
β-Catenin Drives the FOXC2-Mediated Epithelial–Mesenchymal Transition and Acquisition of Stem Cell Properties

 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. β-Catenin Positively Correlates and Interacts with FOXC2
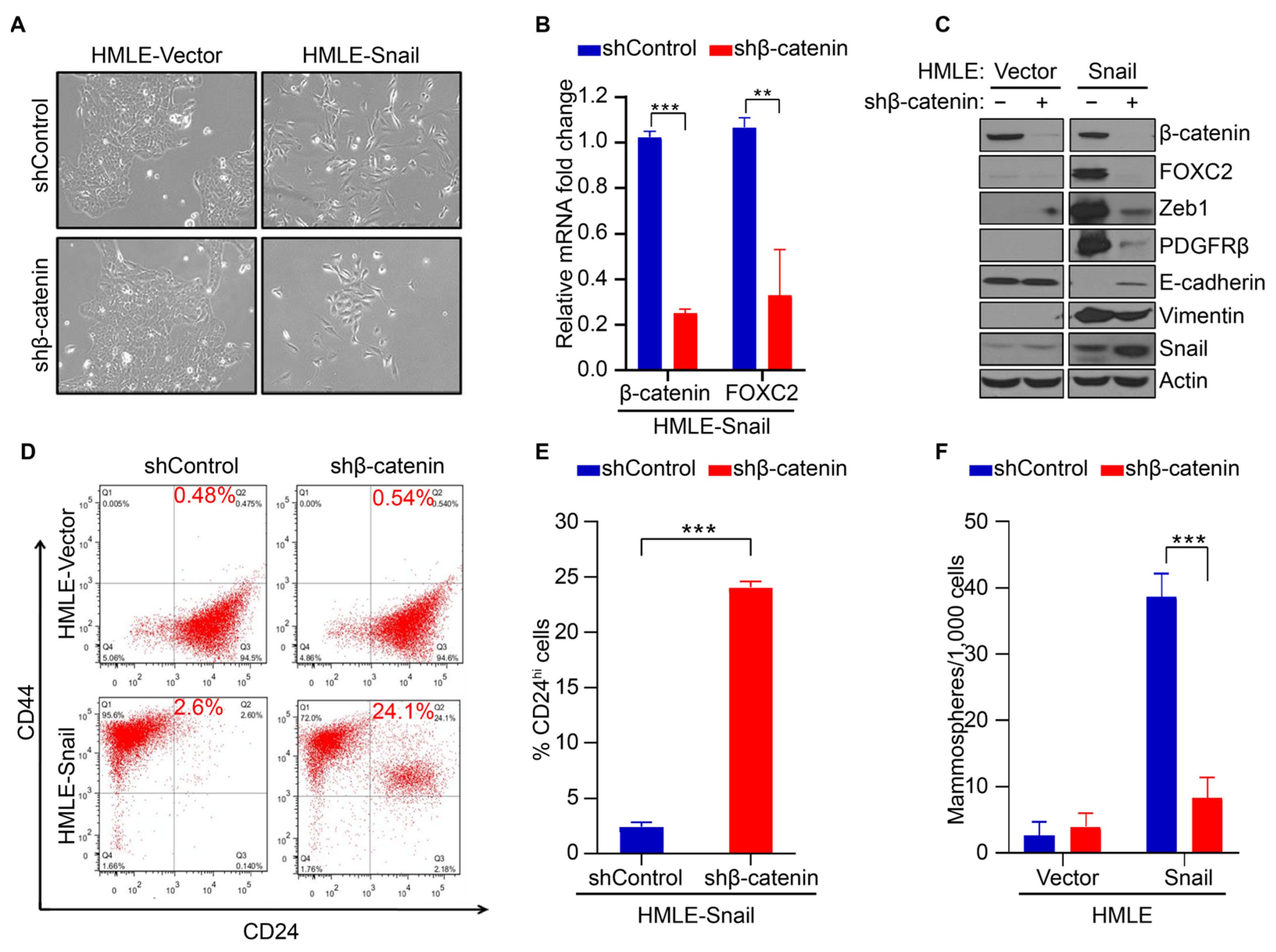
3.2. β-Catenin Mediates FOXC2-Associated EMT and Acquisition of Stem Cell-like Properties
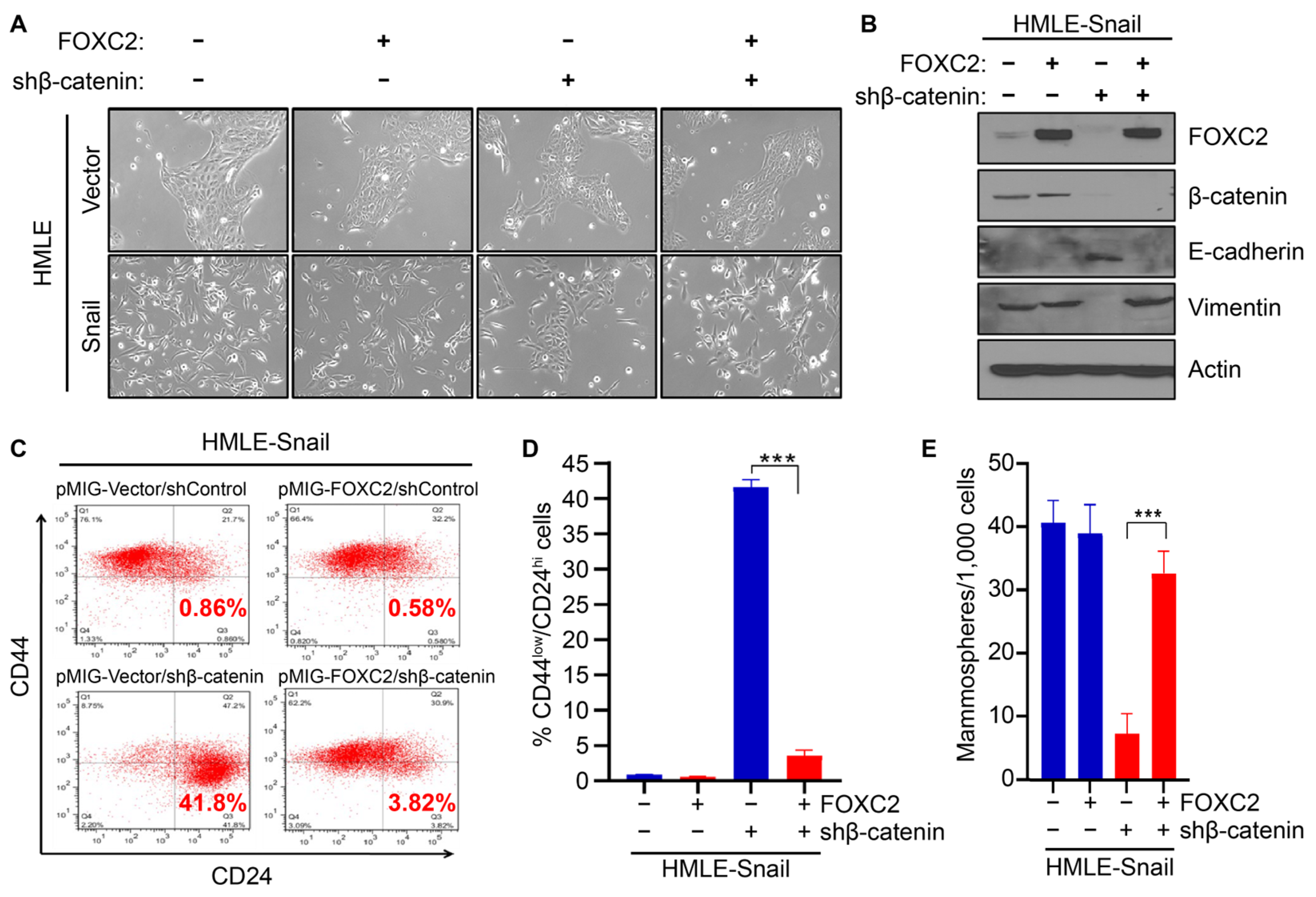
3.3. Expression of FOXC2 in the Absence of β-Catenin Restores EMT and Stem Cell Properties
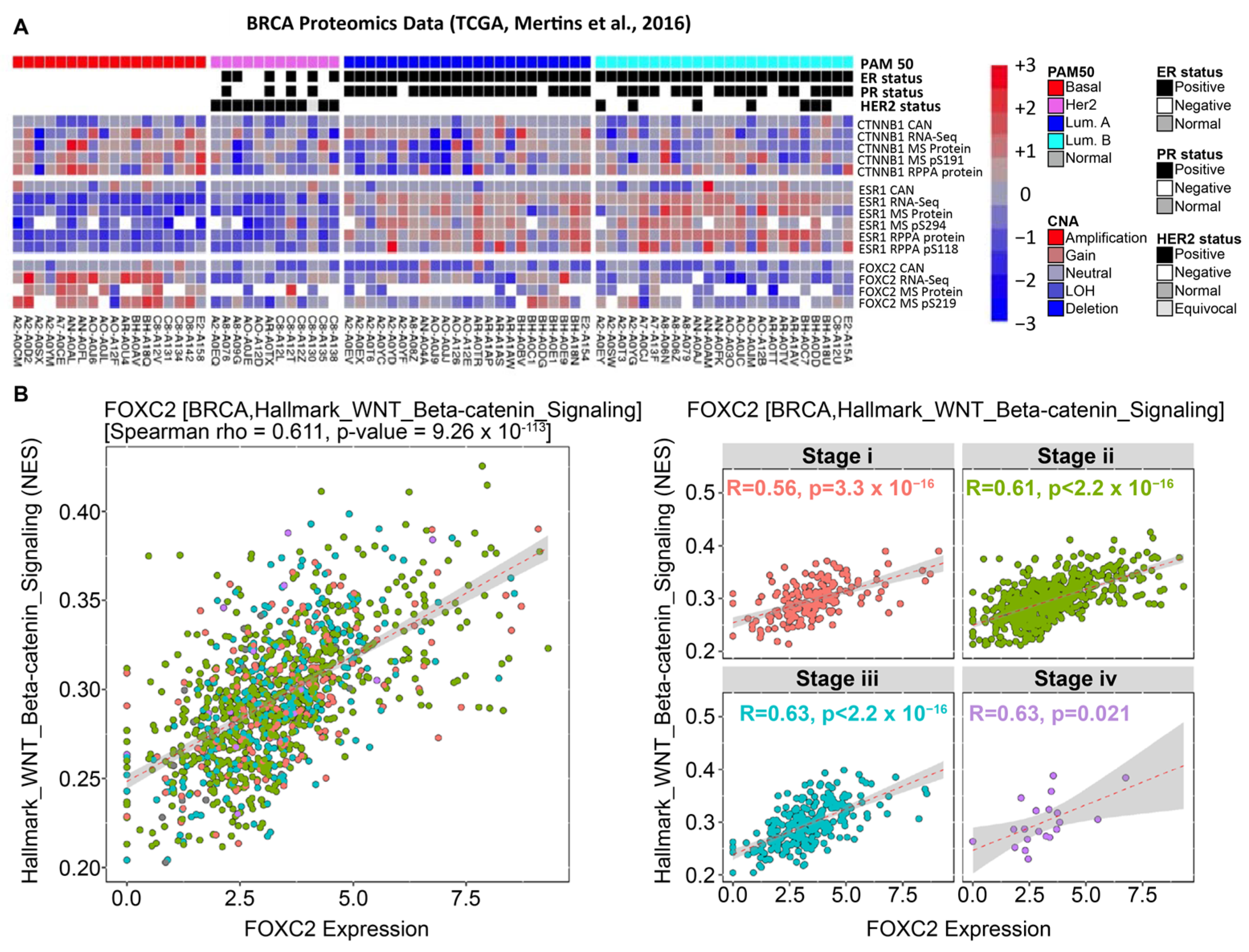
3.4. β-Catenin and FOXC2 Are Directly Correlated in Clinical Datasets of Cancer Patients
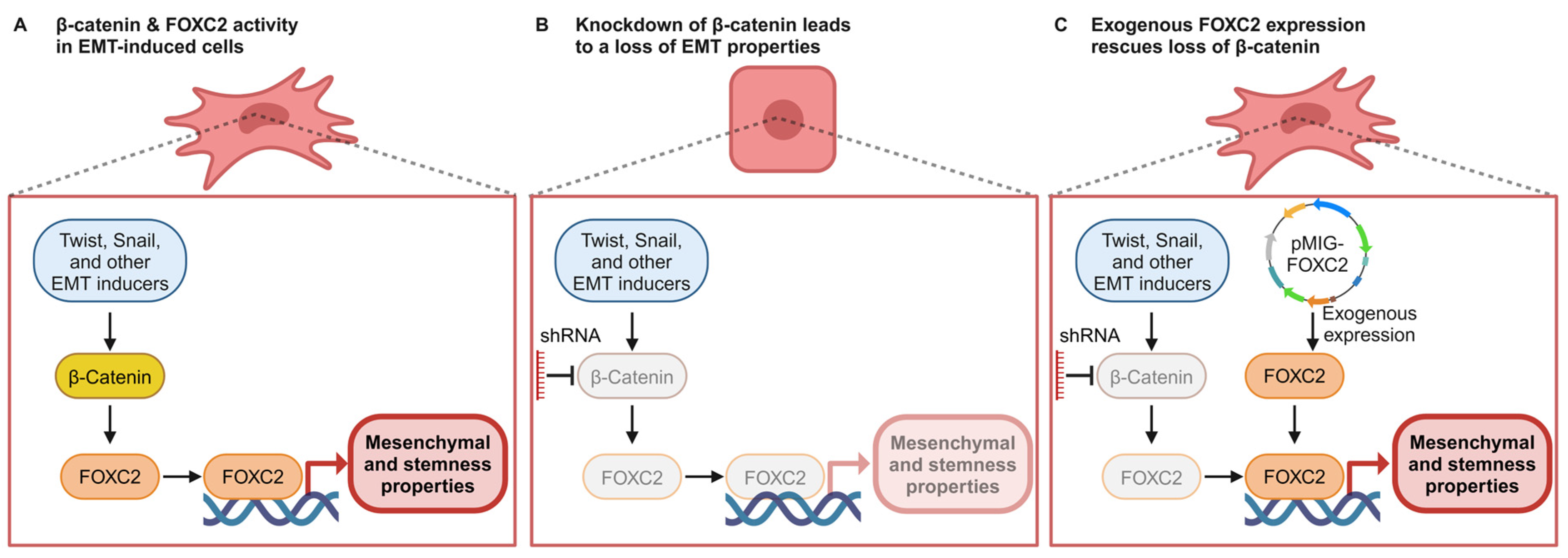
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Neve, R.M.; Chin, K.; Fridlyand, J.; Yeh, J.; Baehner, F.L.; Fevr, T.; Clark, L.; Bayani, N.; Coppe, J.-P.; Tong, F.; et al. A Collection of Breast Cancer Cell Lines for the Study of Functionally Distinct Cancer Subtypes. Cancer Cell 2006, 10, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-Negative Breast Cancer: Challenges and Opportunities of a Heterogeneous Disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of Human Triple-Negative Breast Cancer Subtypes and Preclinical Models for Selection of Targeted Therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.; Eaton, E.N.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Campbell, L.L.; Polyak, K.; et al. Epithelial-Mesenchymal Transition Creates Cells with the Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Den Hollander, P.; Maddela, J.J.; Mani, S.A. Spatial and Temporal Relationship between Epithelial-Mesenchymal Transition (EMT) and Stem Cells in Cancer. Clin. Chem. 2024, 70, 190–205. [Google Scholar] [PubMed]
- Najafi, M.; Farhood, B.; Mortezaee, K.; Kharazinejad, E.; Majidpoor, J.; Ahadi, R. Hypoxia in Solid Tumors: A Key Promoter of Cancer Stem Cell (CSC) Resistance. J. Cancer Res. Clin. Oncol. 2020, 146, 19–31. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and Drug Resistance: The Mechanistic Link and Clinical Implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef]
- Beziaud, L.; Young, C.M.; Alonso, A.M.; Norkin, M.; Minafra, A.R.; Huelsken, J. IFNγ-Induced Stem-like State of Cancer Cells as a Driver of Metastatic Progression Following Immunotherapy. Cell Stem Cell 2023, 30, 818–831.e6. [Google Scholar] [CrossRef]
- Mani, S.A.; Yang, J.; Brooks, M.; Schwaninger, G.; Zhou, A.; Miura, N.; Kutok, J.L.; Hartwell, K.; Richardson, A.L.; Weinberg, R.A. Mesenchyme Forkhead 1 (FOXC2) Plays a Key Role in Metastasis and Is Associated with Aggressive Basal-like Breast Cancers. Proc. Natl. Acad. Sci. USA 2007, 104, 10069–10074. [Google Scholar] [CrossRef]
- Hollier, B.G.; Tinnirello, A.A.; Werden, S.J.; Evans, K.W.; Taube, J.H.; Sarkar, T.R.; Sphyris, N.; Shariati, M.; Kumar, S.V.; Battula, V.L.; et al. FOXC2 Expression Links Epithelial-Mesenchymal Transition and Stem Cell Properties in Breast Cancer. Cancer Res. 2013, 73, 1981–1992. [Google Scholar] [CrossRef]
- Castaneda, M.; den Hollander, P.; Mani, S.A. Forkhead Box Transcription Factors: Double-Edged Swords in Cancer. Cancer Res. 2022, 82, 2057–2065. [Google Scholar] [CrossRef]
- Pietila, M.; Vijay, G.V.; Soundararajan, R.; Yu, X.; Symmans, W.F.; Sphyris, N.; Mani, S.A. FOXC2 Regulates the G2/M Transition of Stem Cell-Rich Breast Cancer Cells and Sensitizes Them to PLK1 Inhibition. Sci. Rep. 2016, 6, 23070. [Google Scholar] [CrossRef] [PubMed]
- Werden, S.J.; Sphyris, N.; Sarkar, T.R.; Paranjape, A.N.; Labaff, A.M.; Taube, J.H.; Hollier, B.G.; Ramirez-Penã, E.Q.; Soundararajan, R.; Den Hollander, P.; et al. Phosphorylation of Serine 367 of FOXC2 by P38 Regulates ZEB1 and Breast Cancer Metastasis, without Impacting Primary Tumor Growth. Oncogene 2016, 35, 5977–5988. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, P.; Mahlapuu, M. Forkhead Transcription Factors: Key Players in Development and Metabolism. Dev. Biol. 2002, 250, 1–23. [Google Scholar] [CrossRef]
- Kim, S.H.; Cho, K.W.; Choi, H.S.; Park, S.J.; Rhee, Y.; Jung, H.S.; Lim, S.K. The Forkhead Transcription Factor Foxc2 Stimulates Osteoblast Differentiation. Biochem. Biophys. Res. Commun. 2009, 386, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xiao, Q.; Xiao, J.; Niu, C.; Li, Y.; Zhang, X.; Zhou, Z.; Shu, G.; Yin, G. Wnt/β-Catenin Signalling: Function, Biological Mechanisms, and Therapeutic Opportunities. Signal Transduct. Target. Ther. 2022, 7, 3. [Google Scholar] [CrossRef]
- Gozo, M.C.; Aspuria, P.J.; Cheon, D.J.; Walts, A.E.; Berel, D.; Miura, N.; Karlan, B.Y.; Orsulic, S. Foxc2 Induces Wnt4 and Bmp4 Expression during Muscle Regeneration and Osteogenesis. Cell Death Differ. 2013, 20, 1031–1042. [Google Scholar] [CrossRef]
- Zhou, P.; Li, Y.; Di, R.; Yang, Y.; Meng, S.; Song, F.; Ma, L. H19 and Foxc2 Synergistically Promotes Osteogenic Differentiation of BMSCs via Wnt-β-Catenin Pathway. J. Cell Physiol. 2019, 234, 13799–13806. [Google Scholar] [CrossRef]
- You, W.; Fan, L.; Duan, D.; Tian, L.; Dang, X.; Wang, C.; Wang, K. Foxc2 Over-Expression in Bone Marrow Mesenchymal Stem Cells Stimulates Osteogenic Differentiation and Inhibits Adipogenic Differentiation. Mol. Cell Biochem. 2014, 386, 125–134. [Google Scholar] [CrossRef]
- Cui, L.; Dang, S.; Qu, J.; Mao, Z.; Wang, X.; Zhang, J.; Chen, J. FOXC2 Is Up-Regulated in Pancreatic Ductal Adenocarcinoma and Promotes the Growth and Migration of Cancer Cells. Tumor Biol. 2016, 37, 8579–8585. [Google Scholar] [CrossRef]
- Wang, R.; Sun, Q.; Wang, P.; Liu, M.; Xiong, S.; Luo, J.; Huang, H.; Du, Q.; Geller, D.A.; Cheng, B. Notch and Wnt/β-Catenin Signaling Pathway Play Important Roles in Activating Liver Cancer Stem Cells. Oncotarget 2016, 7, 5754–5768. [Google Scholar] [CrossRef] [PubMed]
- Bisson, I.; Prowse, D.M. WNT Signaling Regulates Self-Renewal and Differentiation of Prostate Cancer Cells with Stem Cell Characteristics. Cell Res. 2009, 19, 683–697. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.; Wang, X.; Wang, Y.; Ma, D. Wnt/β-Catenin Signaling Regulates Cancer Stem Cells in Lung Cancer A549 Cells. Biochem. Biophys. Res. Commun. 2010, 392, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, L.; De Sousa E Melo, F.; van der Heijden, M.; Cameron, K.; de Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt Activity Defines Colon Cancer Stem Cells and Is Regulated by the Microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef]
- Hoseong Yang, S.; Andl, T.; Grachtchouk, V.; Wang, A.; Liu, J.; Syu, L.-J.; Ferris, J.; Wang, T.S.; Glick, A.B.; Millar, S.E.; et al. Pathological Responses to Oncogenic Hedgehog Signaling in Skin Are Dependent on Canonical Wnt/β-Catenin Signaling. Nat. Genet. 2008, 40, 1130–1135. [Google Scholar] [CrossRef]
- Malanchi, I.; Peinado, H.; Kassen, D.; Hussenet, T.; Metzger, D.; Chambon, P.; Huber, M.; Hohl, D.; Cano, A.; Birchmeier, W.; et al. Cutaneous Cancer Stem Cell Maintenance Is Dependent on β-Catenin Signalling. Nature 2008, 452, 650–653. [Google Scholar] [CrossRef]
- De Sousa e Melo, F.; Vermeulen, L. Wnt Signaling in Cancer Stem Cell Biology. Cancers 2016, 8, 60. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/β-Catenin Signaling: Components, Mechanisms, and Diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef]
- Reya, T.; Clevers, H. Wnt Signalling in Stem Cells and Cancer. Nature 2005, 434, 843–850. [Google Scholar] [CrossRef]
- Logan, C.Y.; Nusse, R. The Wnt Signaling Pathway in Development and Disease. Annu. Rev. Cell Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef]
- Aktary, Z.; Bertrand, J.U.; Larue, L. The WNT-Less Wonder: WNT-Independent β-Catenin Signaling. Pigment. Cell Melanoma Res. 2016, 29, 524–540. [Google Scholar] [CrossRef] [PubMed]
- Doumpas, N.; Lampart, F.; Robinson, M.D.; Lentini, A.; Nestor, C.E.; Cantù, C.; Basler, K. TCF/LEF Dependent and Independent Transcriptional Regulation of Wnt/Β-catenin Target Genes. EMBO J. 2019, 38, e98873. [Google Scholar] [CrossRef]
- Freihen, V.; Rönsch, K.; Mastroianni, J.; Frey, P.; Rose, K.; Boerries, M.; Zeiser, R.; Busch, H.; Hecht, A. SNAIL1 Employs β-Catenin-LEF1 Complexes to Control Colorectal Cancer Cell Invasion and Proliferation. Int. J. Cancer 2020, 146, 2229–2242. [Google Scholar] [CrossRef]
- Chen, X.; Wei, H.; Li, J.; Liang, X.; Dai, S.; Jiang, L.; Guo, M.; Qu, L.; Chen, Z.; Chen, L.; et al. Structural Basis for DNA Recognition by FOXC2. Nucleic Acids Res. 2019, 47, 3752–3764. [Google Scholar] [CrossRef]
- Cao, S.; Wang, Z.; Gao, X.; He, W.; Cai, Y.; Chen, H.; Xu, R. FOXC1 Induces Cancer Stem Cell-like Properties through Upregulation of Beta-Catenin in NSCLC. J. Exp. Clin. Cancer Res. 2018, 37, 220. [Google Scholar] [CrossRef] [PubMed]
- Lam, E.W.-F.; Brosens, J.J.; Gomes, A.R.; Koo, C.-Y. Forkhead Box Proteins: Tuning Forks for Transcriptional Harmony. Nat Rev Cancer 2013, 13, 482. [Google Scholar] [CrossRef]
- Elenbaas, B.; Spirio, L.; Koerner, F.; Fleming, M.D.; Zimonjic, D.B.; Donaher, J.L.; Popescu, N.C.; Hahn, W.C.; Weinberg, R.A. Human Breast Cancer Cells Generated by Oncogenic Transformation of Primary Mammary Epithelial Cells. Genes. Dev. 2001, 15, 50–65. [Google Scholar] [CrossRef] [PubMed]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics Connects Somatic Mutations to Signalling in Breast Cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef]
- Vasaikar, S.V.; Deshmukh, A.P.; den Hollander, P.; Addanki, S.; Kuburich, N.A.; Kudaravalli, S.; Joseph, R.; Chang, J.T.; Soundararajan, R.; Mani, S.A. EMTome: A Resource for Pan-Cancer Analysis of Epithelial-Mesenchymal Transition Genes and Signatures. Br. J. Cancer 2020, 124, 259–269. [Google Scholar] [CrossRef]
- Prat, A.; Karginova, O.; Parker, J.S.; Fan, C.; He, X.; Bixby, L.; Harrell, J.C.; Roman, E.; Adamo, B.; Troester, M.; et al. Characterization of Cell Lines Derived from Breast Cancers and Normal Mammary Tissues for the Study of the Intrinsic Molecular Subtypes. Breast Cancer Res. Treat. 2013, 142, 237–255. [Google Scholar] [CrossRef]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A Reciprocal Repression between ZEB1 and Members of the MiR-200 Family Promotes EMT and Invasion in Cancer Cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef]
- Bai, F.; Liu, S.; Liu, X.; Hollern, D.P.; Scott, A.; Wang, C.; Zhang, L.; Fan, C.; Fu, L.; Perou, C.M.; et al. PDGFRβ Is an Essential Therapeutic Target for BRCA1-Deficient Mammary Tumors. Breast Cancer Res. 2021, 23, 10. [Google Scholar] [CrossRef] [PubMed]
- Berx, G.; Cleton-Jansen, A.M.; Nollet, F.; De Leeuw, W.J.F.; Van De Vijver, M.J.; Cornelisse, C.; Van Roy, F. E-Cadherin Is a Tumour/Invasion Suppressor Gene Mutated in Human Lobular Breast Cancers. EMBO J. 1995, 14, 6107–6115. [Google Scholar] [CrossRef]
- Batlle, E.; Sancho, E.; Francí, C.; Domínguez, D.; Monfar, M.; Baulida, J.; De Herreros, A.G. The Transcription Factor Snail Is a Repressor of E-Cadherin Gene Expression in Epithelial Tumour Cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef]
- Frixen, U.H.; Behrens, J.; Sachs, M.; Eberle, G.; Voss, B.; Warda, A.; Ltehner, D.; Bircluneier, W. E-Cadherin-Mediated Cell-Cell Adhesion Prevents Invasiveness of Human Carcinoma Cells. J. Cell Biol. 1991, 113, 173–185. [Google Scholar]
- Loh, C.Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and n-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef]
- Sánchez-Tilló, E.; De Barrios, O.; Siles, L.; Cuatrecasas, M.; Castells, A.; Postigo, A. β-Catenin/TCF4 Complex Induces the Epithelial-to-Mesenchymal Transition (EMT)-Activator ZEB1 to Regulate Tumor Invasiveness. Proc. Natl. Acad. Sci. USA 2011, 108, 19204–19209. [Google Scholar] [CrossRef]
- Kim, K.; Lu, Z.; Hay, E.D. Direct Evidence for a Role of β-Catenin/LEF-1 Signaling Pathway in Induction of EMT. Cell Biol. Int. 2002, 26, 463–476. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective Identification of Tumorigenic Breast Cancer Cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Sleeman, K.E.; Kendrick, H.; Ashworth, A.; Isacke, C.M.; Smalley, M.J. CD24 Staining of Mouse Mammary Gland Cells Defines Luminal Epithelial, Myoepithelial/Basal and Non-Epithelial Cells. Breast Cancer Res. 2005, 8, R7. [Google Scholar] [CrossRef] [PubMed]
- Dontu, G.; Abdallah, W.M.; Foley, J.M.; Jackson, K.W.; Clarke, M.F.; Kawamura, M.J.; Wicha, M.S. In Vitro Propagation and Transcriptional Profiling of Human Mammary Stem/Progenitor Cells. Genes. Dev. 2003, 17, 1253–1270. [Google Scholar] [CrossRef]
- Liao, M.J.; Cheng, C.Z.; Zhou, B.; Zimonjic, D.B.; Mani, S.A.; Kaba, M.; Gifford, A.; Reinhardt, F.; Popescu, N.C.; Guo, W.; et al. Enrichment of a Population of Mammary Gland Cells That Form Mammospheres and Have in Vivo Repopulating Activity. Cancer Res. 2007, 67, 8131–8138. [Google Scholar] [CrossRef] [PubMed]
- Kuburich, N.A.; Den Hollander, P.; Deshmukh, A.P.; Vasaikar, S.; Joseph, R.; Wicha, M.S.; Mani, S.A. In Vitro Quantification of Cancer Stem Cells Using a Mammosphere Formation Assay. Methods Mol. Biol. 2022, 2429, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in Cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef]
- Dhanasekaran, R.; Deutzmann, A.; Mahauad-Fernandez, W.D.; Hansen, A.S.; Gouw, A.M.; Felsher, D.W. The MYC Oncogene—The Grand Orchestrator of Cancer Growth and Immune Evasion. Nat. Rev. Clin. Oncol. 2021, 19, 23–36. [Google Scholar] [CrossRef]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA Damage and the Balance between Survival and Death in Cancer Biology. Nat. Rev. Cancer 2015, 16, 20–33. [Google Scholar] [CrossRef]
- Ashton, T.M.; Gillies McKenna, W.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Itou, J.; Sato, F.; Toi, M. SALL4—KHDRBS3 Network Enhances Stemness by Modulating CD44 Splicing in Basal-like Breast Cancer. Cancer Med. 2018, 7, 454–462. [Google Scholar] [CrossRef]
- Jackstadt, R.; Hodder, M.C.; Sansom, O.J. WNT and β-Catenin in Cancer: Genes and Therapy. Annu. Rev. Cancer Biol. 2020, 4, 177–196. [Google Scholar] [CrossRef]
- Savage, J.; Voronova, A.; Mehta, V.; Sendi-Mukasa, F.; Skerjanc, I.S. Canonical Wnt Signaling Regulates Foxc1/2 Expression in P19 Cells. Differentiation 2010, 79, 31–40. [Google Scholar] [CrossRef]
- Fishilevich, S.; Nudel, R.; Rappaport, N.; Hadar, R.; Plaschkes, I.; Stein, T.I.; Rosen, N.; Kohn, A.; Twik, M.; Safran, M.; et al. GeneHancer: Genome-Wide Integration of Enhancers and Target Genes in GeneCards. Database 2017, 2017, bax028. [Google Scholar] [CrossRef] [PubMed]
- Craene, B.D.; Berx, G. Regulatory Networks Defining EMT during Cancer Initiation and Progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.W.; Weinberg, R.A. Linking EMT Programmes to Normal and Neoplastic Epithelial Stem Cells. Nat. Rev. Cancer 2021, 21, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Iwatsuki, M.; Mimori, K.; Yokobori, T.; Ishi, H.; Beppu, T.; Nakamori, S.; Baba, H.; Mori, M. Epithelial-Mesenchymal Transition in Cancer Development and Its Clinical Significance. Cancer Sci. 2010, 101, 293–299. [Google Scholar] [CrossRef]
- Korkaya, H.; Paulson, A.; Charafe-Jauffret, E.; Ginestier, C.; Brown, M.; Dutcher, J.; Clouthier, S.G.; Wicha, M.S. Regulation of Mammary Stem/Progenitor Cells by PTEN/Akt/β-Catenin Signaling. PLoS Biol. 2009, 7, e1000121. [Google Scholar] [CrossRef]
- Lamb, R.; Ablett, M.P.; Spence, K.; Landberg, G.; Sims, A.H.; Clarke, R.B. Wnt Pathway Activity in Breast Cancer Sub-Types and Stem-Like Cells. PLoS ONE 2013, 8, e67811. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castaneda, M.; den Hollander, P.; Werden, S.; Ramirez-Peña, E.; Vasaikar, S.V.; Kuburich, N.A.; Gould, C.; Soundararajan, R.; Mani, S.A. β-Catenin Drives the FOXC2-Mediated Epithelial–Mesenchymal Transition and Acquisition of Stem Cell Properties. Cancers 2025, 17, 1114. https://doi.org/10.3390/cancers17071114
Castaneda M, den Hollander P, Werden S, Ramirez-Peña E, Vasaikar SV, Kuburich NA, Gould C, Soundararajan R, Mani SA. β-Catenin Drives the FOXC2-Mediated Epithelial–Mesenchymal Transition and Acquisition of Stem Cell Properties. Cancers. 2025; 17(7):1114. https://doi.org/10.3390/cancers17071114
Chicago/Turabian StyleCastaneda, Maria, Petra den Hollander, Steve Werden, Esmeralda Ramirez-Peña, Suhas V. Vasaikar, Nick A. Kuburich, Claire Gould, Rama Soundararajan, and Sendurai A. Mani. 2025. "β-Catenin Drives the FOXC2-Mediated Epithelial–Mesenchymal Transition and Acquisition of Stem Cell Properties" Cancers 17, no. 7: 1114. https://doi.org/10.3390/cancers17071114
APA StyleCastaneda, M., den Hollander, P., Werden, S., Ramirez-Peña, E., Vasaikar, S. V., Kuburich, N. A., Gould, C., Soundararajan, R., & Mani, S. A. (2025). β-Catenin Drives the FOXC2-Mediated Epithelial–Mesenchymal Transition and Acquisition of Stem Cell Properties. Cancers, 17(7), 1114. https://doi.org/10.3390/cancers17071114