V.; Ghedini, G.C.; Rezzola, S.; Giacomini, A.; Triggiani, L.; Kostrzewa, M.; et al. Modeling Acquired Resistance to the Second-Generation Androgen Receptor Antagonist Enzalutamide in the TRAMP Model of Prostate Cancer. Cancer Res. 2020, 80, 1564–1577. [Google Scholar] [CrossRef]
- Khalil, M.I.; De Benedetti, A. Tousled-like kinase 1: A novel factor with multifaceted role in mCRPC progression and development of therapy resistance. Cancer Drug Resist. 2022, 5, 93–101. [Google Scholar] [CrossRef]
- Khalil, M.I.; Singh, V.; King, J.; De Benedetti, A. TLK1-mediated MK5-S354 phosphorylation drives prostate cancer cell motility and may signify distinct pathologies. Mol. Oncol. 2022, 16, 2537–2557. [Google Scholar] [CrossRef] [PubMed]
- Ronkina, N.; Gaestel, M. MAPK-Activated Protein Kinases: Servant or Partner? Annu. Rev. Biochem. 2022, 91, 505–540. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, S.; Laass, K.; Kant, S.; Shi, Y.; Visel, A.; Gruber, A.D.; Kotlyarov, A.; Gaestel, M. Scaffolding by ERK3 regulates MK5 in development. EMBO J. 2004, 23, 4770–4779. [Google Scholar] [CrossRef] [PubMed]
- Kant, S.; Schumacher, S.; Singh, M.K.; Kispert, A.; Kotlyarov, A.; Gaestel, M. Characterization of the atypical MAPK ERK4 and its activation of the MAPK-activated protein kinase MK5. J. Biol. Chem. 2006, 281, 35511–35519. [Google Scholar] [CrossRef]
- Seternes, O.M.; Mikalsen, T.; Johansen, B.; Michaelsen, E.; Armstrong, C.G.; Morrice, N.A.; Turgeon, B.; Meloche, S.; Moens, U.; Keyse, S.M. Activation of MK5/PRAK by the atypical MAP kinase ERK3 defines a novel signal transduction pathway. Embo. J. 2004, 23, 4780–4791. [Google Scholar] [CrossRef]
- Åberg, E.; Perander, M.; Johansen, B.; Julien, C.; Meloche, S.; Keyse, S.M.; Seternes, O.-M. Regulation of MAPK-activated Protein Kinase 5 Activity and Subcellular Localization by the Atypical MAPK ERK4/MAPK4*. J. Biol. Chem. 2006, 281, 35499–35510. [Google Scholar] [CrossRef]
- Boudghene-Stambouli, F.; Soulez, M.; Ronkina, N.; Dörrie, A.; Kotlyarov, A.; Seternes, O.M.; Gaestel, M.; Meloche, S. On the Therapeutic Potential of ERK4 in Triple-Negative Breast Cancer. Cancers 2022, 15, 25. [Google Scholar] [CrossRef]
- Coulombe, P.; Meloche, S. Atypical mitogen-activated protein kinases: Structure, regulation and functions. Biochim. Biophys. Acta 2007, 1773, 1376–1387. [Google Scholar] [CrossRef]
- Sun, P.; Yoshizuka, N.; New, L.; Moser, B.A.; Li, Y.; Liao, R.; Xie, C.; Chen, J.; Deng, Q.; Yamout, M.; et al. PRAK is essential for ras-induced senescence and tumor suppression. Cell 2007, 128, 295–308. [Google Scholar] [CrossRef]
- Yoshizuka, N.; Chen, R.M.; Xu, Z.; Liao, R.; Hong, L.; Hu, W.Y.; Yu, G.; Han, J.; Chen, L.; Sun, P. A novel function of p38-regulated/activated kinase in endothelial cell migration and tumor angiogenesis. Mol. Cell. Biol. 2012, 32, 606–618. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, W.; Wu, H.; Zhou, Y.; Qin, X.; Wang, Y.; Wu, J.; Sun, X.Y.; Yang, Y.; Xu, H.; et al. The essential role of PRAK in tumor metastasis and its therapeutic potential. Nat. Commun. 2021, 12, 1736. [Google Scholar] [CrossRef] [PubMed]
- Ronkina, N.; Johansen, C.; Bohlmann, L.; Lafera, J.; Menon, M.B.; Tiedje, C.; Laaß, K.; Turk, B.E.; Iversen, L.; Kotlyarov, A.; et al. Comparative Analysis of Two Gene-Targeting Approaches Challenges the Tumor-Suppressive Role of the Protein Kinase MK5/PRAK. PLoS ONE 2015, 10, e0136138. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Kim, M.H.; Hong, H.; Cho, H.; Park, S.; Kim, S.K.; Kim, J. MK5 Regulates YAP Stability and Is a Molecular Target in YAP-Driven Cancers. Cancer Res. 2019, 79, 6139–6152. [Google Scholar] [CrossRef]
- Khalil, M.I.; Ghosh, I.; Singh, V.; Chen, J.; Zhu, H.; De Benedetti, A. NEK1 Phosphorylation of YAP Promotes Its Stabilization and Transcriptional Output. Cancers 2020, 12, 3666. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Wang, Y.H.; Wu, X.N.; Wu, S.Q.; Lu, B.J.; Dong, M.Q.; Zhang, H.; Sun, P.; Lin, S.C.; Guan, K.L.; et al. Inactivation of Rheb by PRAK-mediated phosphorylation is essential for energy-depletion-induced suppression of mTORC1. Nat. Cell. Biol. 2011, 13, 263–272. [Google Scholar] [CrossRef]
- Zhou, H.; Huang, S. Role of mTOR signaling in tumor cell motility, invasion and metastasis. Curr. Protein Pept. Sci. 2011, 12, 30–42. [Google Scholar] [CrossRef]
- Jiang, J.; Jia, P.; Zhao, Z.; Shen, B. Key regulators in prostate cancer identified by co-expression module analysis. BMC Genom. 2014, 15, 1015. [Google Scholar] [CrossRef]
- Singh, V.; Connelly, Z.M.; Shen, X.; De Benedetti, A. Identification of the proteome complement of humanTLK1 reveals it binds and phosphorylates NEK1 regulating its activity. Cell Cycle 2017, 16, 915–926. [Google Scholar] [CrossRef]
- Akhmetkaliyev, A.; Alibrahim, N.; Shafiee, D.; Tulchinsky, E. EMT/MET plasticity in cancer and Go-or-Grow decisions in quiescence: The two sides of the same coin? Mol. Cancer 2023, 22, 90. [Google Scholar] [CrossRef]
- Bornes, L.; Belthier, G.; van Rheenen, J. Epithelial-to-Mesenchymal Transition in the Light of Plasticity and Hybrid E/M States. J. Clin. Med. 2021, 10, 2403. [Google Scholar] [CrossRef]
- Elkin, M.; Vlodavsky, I. Tail vein assay of cancer metastasis. Curr. Protoc. Cell Biol. 2001, 12, 19.12.11–19.12.17. [Google Scholar]
- Namour, F.; Vanhoutte, F.P.; Beetens, J.; Blockhuys, S.; De Weer, M.; Wigerinck, P. Pharmacokinetics, safety, and tolerability of GLPG0259, a mitogen-activated protein kinase-activated protein kinase 5 (MAPKAPK5) inhibitor, given as single and multiple doses to healthy male subjects. Drugs RD 2012, 12, 141–163. [Google Scholar]
- Westhovens, R.; Keyser, F.D.; Rekalov, D.; Nasonov, E.L.; Beetens, J.; Van der Aa, A.; Wigerinck, P.; Namour, F.; Vanhoutte, F.; Durez, P. Oral administration of GLPG0259, an inhibitor of MAPKAPK5, a new target for the treatment of rheumatoid arthritis: A phase II, randomised, double-blind, placebo-controlled, multicentre trial. Ann. Rheum. Dis. 2013, 72, 741–744. [Google Scholar] [CrossRef] [PubMed]
- Olatunde, D.; De Benedetti, A. TLK1>Nek1 Axis Promotes Nuclear Retention and Activation of YAP with Implications for Castration-Resistant Prostate Cancer. Cancers 2024, 16, 2918. [Google Scholar] [CrossRef]
- Stewart, S.B.; Cheville, J.C.; Sebo, T.J.; Frank, I.; Boorjian, S.A.; Thompson, R.H.; Gettman, M.T.; Tollefson, M.K.; Umbriet, E.C.; Psutka, S.P.; et al. Gleason grading after neoadjuvant hormonal therapy retains prognostic value for systemic progression following radical prostatectomy. Prostate Cancer Prostatic Dis. 2014, 17, 332–337. [Google Scholar] [CrossRef]
- Snow, A.; Chen, D.; Lang, J.E. The current status of the clinical utility of liquid biopsies in cancer. Expert Rev. Mol. Diagn. 2019, 19, 1031–1041. [Google Scholar] [CrossRef]
- Zeng, Z.; Yi, Z.; Xu, B. The biological and technical challenges facing utilizing circulating tumor DNA in non-metastatic breast cancer patients. Cancer Lett. 2025, 616, 217574. [Google Scholar] [CrossRef]
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef]
- Cifone, M.A. In vitro growth characteristics associated with benign and metastatic variants of tumor cells. Cancer Metastasis Rev. 1982, 1, 335–347. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Hartung, N.; Mollard, S.; Barbolosi, D.; Benabdallah, A.; Chapuisat, G.; Henry, G.; Giacometti, S.; Iliadis, A.; Ciccolini, J.; Faivre, C.; et al. Mathematical Modeling of Tumor Growth and Metastatic Spreading: Validation in Tumor-Bearing Mice. Cancer Res. 2014, 74, 6397–6407. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.H.; Kim, C.G.; Kim, S.K.; Shin, S.J.; Choe, E.A.; Park, S.H.; Shin, E.C.; Kim, J. YAP-Induced PD-L1 Expression Drives Immune Evasion in BRAFi-Resistant Melanoma. Cancer Immunol Res 2018, 6, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R. Neoantigens and genome instability: Impact on immunogenomic phenotypes and immunotherapy response. Genome Med. 2019, 11, 71. [Google Scholar] [CrossRef] [PubMed]
- Sahadevan, P.; Allen, B.G. MK5: A novel regulator of cardiac fibroblast function? IUBMB Life 2017, 69, 785–794. [Google Scholar] [CrossRef]
- Perander, M.; Keyse, S.M.; Seternes, O.M. New insights into the activation, interaction partners and possible functions of MK5/PRAK. Front. Biosci. 2016, 21, 374–384. [Google Scholar] [CrossRef]
- Moens, U.; Kostenko, S. Structure and function of MK5/PRAK: The loner among the mitogen-activated protein kinase-activated protein kinases. Biol. Chem. 2013, 394, 1115–1132. [Google Scholar] [CrossRef]
- Kress, T.R.; Cannell, I.G.; Brenkman, A.B.; Samans, B.; Gaestel, M.; Roepman, P.; Burgering, B.M.; Bushell, M.; Rosenwald, A.; Eilers, M. The MK5/PRAK Kinase and Myc Form a Negative Feedback Loop that Is Disrupted during Colorectal Tumorigenesis. Mol. Cell 2011, 41, 445–457. [Google Scholar] [CrossRef]
- Dwyer, S.F.; Gelman, I.H. Cross-Phosphorylation and Interaction between Src/FAK and MAPKAP5/PRAK in Early Focal Adhesions Controls Cell Motility. J. Cancer Biol. Res. 2014, 2, 1045. [Google Scholar]
- Rajah, A.; Boudreau, C.G.; Ilie, A.; Wee, T.-L.; Tang, K.; Borisov, A.Z.; Orlowski, J.; Brown, C.M. Paxillin S273 Phosphorylation Regulates Adhesion Dynamics and Cell Migration through a Common Protein Complex with PAK1 and βPIX. Sci. Rep. 2019, 9, 11430. [Google Scholar] [CrossRef]
- Rauch, J.; Volinsky, N.; Romano, D.; Kolch, W. The secret life of kinases: Functions beyond catalysis. Cell Commun. Signal. 2011, 9, 23. [Google Scholar] [CrossRef]
- Stefano, C.D.; Grazioli, P.; Anna Fontanella, R.; Cesaris, P.D.; D’Amore, A.; Regno, M.; Starace, D.; Padula, F.; Elena Fiori, M.; Canipari, R.; et al. Stem-like and highly invasive prostate cancer cells expressing CD44v8-10 marker originate from CD44-negative cells. Oncotarget 2018, 9, 30905–30918. [Google Scholar] [PubMed]
- Mulholland, D.J.; Tran, L.M.; Li, Y.; Cai, H.; Morim, A.; Wang, S.; Plaisier, S.; Garraway, I.P.; Huang, J.; Graeber, T.G.; et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell. 2011, 19, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Li, S.; Wang, F.; Fan, C.; Wang, J. Identification of key pathways and genes in PTEN mutation prostate cancer by bioinformatics analysis. BMC Med. Genet. 2019, 20, 191. [Google Scholar] [CrossRef]
- Phin, S.; Moore, M.W.; Cotter, P.D. Genomic Rearrangements of PTEN in Prostate Cancer. Front. Oncol. 2013, 3, 240. [Google Scholar] [CrossRef]
- Dong, J.-T.; Li, C.-L.; Sipe, T.W.; Frierson, H.F., Jr. Mutations of PTEN/MMAC1 in Primary Prostate Cancers from Chinese Patients1. Clin. Cancer Res. 2001, 7, 304–308. [Google Scholar]
- Jamaspishvili, T.; Berman, D.M.; Ross, A.E.; Scher, H.I.; De Marzo, A.M.; Squire, J.A.; Lotan, T.L. Clinical implications of PTEN loss in prostate cancer. Nat. Rev. Urol. 2018, 15, 222–234. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Egawa, M.; Fukuda, M.; Takashima, H.; Misaki, T.; Kinuya, K.; Terahata, S. The sentinel node concept in prostate cancer: Present reality and future prospects. Indian J. Urol. 2008, 24, 451–456. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olatunde, D.; Franco, O.C.; Gaestel, M.; De Benedetti, A. Targeting the TLK1-MK5 Axis Suppresses Prostate Cancer Metastasis. Cancers 2025, 17, 1187. https://doi.org/10.3390/cancers17071187
Olatunde D, Franco OC, Gaestel M, De Benedetti A. Targeting the TLK1-MK5 Axis Suppresses Prostate Cancer Metastasis. Cancers. 2025; 17(7):1187. https://doi.org/10.3390/cancers17071187
Chicago/Turabian StyleOlatunde, Damilola, Omar Coronel Franco, Matthias Gaestel, and Arrigo De Benedetti. 2025. "Targeting the TLK1-MK5 Axis Suppresses Prostate Cancer Metastasis" Cancers 17, no. 7: 1187. https://doi.org/10.3390/cancers17071187
APA StyleOlatunde, D., Franco, O. C., Gaestel, M., & De Benedetti, A. (2025). Targeting the TLK1-MK5 Axis Suppresses Prostate Cancer Metastasis. Cancers, 17(7), 1187. https://doi.org/10.3390/cancers17071187