

PPM1D Mutation as a Distinct Feature of Myeloid Neoplasms in B-Cell Non-Hodgkin Lymphoma Patients: A Retrospective Analysis
 , , ,
, , ,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient
2.2. Study Design
2.3. Samples Collection and Processing for NGS Analysis
2.4. Next-Generation Sequencing Analysis
2.5. Statistical Analysis
3. Results
3.1. Characteristics of Patients with B-Cell Non-Hodgkin Lymphoma
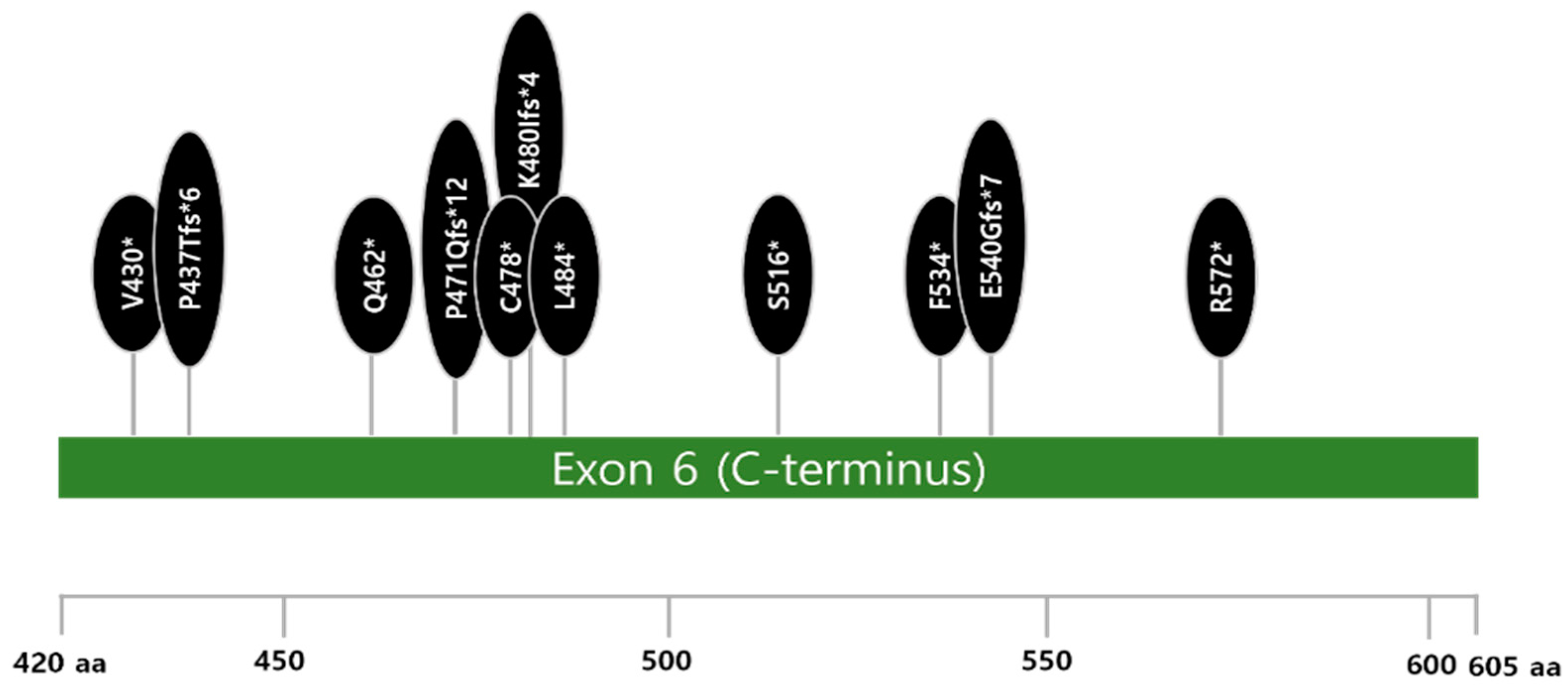
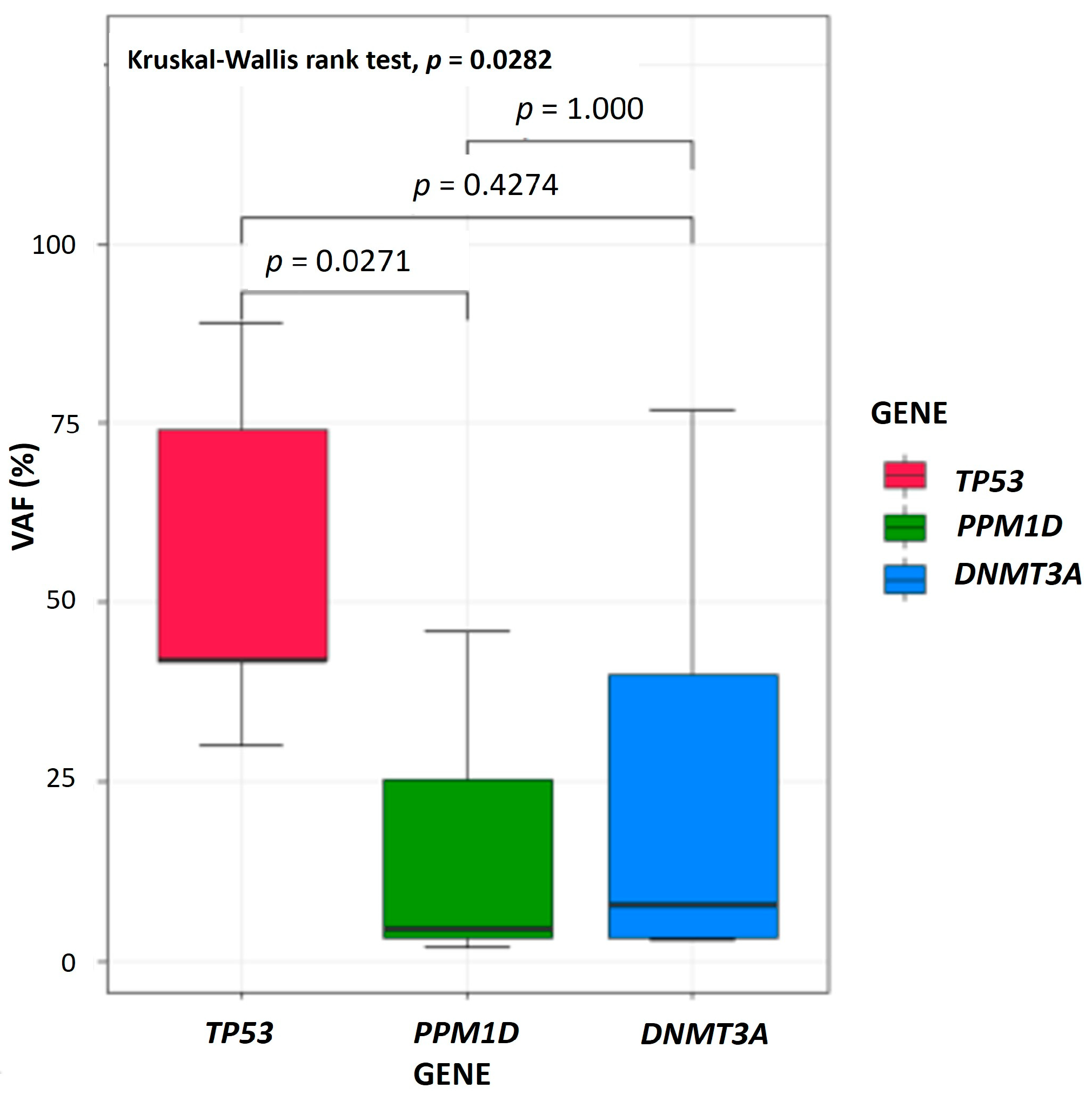
3.2. Genetic Features of Myeloid Neoplasms After B-Cell Non-Hodgkin Lymphoma
3.3. Comparison of Clinicopathologic and Genetic Features of Myeloid Neoplasms After B-Cell Non-Hodgkin Lymphoma and Solid Cancer
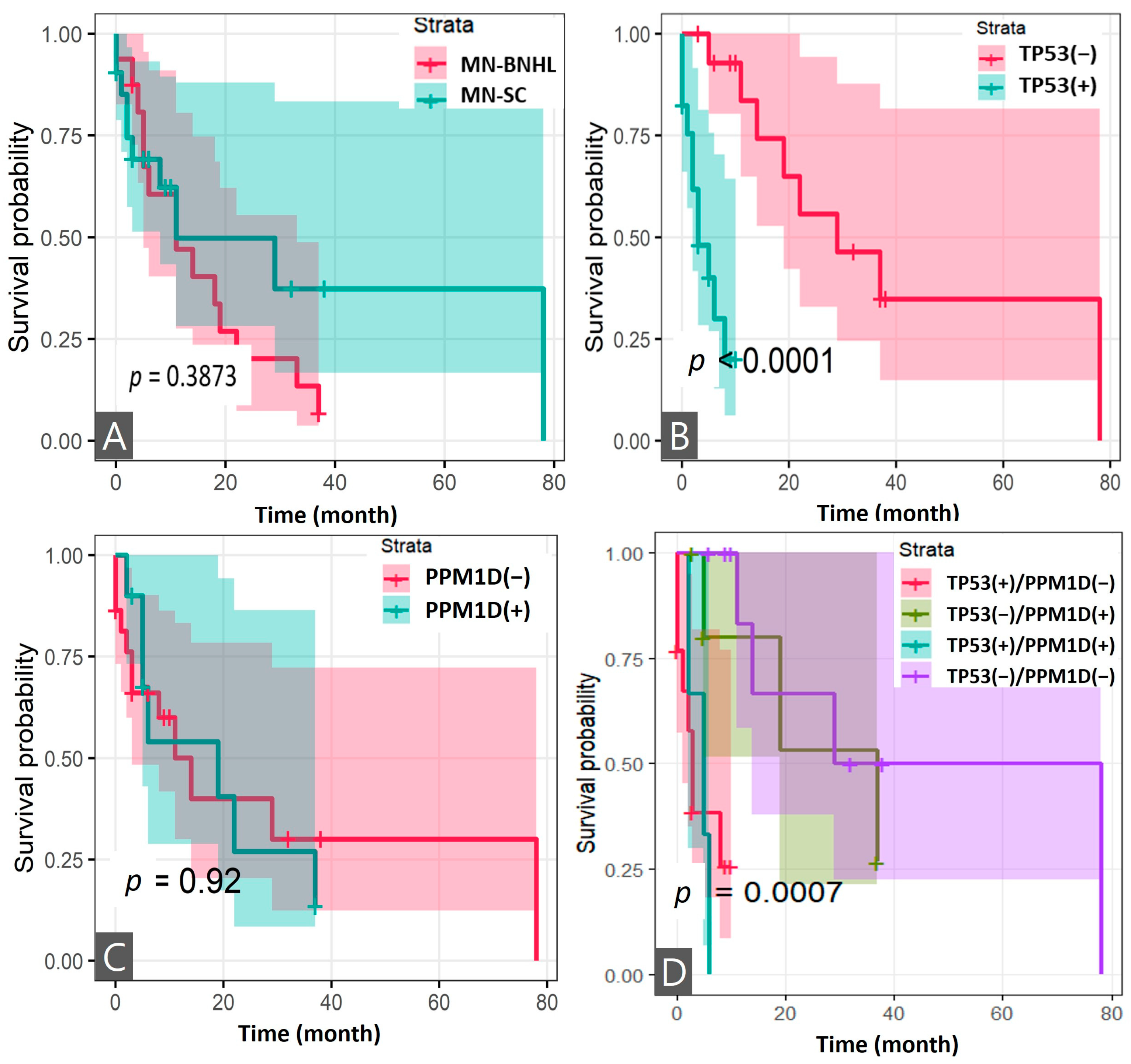
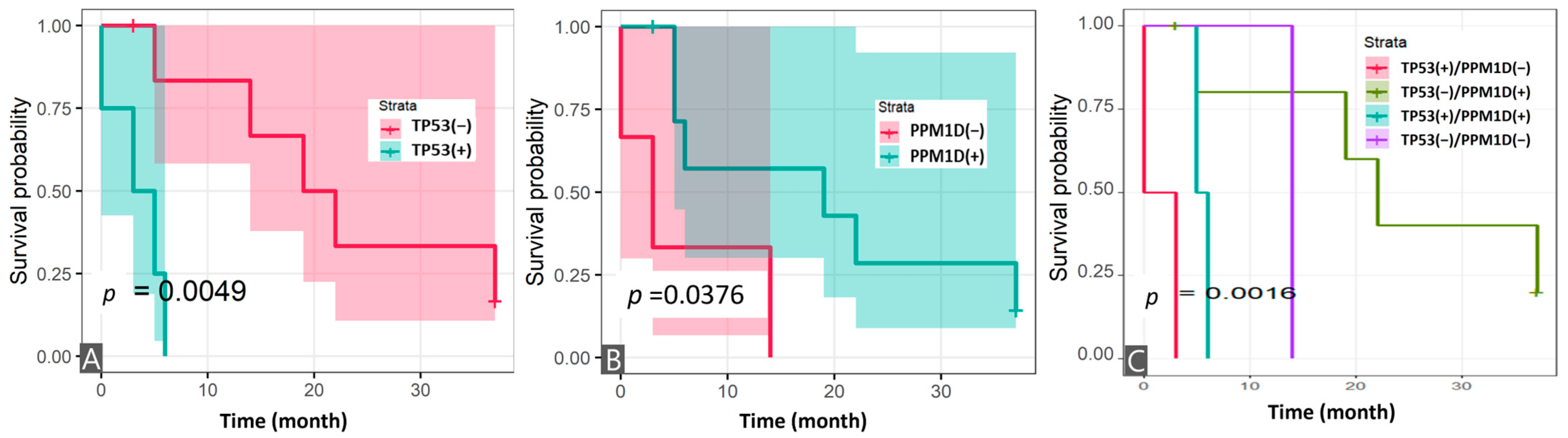
3.4. Survival Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef]
- Usui, Y.; Ito, H.; Katanoda, K.; Matsuda, T.; Maeda, Y.; Matsuo, K. Trends in non-Hodgkin lymphoma mortality rate in Japan and the United States: A population-based study. Cancer Sci. 2023, 114, 4073–4080. [Google Scholar] [CrossRef]
- Kim, J.S.; Liu, Y.; Ha, K.H.; Qiu, H.; Rothwell, L.A.; Kim, H.C. Increasing Incidence of B-Cell Non-Hodgkin Lymphoma and Occurrence of Second Primary Malignancies in South Korea: 10-Year Follow-up Using the Korean National Health Information Database. Cancer Res. Treat. 2020, 52, 1262–1272. [Google Scholar] [CrossRef]
- Larouche, J.F.; Berger, F.; Chassagne-Clement, C.; Ffrench, M.; Callet-Bauchu, E.; Sebban, C.; Ghesquieres, H.; Broussais-Guillaumot, F.; Salles, G.; Coiffier, B. Lymphoma recurrence 5 years or later following diffuse large B-cell lymphoma: Clinical characteristics and outcome. J. Clin. Oncol. 2010, 28, 2094–2100. [Google Scholar] [CrossRef] [PubMed]
- McNerney, M.E.; Godley, L.A.; Le Beau, M.M. Therapy-related myeloid neoplasms: When genetics and environment collide. Nat. Rev. Cancer 2017, 17, 513–527. [Google Scholar] [CrossRef] [PubMed]
- Bari, A.; Marcheselli, L.; Marcheselli, R.; Liardo, E.V.; Pozzi, S.; Ferri, P.; Sacchi, S. Therapy-related myeloid neoplasm in non-hodgkin lymphoma survivors. Mediterr. J. Hematol. Infect. Dis. 2011, 3, e2011065. [Google Scholar] [CrossRef]
- Joelsson, J.; Wasterlid, T.; Rosenquist, R.; Jakobsen, L.H.; El-Galaly, T.C.; Smedby, K.E.; Eloranta, S. Incidence and time trends of second primary malignancies after non-Hodgkin lymphoma: A Swedish population-based study. Blood Adv. 2022, 6, 2657–2666. [Google Scholar] [CrossRef] [PubMed]
- Lupo-Stanghellini, M.T.; Piemontese, S.; Assanelli, A.; Serpenti, F.; Mastaglio, S.; Clerici, D.; Giglio, F.; Greco, R.; Lorentino, F.; Pavesi, F.; et al. Second Solid Cancers After Hematopoietic Stem Cell Transplantation: Active Surveillance During Long-term Follow-up. Hemasphere 2021, 5, e654. [Google Scholar] [CrossRef]
- Singhal, D.; Kutyna, M.M.; Hahn, C.N.; Shah, M.V.; Hiwase, D.K. Therapy-Related Myeloid Neoplasms: Complex Interactions among Cytotoxic Therapies, Genetic Factors, and Aberrant Microenvironment. Blood Cancer Discov. 2024, 5, 400–416. [Google Scholar] [CrossRef]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Kuzmanovic, T.; Patel, B.J.; Sanikommu, S.R.; Nagata, Y.; Awada, H.; Kerr, C.M.; Przychodzen, B.P.; Jha, B.K.; Hiwase, D.; Singhal, D.; et al. Genomics of therapy-related myeloid neoplasms. Haematologica 2020, 105, e98–e101. [Google Scholar] [CrossRef] [PubMed]
- Radivoyevitch, T.; Dean, R.M.; Shaw, B.E.; Brazauskas, R.; Tecca, H.R.; Molenaar, R.J.; Battiwalla, M.; Savani, B.N.; Flowers, M.E.D.; Cooke, K.R.; et al. Risk of acute myeloid leukemia and myelodysplastic syndrome after autotransplants for lymphomas and plasma cell myeloma. Leuk. Res. 2018, 74, 130–136. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, P.; Estey, E.; Glassman, A.; Romaguera, J.; Samaniego, F.; Ayala, A.; Hayes, K.; Maddox, A.M.; Preti, H.A.; Hagemeister, F.B. Myelodysplasia and acute myeloid leukemia following therapy for indolent lymphoma with fludarabine, mitoxantrone, and dexamethasone (FND) plus rituximab and interferon alpha. Blood 2005, 105, 4573–4575. [Google Scholar] [CrossRef] [PubMed]
- Farina, M.; Bernardi, S.; Malagola, M.; Re, A.; Galli, E.; Riva, M.; Cutini, I.; Leoni, A.; Martino, M.; Ferrari, S.; et al. Real-world collection of secondary myeloid neoplasms after CD19 CAR-T cell therapy: First report of the ClonHema study. Bone Marrow Transplant. 2025. [Google Scholar] [CrossRef]
- Hou, H.A.; Liu, C.Y.; Kuo, Y.Y.; Chou, W.C.; Tsai, C.H.; Lin, C.C.; Lin, L.I.; Tseng, M.H.; Chiang, Y.C.; Liu, M.C.; et al. Splicing factor mutations predict poor prognosis in patients with de novo acute myeloid leukemia. Oncotarget 2016, 7, 9084–9101. [Google Scholar] [CrossRef]
- Rombaut, D.; Sandmann, S.; Tekath, T.; Crouch, S.; de Graaf, A.O.; Smith, A.; Painter, D.; Kosmider, O.; Tobiasson, M.; Lennartsson, A.; et al. Somatic mutations and DNA methylation identify a subgroup of poor prognosis within lower-risk myelodysplastic syndromes. Hemasphere 2025, 9, e70073. [Google Scholar] [CrossRef] [PubMed]
- Joshi, U.; Bhattarai, A.; Gaire, S.; Budhathoki, P.; Agrawal, V.; Subedi, R.; Poudyal, B.S.; Dhakal, P.; Sham, R.; Bhatt, V.R. Therapy-related Acute Myeloid Leukemia in Non-Hodgkin Lymphoma Survivors: Risk, Survival Outcomes and Prognostic Factor Analysis. Hematol. Oncol. Stem Cell Ther. 2023, 17, 79–87. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef]
- Ye, K.; Schulz, M.H.; Long, Q.; Apweiler, R.; Ning, Z. Pindel: A pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 2009, 25, 2865–2871. [Google Scholar] [CrossRef] [PubMed]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef]
- Kwon, J.H.; Kim, H.; Lee, J.K.; Hong, Y.J.; Kang, H.J.; Jang, Y.J. Incidence and Characteristics of Multiple Primary Cancers: A 20-Year Retrospective Study of a Single Cancer Center in Korea. Cancers 2024, 16, 2346. [Google Scholar] [CrossRef]
- Deng, W.; Li, J.; Dorrah, K.; Jimenez-Tapia, D.; Arriaga, B.; Hao, Q.; Cao, W.; Gao, Z.; Vadgama, J.; Wu, Y. The role of PPM1D in cancer and advances in studies of its inhibitors. Biomed. Pharmacother. 2020, 125, 109956. [Google Scholar] [CrossRef]
- Seipel, K.; Frey, M.; Nilius, H.; Akhoundova, D.; Banz, Y.; Bacher, U.; Pabst, T. Low-Frequency PPM1D Gene Mutations Affect Treatment Response to CD19-Targeted CAR T-Cell Therapy in Large B-Cell Lymphoma. Curr. Oncol. 2023, 30, 10463–10476. [Google Scholar] [CrossRef]
- Hsu, J.I.; Dayaram, T.; Tovy, A.; De Braekeleer, E.; Jeong, M.; Wang, F.; Zhang, J.; Heffernan, T.P.; Gera, S.; Kovacs, J.J.; et al. PPM1D Mutations Drive Clonal Hematopoiesis in Response to Cytotoxic Chemotherapy. Cell Stem Cell 2018, 23, 700–713.e6. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.N.; Miller, C.A.; Jotte, M.R.M.; Bagegni, N.; Baty, J.D.; Schmidt, A.P.; Cashen, A.F.; Duncavage, E.J.; Helton, N.M.; Fiala, M.; et al. Cellular stressors contribute to the expansion of hematopoietic clones of varying leukemic potential. Nat. Commun. 2018, 9, 455. [Google Scholar] [CrossRef] [PubMed]
- Pich, O.; Reyes-Salazar, I.; Gonzalez-Perez, A.; Lopez-Bigas, N. Discovering the drivers of clonal hematopoiesis. Nat. Commun. 2022, 13, 4267. [Google Scholar] [CrossRef]
- Kahn, J.D.; Miller, P.G.; Silver, A.J.; Sellar, R.S.; Bhatt, S.; Gibson, C.; McConkey, M.; Adams, D.; Mar, B.; Mertins, P.; et al. PPM1D-truncating mutations confer resistance to chemotherapy and sensitivity to PPM1D inhibition in hematopoietic cells. Blood 2018, 132, 1095–1105. [Google Scholar] [CrossRef]
- Zhang, L.; Hsu, J.I.; Goodell, M.A. PPM1D in Solid and Hematologic Malignancies: Friend and Foe? Mol. Cancer Res. 2022, 20, 1365–1378. [Google Scholar] [CrossRef]
- Burocziova, M.; Danek, P.; Oravetzova, A.; Chalupova, Z.; Alberich-Jorda, M.; Macurek, L. Ppm1d truncating mutations promote the development of genotoxic stress-induced AML. Leukemia 2023, 37, 2209–2220. [Google Scholar] [CrossRef] [PubMed]
- Spurr, L.; Li, M.; Alomran, N.; Zhang, Q.; Restrepo, P.; Movassagh, M.; Trenkov, C.; Tunnessen, N.; Apanasovich, T.; Crandall, K.A.; et al. Systematic pan-cancer analysis of somatic allele frequency. Sci. Rep. 2018, 8, 7735. [Google Scholar] [CrossRef] [PubMed]
- Bomben, R.; Rossi, F.M.; Vit, F.; Bittolo, T.; D’Agaro, T.; Zucchetto, A.; Tissino, E.; Pozzo, F.; Vendramini, E.; Degan, M.; et al. TP53 Mutations with Low Variant Allele Frequency Predict Short Survival in Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2021, 27, 5566–5575. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Derkach, A.; Lewis, N.; Zhu, M.; Zhang, Y.; Arcila, M.; Salles, G.; Dogan, A.; Xiao, W. Clonal hematopoiesis in diffuse large B-cell lymphoma: Clinical impact and genetic relatedness to lymphoma and therapy-related myeloid neoplasm. Haematologica 2023, 108, 917–922. [Google Scholar] [CrossRef]
- Tuval, A.; Shlush, L.I. Evolutionary trajectory of leukemic clones and its clinical implications. Haematologica 2019, 104, 872–880. [Google Scholar] [CrossRef]
- Gibson, C.J.; Lindsley, R.C.; Tchekmedyian, V.; Mar, B.G.; Shi, J.; Jaiswal, S.; Bosworth, A.; Francisco, L.; He, J.; Bansal, A.; et al. Clonal Hematopoiesis Associated with Adverse Outcomes After Autologous Stem-Cell Transplantation for Lymphoma. J. Clin. Oncol. 2017, 35, 1598–1605. [Google Scholar] [CrossRef]
- Schwartz, J.R.; Ma, J.; Kamens, J.; Westover, T.; Walsh, M.P.; Brady, S.W.; Robert Michael, J.; Chen, X.; Montefiori, L.; Song, G.; et al. The acquisition of molecular drivers in pediatric therapy-related myeloid neoplasms. Nat. Commun. 2021, 12, 985. [Google Scholar] [CrossRef]
- Nishiyama, T.; Ishikawa, Y.; Kawashima, N.; Akashi, A.; Adachi, Y.; Hattori, H.; Ushijima, Y.; Kiyoi, H. Mutation analysis of therapy-related myeloid neoplasms. Cancer Genet. 2018, 222–223, 38–45. [Google Scholar] [CrossRef]
- Andrysik, Z.; Sullivan, K.D.; Kieft, J.S.; Espinosa, J.M. PPM1D suppresses p53-dependent transactivation and cell death by inhibiting the Integrated Stress Response. Nat. Commun. 2022, 13, 7400. [Google Scholar] [CrossRef]
- Milosevic, J.; Fransson, S.; Gulyas, M.; Olsen, T.K.; Gallo-Oller, G.; Treis, D.; Elfman, L.H.M.; Wilhelm, M.; Martinsson, T.; Baryawno, N.; et al. High Expression of PPM1D Induces Tumors Phenotypically Similar to TP53 Loss-of-Function Mutations in Mice. Cancers 2021, 13, 5493. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | BNHL (n = 16) |
|---|---|
| Age of diagnosis of BNHL, years | 56.7 ± 11.5 |
| Male gender, n (%) | 10 (63%) |
| Stage * | |
| I | 1 (6%) |
| II | 2 (13%) |
| III | 3 (19%) |
| IV | 10 (63%) |
| Pathologic diagnosis | |
| DLBL | 7 (44%) |
| FL | 4 (25%) |
| MZBL CLL MCL | 2 (13%) 1 (6%) 2 (13%) |
| Cytotoxic therapy exposure | 16 (100%) |
| Chemotherapy only | 2 (13%) |
| Chemotherapy + RIT | 1 (6%) |
| Chemotherapy + RIT + RT | 1 (6%) |
| Immunochemotherapy (rituximab) combination | 12 (75%) |
| Immunochemotherapy | 2 (13%) |
| Immunochemotherapy + RIT | 7 (44%) |
| Immunochemotherapy + RIT + RT | 3 (19%) |
| Treatment response (IWC + PET criteria) | |
| CR | 10 (63%) |
| PR | 2 (13%) |
| SD | 2 (13%) |
| PD | 1 (6%) |
| NA | 1 (6%) |
| Relapse | |
| NA | 1 (8%) |
| CR or PR | 3 (25%) |
| Relapse | 8 (67%) |
| Characteristics | MN-BNHL (n = 16) | MN-SC † (n = 21) | p-Value |
|---|---|---|---|
| Age, years at diagnosis of MN | 66.5 (57.5, 72) | 63 (57, 67) | 0.3735 |
| Exposure to cytotoxic therapies | |||
| Chemotherapy | 16 (100%) | 19 (90%) | 0.4955 |
| Rituximab | 12 (75%) | 0 (0%) | <0.001 |
| Radiotherapy | 4 (25%) | 12 (57%) | 0.1052 |
| RIT/RAI | 12 (75%) | 1 (5%) | <0.001 |
| Interval period, year (range) | 7.0 ± 4.0 (2.4 to 17.3) | 11.7 ± 8.3 (1.3 to 28) | 0.0305 |
| Survival state | |||
| Death | 14 (88%) | 10 (48%) | 0.03 |
| Survival period after MN, month (range) | 11 (4.8–19.8) | 6 (2–10) | 0.1574 |
| Follow-up period, year (range) | 9.0 ± 4.3 (2.8–18.6) | 12.4 ± 8.4 (1.3–28.4) | 0.1183 |
| BM study | |||
| Blast count (%) | 6.9 (4.5, 16.9) | 14.6 (6, 46.6) | 0.2696 |
| Dysplasia (>10%) | |||
| Dyserythropoiesis | 10 (63%) | 8 (42%) | 0.388 |
| Dysgranulopoiesis | 2 (13%) | 4 (21%) | 0.6657 |
| Dysmegakaryopoiesis | 6 (43%) | 15 (83%) | 0.0265 |
| BM Fibrosis (≥MF − 1) | 0 (0%) | 10 (59%) | <0.001 |
| Chromosomal aberrations | |||
| Complex karyotype | 11 (69%) | 13 (62%) | 0.9326 |
| –7/del7q | 10 (63%) | 8 (38%) | 0.2545 |
| –5/del5q | 4 (25%) | 9 (45%) | 0.3722 |
| i (17q), –17/add(17p) or del(17p) | 4 (25%) | 1 (5%) | 0.1444 |
| –20/del20q | 1 (6%) | 3 (15%) | 0.6129 |
| Somatic mutations associated with myeloid neoplasms * (3 dominant tier-1-mutated genes) | |||
| PPM1D | 8 (73%) | 2 (10%) | <0.001 |
| TP53 | 4 (36%) | 13 (62%) | 0.2818 |
| DNMT3A | 5 (45%) | 2 (10%) | 0.0318 |
| Treatment/Gene Mutation | Relative Risk (95% CI) | |||
|---|---|---|---|---|
| Chemotherapy | Rituximab (Immunochemotherapy) | RIT/RAI | Radiotherapy | |
| PPM1Dm (+) | 1.07 (0.88, 1.30) | 3.67 (1.08, 12.43) | 3.3 (1.19, 9.16) | 0.51 (0.19, 1.39) |
| TP53m (+) | 1.15 (0.92, 1.44) | 0.88 (0.26, 2.92) | 0.59 (0.20, 1.69) | 1.13 (0.56, 2.29) |
| DNMT3Am (+) | 1.03 (0.83, 1.29) | 2.14 (0.67, 6.83) | 3.57 (1.43, 8.90) | 0.51 (0.15, 1.73) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.; Lee, J.K.; Hong, Y.J.; Kang, H.J.; Byun, B.H.; Lee, S.-S. PPM1D Mutation as a Distinct Feature of Myeloid Neoplasms in B-Cell Non-Hodgkin Lymphoma Patients: A Retrospective Analysis. Cancers 2025, 17, 1185. https://doi.org/10.3390/cancers17071185
Kim H, Lee JK, Hong YJ, Kang HJ, Byun BH, Lee S-S. PPM1D Mutation as a Distinct Feature of Myeloid Neoplasms in B-Cell Non-Hodgkin Lymphoma Patients: A Retrospective Analysis. Cancers. 2025; 17(7):1185. https://doi.org/10.3390/cancers17071185
Chicago/Turabian StyleKim, Heyjin, Jin Kyung Lee, Young Jun Hong, Hye Jin Kang, Byung Hyun Byun, and Seung-Sook Lee. 2025. "PPM1D Mutation as a Distinct Feature of Myeloid Neoplasms in B-Cell Non-Hodgkin Lymphoma Patients: A Retrospective Analysis" Cancers 17, no. 7: 1185. https://doi.org/10.3390/cancers17071185
APA StyleKim, H., Lee, J. K., Hong, Y. J., Kang, H. J., Byun, B. H., & Lee, S.-S. (2025). PPM1D Mutation as a Distinct Feature of Myeloid Neoplasms in B-Cell Non-Hodgkin Lymphoma Patients: A Retrospective Analysis. Cancers, 17(7), 1185. https://doi.org/10.3390/cancers17071185