

Addressing Challenges in Targeted Therapy for Metastatic Colorectal Cancer


Simple Summary
Abstract
1. Introduction
- -
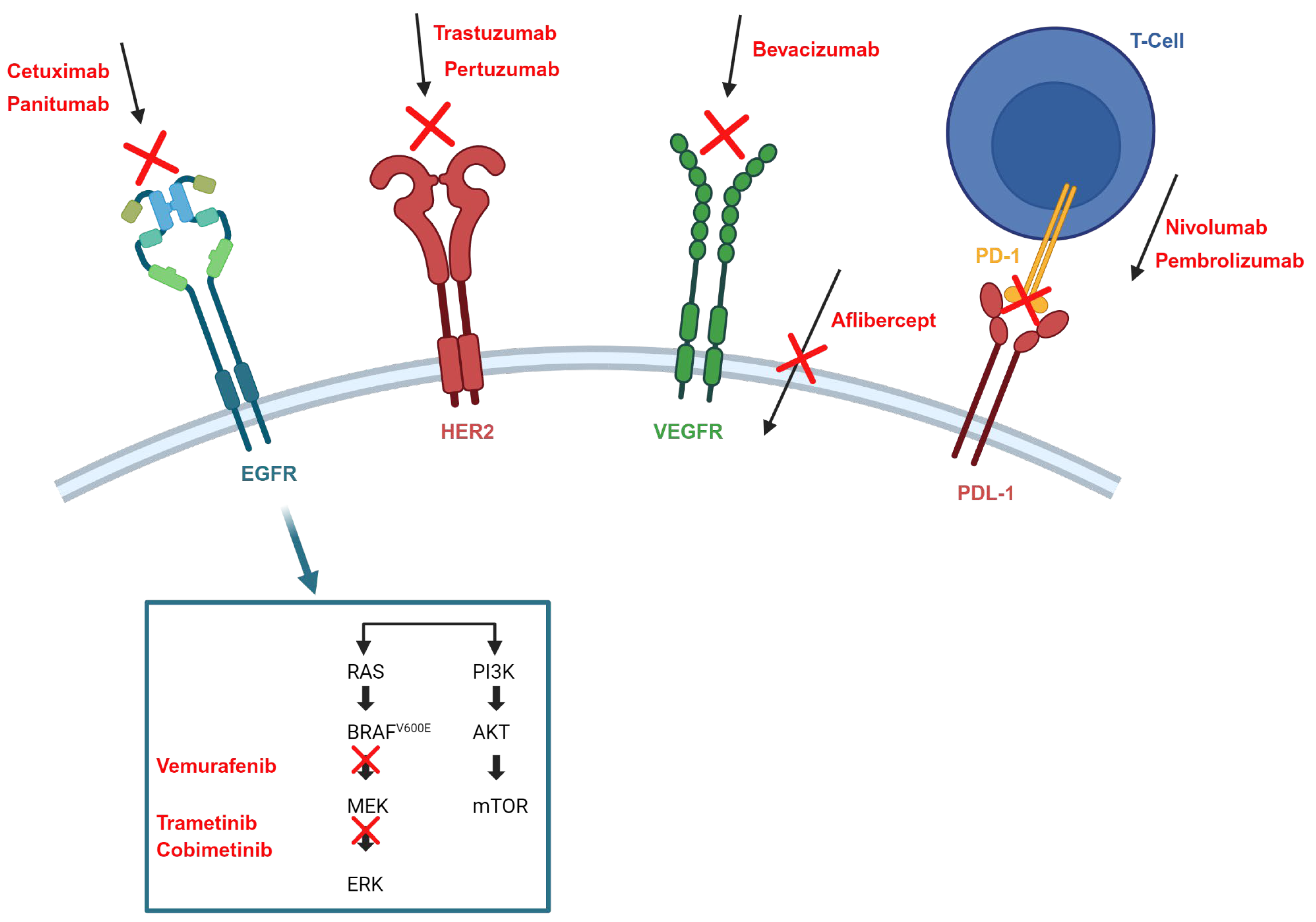
- Among patients diagnosed with mCRC, the presence or absence of RAS mutations is a key determinant in identifying patients who may benefit from EGFR-targeted treatment strategies. In particular, the administration of anti-EGFR monoclonal antibodies (cetuximab, panitumumab) should be limited to patients with RAS wild-type tumors. Conversely, patients with BRAF-mutated tumors generally do not respond to anti-EGFR antibodies as monotherapy. However, in selected cases, the introduction of a BRAF inhibitor has shown some positive responses, although this therapeutic approach is typically reserved for advanced stages of treatment [4].
- -
- For individuals with tumors that exhibit deficient mismatch repair (dMMR), typically characterized by high levels of microsatellite instability (MSI-H), initiation of treatment with an immune checkpoint inhibitor should be considered as a first-line therapeutic approach [4].
2. Drugs Used in Targeted Therapy for CRC
- -
- EGFR inhibitors
- -
- Targeting BRAFV600E
- -
- Human epidermal growth factor receptor 2 (HER2) inhibitor
- -
- MEK inhibitors
- -
- Immune checkpoint inhibitors
Challenges in Targeted Therapy
3. Drug Resistance
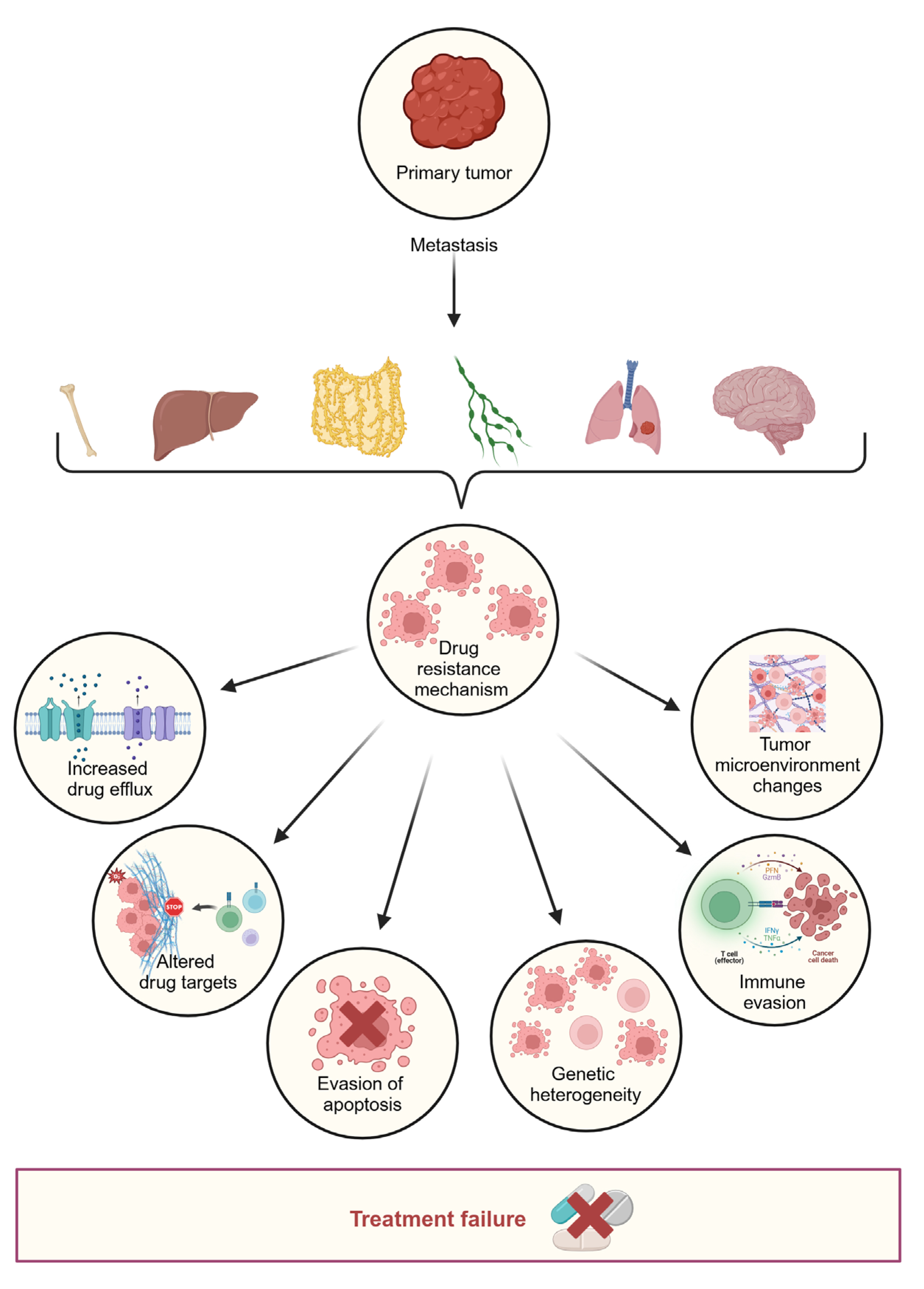
3.1. Primary Resistance
- a.
- Inter-patient heterogeneity
- b.
- Spatial heterogeneity
3.2. Acquired Resistance
- i.
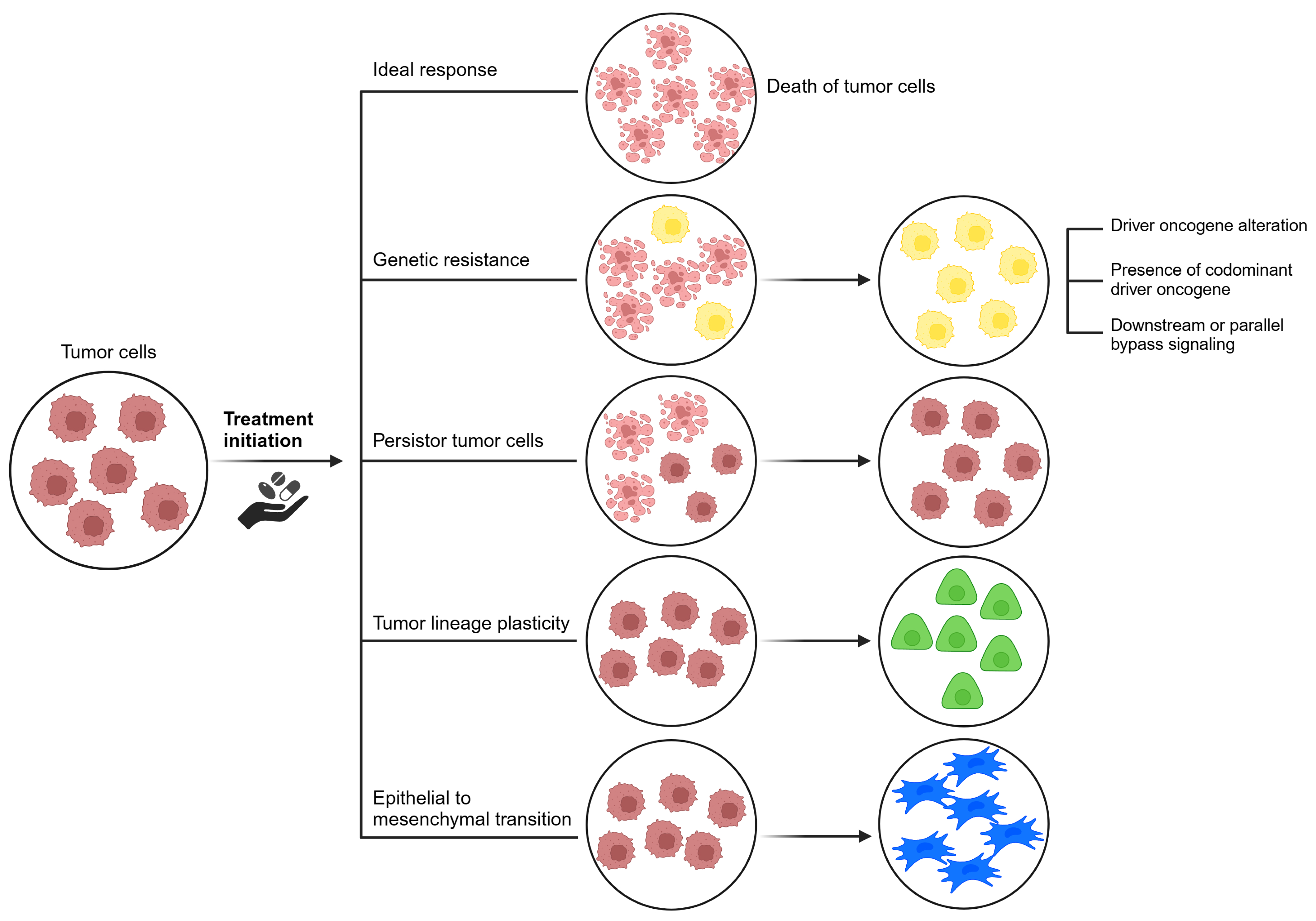
- Driver oncogene alterations: Alterations in the target gene, such as mutations and amplifications, allow cells to proliferate despite the presence of inhibitors. Selective pressure from targeted therapies can induce aberrations that reactivate the driver oncogene. Initially, malignancies with these aberrations show substantial responses to selective inhibitors. However, resistance tends to develop over time. Genetic aberrations in the target gene can be categorized as on-target or off-target resistance [23]. On-target effects correspond to enhanced and adverse pharmacological effects specifically at the intended target in the test system. Off-target effects, on the other hand, involve adverse effects due to modulation of other targets that may be biologically related or completely unrelated to the intended target [57]. Off-target resistance involves abnormal activation of alternative signaling pathways, coexisting driver oncogenes, lineage plasticity, epithelial-mesenchymal transition, and persistent cancer cells [58].
- ii.
- iii.
- Abnormal signaling activation processes often result from gain-of-function mutations, genomic amplification, chromosomal rearrangements, or autocrine activation. Gain-of-function mutations occur when downstream signaling persists despite upstream blockade by targeted agents. Gene amplification refers to an increase in the number of gene copies [23]. In cancer cells, gene amplification occurs when signals from surrounding cells or the environment induce the production of multiple gene copies [58]. Chromosomal rearrangements can induce cancer either by creating a fusion gene or by disrupting gene regulation [60]. Autocrine signaling refers to the production and release of a signaling molecule by a cell, which then binds to receptors on the same cell to initiate signaling [61].
- iv.
- Presence of co-dominant driver oncogenes: It is evident that a number of non-responsive mutations coexist, resulting in resistance to targeted therapies [23].
- v.
- Tumor lineage plasticity: Lineage plasticity refers to the ability of a cell to undergo phenotypic transformation toward a different developmental lineage [62]. Cancer cell plasticity arises due to exposure and selective pressure of targeted therapeutic agents [62,63]. This transformation allows tumor cells to adapt to challenging conditions, such as a hypoxic tumor microenvironment [62]. Although the driver mutation is retained, transformed cells no longer rely on it for proliferation, leading to therapeutic resistance. Addressing the epigenetic, genomic, and microenvironmental factors that drive lineage plasticity will be critical to the development of innovative treatment approaches [23].
- vi.
- Epithelial-mesenchymal transition: Although the driver mutation is retained, the transformed cells no longer rely on it for proliferation, leading to therapeutic resistance. Addressing the epigenetic, genomic, and microenvironmental factors that drive lineage plasticity will be critical to the development of innovative treatment approaches [58]. It is hypothesized that the process involves the stimulation and activation of intracellular signaling pathways, resulting in the reduction of E-cadherin. EGFR TKIs may induce cells to transition to a mesenchymal phenotype characterized by decreased E-cadherin expression and expression of mesenchymal markers such as N-cadherin [64].
- vii.
- Persister cancer cells: The concept of drug-tolerant persister cells has emerged as an important concept. Persister cancer cells refer to a group of cells that can survive systemic treatments by entering a reversible and sluggish proliferative state [65,66] and are thought to be distinct from cancer stem cells. Persister cells typically do not possess conventional driver alterations associated with drug resistance, and their resistant properties may be transient and reversible upon cessation of drug treatment [67]. In contrast, cancer stem cells present in a tumor possess the capacity for self-renewal and the generation of diverse cancer cell lineages [68].
- viii.
- The survival of persister cells is attributed to either pre-existing drug-resistant cells or the induction of intrinsic changes that facilitate phenotypic variation [23]. Malignant cells experience various stresses during proliferation, including metabolic, hypoxic, and nutrient limitations. Case reports demonstrate that EGFR inhibitors contribute to chromatin repression, which affects the development and survival of drug-tolerant persister cells [66].
4. Resistance Mechanisms Towards the Different Classes of Drugs
4.1. Resistance to Antiangiogenic Therapy (VEGF and VGFR)
4.2. Mode of Resistance to EGFR Inhibitors
4.3. Primary and Secondary Resistance to BRAFV600E Inhibitors
4.4. Resistance to HER2 Inhibition
4.5. Challenges of ICI in CRC
5. Toxicity
5.1. Combination Therapy Leads to Higher Toxicity
5.2. On-Target Toxicity
5.3. Off-Target Toxicity
6. Cost and Access
7. Conclusions and Future Perspective
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| mCRC | metastatic colorectal cancer |
| VEFG | Endothelial Growth Factor |
| VGFR | Vascular Endothelial Growth Factor Receptor |
| EGFR | Epidermal Growth Factor Receptor |
| HER2 | Human Epidermal Growth Factor Receptor 2 |
| TRK | tropomyosin receptor kinase |
| mismatch repair | MMR |
| Placental Growth Factor | PIGF |
| microsatellite instability | MSI-H |
| OS | Overall Survival |
| PFS | Progression-Free Survival |
| HR | Hazard Ratio |
| ICI | immune checkpoint inhibitors |
| PIGF | Placental Growth Factor |
| TAT | Tumor antigen targeting |
| ctDNA | circulating tumor DNA |
References
- Xie, Y.H.; Chen, Y.X.; Fang, J.Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Di Nicolantonio, F.; Vitiello, P.P.; Marsoni, S.; Siena, S.; Tabernero, J.; Trusolino, L.; Bernards, R.; Bardelli, A. Precision oncology in metastatic colorectal cancer–from biology to medicine. Nat. Rev. Clin. Oncol. 2021, 18, 506–525. [Google Scholar] [CrossRef]
- Lieu, C.H.; Corcoran, R.B.; Overman, M.J. Integrating biomarkers and targeted therapy into colorectal cancer management. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Morris, V.K.; Kennedy, E.B.; Baxter, N.N.; Benson, A.B., 3rd; Cercek, A.; Cho, M.; Ciombor, K.K.; Cremolini, C.; Davis, A.; Deming, D.A.; et al. Treatment of metastatic colorectal cancer: ASCO guideline. J. Clin. Oncol. 2023, 41, 678–700. [Google Scholar] [CrossRef] [PubMed]
- Keefe, D.M.K.; Bateman, E.H. Potential successes and challenges of targeted cancer therapies. J. Natl. Cancer Inst. Monogr. 2019, 2019, lgz008. [Google Scholar] [CrossRef]
- Cunningham, D.; Humblet, Y.; Siena, S.; Khayat, D.; Bleiberg, H.; Santoro, A.; Bets, D.; Mueser, M.; Harstrick, A.; Verslype, C.; et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N. Engl. J. Med. 2004, 351, 337–345. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Peeters, M.; Siena, S.; Humblet, Y.; Hendlisz, A.; Neyns, B.; Canon, J.L.; Van Laethem, J.L.; Maurel, J.; Richardson, G.; et al. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J. Clin. Oncol. 2007, 25, 1658–1664. [Google Scholar] [CrossRef]
- Douillard, J.Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef]
- Amado, R.G.; Wolf, M.; Peeters, M.; Van Cutsem, E.; Siena, S.; Freeman, D.J.; Juan, T.; Sikorski, R.; Suggs, S.; Radinsky, R.; et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 1626–1634. [Google Scholar] [CrossRef]
- Stintzing, S.; Miller-Phillips, L.; Modest, D.P.; Fischer von Weikersthal, L.; Decker, T.; Kiani, A.; Vehling-Kaiser, U.; Al-Batran, S.E.; Heintges, T.; Kahl, C.; et al. Impact of BRAF and RAS mutations on first-line efficacy of FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab: Analysis of the FIRE-3 (AIO KRK-0306) study. Eur. J. Cancer 2017, 79, 50–60. [Google Scholar] [CrossRef]
- García-Foncillas, J.; Sunakawa, Y.; Aderka, D.; Wainberg, Z.; Ronga, P.; Witzler, P.; Stintzing, S. Distinguishing features of cetuximab and panitumumab in colorectal cancer and other solid tumors. Front. Oncol. 2019, 9, 849. [Google Scholar] [CrossRef]
- Planchard, D.; Besse, B.; Groen, H.J.M.; Hashemi, S.M.S.; Mazieres, J.; Kim, T.M.; Quoix, E.; Souquet, P.J.; Barlesi, F.; Baik, C.; et al. Phase 2 study of dabrafenib plus trametinib in patients with BRAF V600E-mutant metastatic NSCLC: Updated 5-year survival rates and genomic analysis. J. Thorac. Oncol. 2022, 17, 103–115. [Google Scholar] [CrossRef]
- Ros, J.; Baraibar, I.; Sardo, E.; Mulet, N.; Salvà, F.; Argilés, G.; Martini, G.; Ciardiello, D.; Cuadra, J.L.; Tabernero, J.; et al. BRAF, MEK and EGFR inhibition as treatment strategies in BRAF V600E metastatic colorectal cancer. Ther. Adv. Med Oncol. 2021, 13, 1758835921992974. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Trunk, A.; Braithwaite, M.; Nevala-Plagemann, C.; Pappas, L.; Haaland, B.; Garrido-Laguna, I. Real-world outcomes of patients with BRAF-mutated metastatic colorectal cancer treated in the United States. J. Natl. Compr. Cancer Netw. 2022, 20, 144–150. [Google Scholar] [CrossRef]
- Takegawa, N.; Yonesaka, K. HER2 as an emerging oncotarget for colorectal cancer treatment after failure of anti-epidermal growth factor receptor therapy. Clin. Color. Cancer 2017, 16, 247–251. [Google Scholar] [CrossRef]
- Oh, D.Y.; Bang, Y.J. HER2-targeted therapies—A role beyond breast cancer. Nat. Rev. Clin. Oncol. 2020, 17, 33–48. [Google Scholar] [CrossRef]
- Ahcene Djaballah, S.; Daniel, F.; Milani, A.; Ricagno, G.; Lonardi, S. HER2 in colorectal cancer: The long and winding road from negative predictive factor to positive actionable target. Am. Soc. Clin. Oncol. Educ. Book 2022, 42, 219–232. [Google Scholar] [CrossRef]
- Chiron, M.; Bagley, R.G.; Pollard, J.; Mankoo, P.K.; Henry, C.; Vincent, L.; Geslin, C.; Baltes, N.; Bergstrom, D.A. Differential antitumor activity of aflibercept and bevacizumab in patient-derived xenograft models of colorectal cancer. Mol. Cancer Ther. 2014, 13, 1636–1644. [Google Scholar] [CrossRef]
- Tang, P.A.; Cohen, S.J.; Kollmannsberger, C.; Bjarnason, G.; Virik, K.; MacKenzie, M.J.; Lourenco, L.; Wang, L.; Chen, A.; Moore, M.J. Phase II clinical and pharmacokinetic study of aflibercept in patients with previously treated metastatic colorectal cancer. Clin. Cancer Res. 2012, 18, 6023–6031. [Google Scholar] [CrossRef]
- Moore, M.; Gill, S.; Asmis, T.; Berry, S.; Burkes, R.; Zbuk, K.; Alcindor, T.; Jeyakumar, A.; Chan, T.; Rao, S.; et al. Randomized phase II study of modified FOLFOX-6 in combination with ramucirumab or icrucumab as second-line therapy in patients with metastatic colorectal cancer after progression on first-line irinotecan-based therapy. Ann. Oncol. 2016, 27, 2216–2224. [Google Scholar] [CrossRef]
- Diaz, L.A., Jr.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab versus chemotherapy for microsatellite instability-high or mismatch repair-deficient metastatic colorectal cancer (KEYNOTE-177): Final analysis of a randomised, open-label, phase 3 study. Lancet Oncol. 2022, 23, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Mielgo-Rubio, X.; Martín, M.; Remon, J.; Higuera, O.; Calvo, V.; Jarabo, J.R.; Conde, E.; Luna, J.; Provencio, M.; De Castro, J.; et al. Targeted therapy moves to earlier stages of non-small-cell lung cancer: Emerging evidence, controversies and future challenges. Future Oncol. 2021, 17, 4011–4025. [Google Scholar] [CrossRef]
- Rivera-Concepcion, J.; Uprety, D.; Adjei, A.A. Challenges in the use of targeted therapies in non-small cell lung cancer. Cancer Res. Treat. 2022, 54, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Michor, F.; Polyak, K. The origins and implications of intratumor heterogeneity. Cancer Prev. Res. 2010, 3, 1361–1364. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
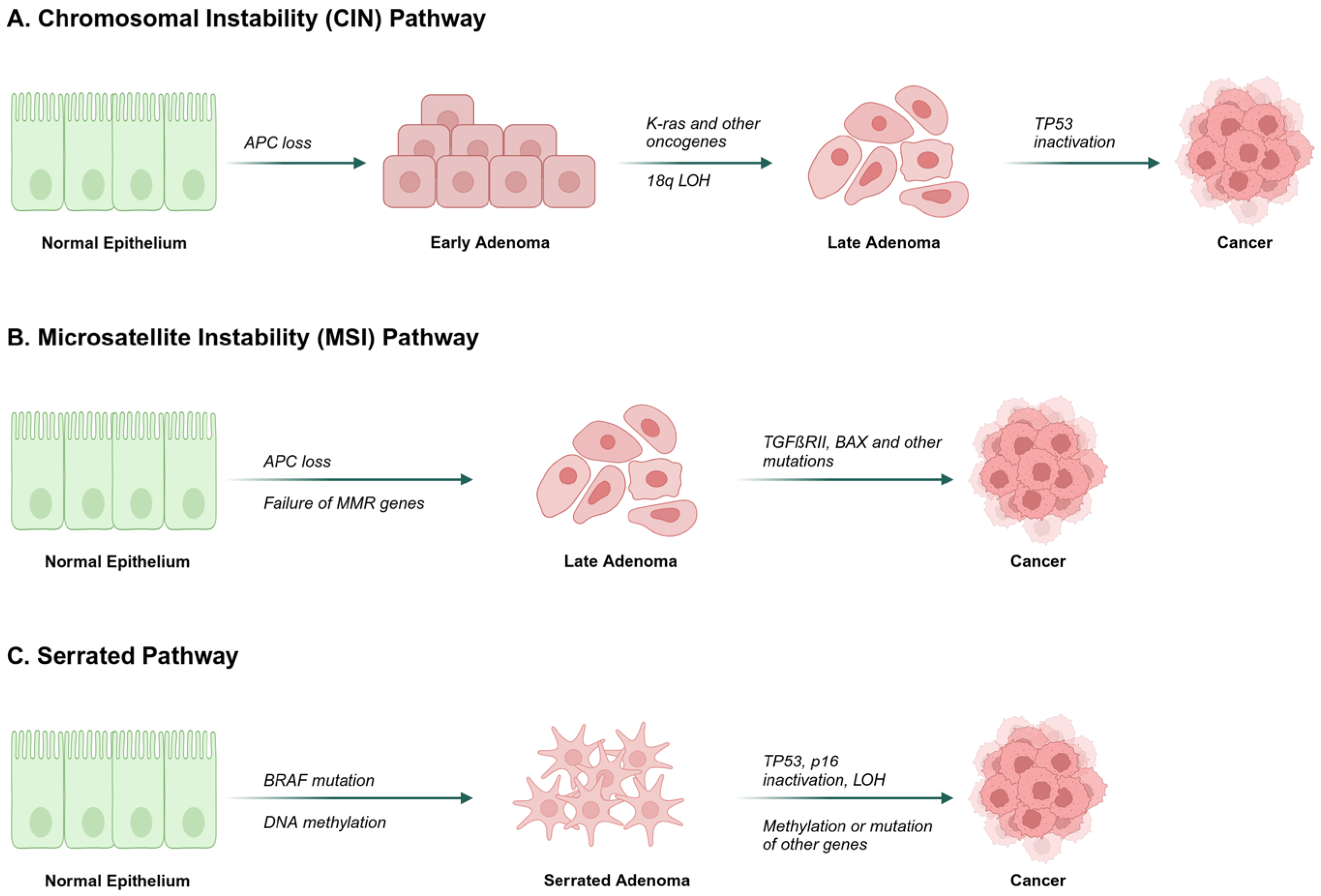
- Grady, W.M.; Carethers, J.M. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology 2008, 135, 1079–1099. [Google Scholar] [CrossRef]
- Bramsen, J.B.; Rasmussen, M.H.; Ongen, H.; Mattesen, T.B.; Ørntoft, M.W.; Árnadóttir, S.S.; Sandoval, J.; Laguna, T.; Vang, S.; Øster, B.; et al. Molecular-subtype-specific biomarkers improve prediction of prognosis in colorectal cancer. Cell Rep. 2017, 19, 1268–1280. [Google Scholar] [CrossRef]
- Ogino, S.; Nowak, J.A.; Hamada, T.; Phipps, A.I.; Peters, U.; Milner, D.A., Jr.; Giovannucci, E.L.; Nishihara, R.; Giannakis, M.; Garrett, W.S.; et al. Integrative analysis of exogenous, endogenous, tumour and immune factors for precision medicine. Gut 2018, 67, 1168–1180. [Google Scholar] [CrossRef]
- Ashique, S.; Bhowmick, M.; Pal, R.; Khatoon, H.; Kumar, P.; Sharma, H.; Garg, A.; Kumar, S.; Das, U. Multi drug resistance in colorectal cancer: Approaches to overcome, advancements and future success. Adv. Cancer Biol. Metastasis 2024, 10, 100114. [Google Scholar] [CrossRef]
- Rebersek, M. Gut microbiome and its role in colorectal cancer. BMC Cancer 2021, 21, 1325. [Google Scholar] [CrossRef]
- Hua, H.; Kong, Q.; Yin, J.; Zhang, J.; Jiang, Y. Insulin-like growth factor receptor signaling in tumorigenesis and drug resistance: A challenge for cancer therapy. J. Hematol. Oncol. 2020, 13, 64. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wu, L.; Yan, G.; Chen, Y.; Zhou, M.; Wu, Y.; Li, Y. Inflammation and tumor progression: Signaling pathways and targeted intervention. Signal Transduct. Target. Ther. 2021, 6, 263. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jung, G.; Hernández-Illán, E.; Moreira, L.; Balaguer, F.; Goel, A. Epigenetics of colorectal cancer: Biomarker and therapeutic potential. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 111–130. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Pan, J.; Chen, W.; Jiang, J.; Huang, J. Chronic stress-induced immune dysregulation in cancer: Implications for initiation, progression, metastasis, and treatment. Am. J. Cancer Res. 2020, 10, 1294–1307. [Google Scholar]
- Fearon, E.F.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar]
- Singh, P.P.; Sharma, P.K.; Krishnan, G.; Lockhart, A.C. Immune checkpoints and immunotherapy for colorectal cancer. Gastroenterol. Rep. 2015, 3, 289–297. [Google Scholar] [CrossRef]
- Molinari, C.; Marisi, G.; Passardi, A.; Matteucci, L.; De Maio, G.; Ulivi, P. Heterogeneity in colorectal cancer: A challenge for personalized medicine? Int. J. Mol. Sci. 2018, 19, 3733. [Google Scholar] [CrossRef]
- Mogensen, M.B.; Rossing, M.; Østrup, O.; Larsen, P.N.; Heiberg Engel, P.J.; Jørgensen, L.N.; Hogdall, E.V.; Eriksen, J.; Ibsen, P.; Jess, P.; et al. Genomic alterations accompanying tumour evolution in colorectal cancer: Tracking the differences between primary tumours and synchronous liver metastases by whole-exome sequencing. BMC Cancer 2018, 18, 752. [Google Scholar] [CrossRef]
- Ulivi, P.; Scarpi, E.; Chiadini, E.; Marisi, G.; Valgiusti, M.; Capelli, L.; Casadei Gardini, A.; Monti, M.; Ruscelli, S.; Frassineti, G.L.; et al. Right- vs. left-sided metastatic colorectal cancer: Differences in tumor biology and bevacizumab efficacy. Int. J. Mol. Sci. 2017, 18, 1240. [Google Scholar] [CrossRef]
- Stintzing, S.; Tejpar, S.; Gibbs, P.; Thiebach, L.; Lenz, H.J. Understanding the role of primary tumour localisation in colorectal cancer treatment and outcomes. Eur. J. Cancer 2017, 84, 69–80. [Google Scholar] [CrossRef]
- Tejpar, S.; Stintzing, S.; Ciardiello, F.; Tabernero, J.; Van Cutsem, E.; Beier, F.; Esser, R.; Lenz, H.J.; Heinemann, V. Prognostic and predictive relevance of primary tumor location in patients with RAS wild-type metastatic colorectal cancer: Retrospective analyses of the CRYSTAL and FIRE-3 trials. JAMA Oncol. 2017, 3, 194–201, Erratum in JAMA Oncol. 2017, 3, 1742. https://doi.org/10.1001/jamaoncol.2017.4136. [Google Scholar] [CrossRef]
- Cremolini, C.; Antoniotti, C.; Stein, A.; Bendell, J.; Gruenberger, T.; Rossini, D.; Masi, G.; Ongaro, E.; Hurwitz, H.; Falcone, A.; et al. Individual patient data meta-analysis of FOLFOXIRI plus bevacizumab versus doublets plus bevacizumab as initial therapy of unresectable metastatic colorectal cancer. J. Clin. Oncol. 2020, 38, 3314–3324. [Google Scholar] [CrossRef]
- Amaro, A.; Chiara, S.; Pfeffer, U. Molecular evolution of colorectal cancer: From multistep carcinogenesis to the big bang. Cancer Metastasis Rev. 2016, 35, 63–74. [Google Scholar] [CrossRef]
- Kreso, A.; O’Brien, C.A.; van Galen, P.; Gan, O.I.; Notta, F.; Brown, A.M.; Ng, K.; Ma, J.; Wienholds, E.; Dunant, C.; et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science 2013, 339, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.; Pusztai, L.; Swanton, C. Cancer heterogeneity: Implications for targeted therapeutics. Br. J. Cancer 2013, 108, 479–485. [Google Scholar] [CrossRef]
- Enriquez-Navas, P.M.; Kam, Y.; Das, T.; Hassan, S.; Silva, A.; Foroutan, P.; Ruiz, E.; Martinez, G.; Minton, S.; Gillies, R.J.; et al. Exploiting evolutionary principles to prolong tumor control in preclinical models of breast cancer. Sci. Transl. Med. 2016, 8, 327ra24. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, S.; Swanton, C.; Taylor, B.S.; Costello, J.F. Treatment-induced mutagenesis and selective pressures sculpt cancer evolution. Cold Spring Harb. Perspect. Med. 2017, 7, a026617. [Google Scholar] [CrossRef]
- Andor, N.; Graham, T.A.; Jansen, M.; Xia, L.C.; Aktipis, C.A.; Petritsch, C.; Ji, H.P.; Maley, C.C. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat. Med. 2016, 22, 105–113. [Google Scholar] [CrossRef]
- Sveen, A.; Løes, I.M.; Alagaratnam, S.; Nilsen, G.; Høland, M.; Lingjærde, O.C.; Sorbye, H.; Berg, K.C.; Horn, A.; Angelsen, J.H.; et al. Intra-patient inter-metastatic genetic heterogeneity in colorectal cancer as a key determinant of survival after curative liver resection. PLoS Genet. 2016, 12, e1006225. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Clonal heterogeneity and tumor evolution: Past, present, and the future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef]
- Wang, W.; Kandimalla, R.; Huang, H.; Zhu, L.; Li, Y.; Gao, F.; Goel, A.; Wang, X. Molecular subtyping of colorectal cancer: Recent progress, new challenges and emerging opportunities. Semin. Cancer Biol. 2019, 55, 37–52. [Google Scholar] [CrossRef]
- Jackman, D.; Pao, W.; Riely, G.J.; Engelman, J.A.; Kris, M.G.; Jänne, P.A.; Lynch, T.; Johnson, B.E.; Miller, V.A. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Wu, Y.L.; Ahn, M.J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef]
- Drilon, A.; Ou, S.I.; Cho, B.C.; Kim, D.W.; Lee, J.; Lin, J.J.; Zhu, V.W.; Ahn, M.J.; Camidge, D.R.; Nguyen, J.; et al. Repotrectinib (TPX-0005) is a next-generation ROS1/TRK/ALK inhibitor that potently inhibits ROS1/TRK/ALK solvent-front mutations. Cancer Discov. 2018, 8, 1227–1236. [Google Scholar] [CrossRef] [PubMed]
- Neel, D.S.; Bivona, T.G. Resistance is futile: Overcoming resistance to targeted therapies in lung adenocarcinoma. npj Precis. Oncol. 2017, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Mikubo, M.; Inoue, Y.; Liu, G.; Tsao, M.S. Mechanism of drug tolerant persister cancer cells: The landscape and clinical implication for therapy. J. Thorac. Oncol. 2021, 16, 1798–1809. [Google Scholar] [CrossRef]
- Rudmann, D.G. On-target and off-target-based toxicologic effects. Toxicol. Pathol. 2012, 41, 310–314. [Google Scholar] [CrossRef]
- Marine, J.C.; Dawson, S.J.; Dawson, M.A. Non-genetic mechanisms of therapeutic resistance in cancer. Nat. Rev. Cancer 2020, 20, 743–756. [Google Scholar] [CrossRef] [PubMed]
- Sudhesh Dev, S.; Zainal Abidin, S.A.; Farghadani, R.; Othman, I.; Naidu, R. Receptor tyrosine kinases and their signaling pathways as therapeutic targets of curcumin in cancer. Front. Pharmacol. 2021, 12, 772510. [Google Scholar] [CrossRef]
- Holland, A.J.; Cleveland, D.W. Chromoanagenesis and cancer: Mechanisms and consequences of localized, complex chromosomal rearrangements. Nat. Med. 2012, 18, 1630–1638. [Google Scholar] [CrossRef]
- Ungefroren, H. Autocrine TGF-β in cancer: Review of the literature and caveats in experimental analysis. Int. J. Mol. Sci. 2021, 22, 977. [Google Scholar] [CrossRef] [PubMed]
- Quintanal-Villalonga, Á.; Chan, J.M.; Yu, H.A.; Pe’er, D.; Sawyers, C.L.; Sen, T.; Rudin, C.M. Lineage plasticity in cancer: A shared pathway of therapeutic resistance. Nat. Rev. Clin. Oncol. 2020, 17, 360–371, Erratum in Nat. Rev. Clin. Oncol. 2020, 17, 382. https://doi.org/10.1038/s41571-020-0355-5. [Google Scholar] [CrossRef] [PubMed]
- Zellmer, V.R.; Zhang, S. Evolving concepts of tumor heterogeneity. Cell Biosci. 2014, 4, 69. [Google Scholar] [CrossRef]
- Weng, C.H.; Chen, L.Y.; Lin, Y.C.; Shih, J.Y.; Lin, Y.C.; Tseng, R.Y.; Chiu, A.C.; Yeh, Y.H.; Liu, C.; Lin, Y.T.; et al. Epithelial-mesenchymal transition (EMT) beyond EGFR mutations per se is a common mechanism for acquired resistance to EGFR TKI. Oncogene 2019, 38, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Oren, Y.; Tsabar, M.; Cuoco, M.S.; Amir-Zilberstein, L.; Cabanos, H.F.; Hütter, J.C.; Hu, B.; Thakore, P.I.; Tabaka, M.; Fulco, C.P.; et al. Cycling cancer persister cells arise from lineages with distinct programs. Nature 2021, 596, 576–582. [Google Scholar] [CrossRef]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef]
- Cabanos, H.F.; Hata, A.N. Emerging insights into targeted therapy-tolerant persister cells in cancer. Cancers 2021, 13, 2666. [Google Scholar] [CrossRef]
- Zakaria, N.; Satar, N.A.; Abu Halim, N.H.; Ngalim, S.H.; Yusoff, N.M.; Lin, J.; Yahaya, B.H. Targeting lung cancer stem cells: Research and clinical impacts. Front. Oncol. 2017, 7, 80. [Google Scholar] [CrossRef]
- Kopetz, S.; Hoff, P.M.; Morris, J.S.; Wolff, R.A.; Eng, C.; Glover, K.Y.; Adinin, R.; Overman, M.J.; Valero, V.; Wen, S.; et al. Phase II trial of infusional fluorouracil, irinotecan, and bevacizumab for metastatic colorectal cancer: Efficacy and circulating angiogenic biomarkers associated with therapeutic resistance. J. Clin. Oncol. 2010, 28, 453–459. [Google Scholar] [CrossRef]
- Goede, V.; Coutelle, O.; Neuneier, J.; Reinacher-Schick, A.; Schnell, R.; Koslowsky, T.C.; Weihrauch, M.R.; Cremer, B.; Kashkar, H.; Odenthal, M.; et al. Identification of serum angiopoietin-2 as a biomarker for clinical outcome of colorectal cancer patients treated with bevacizumab-containing therapy. Br. J. Cancer 2010, 103, 1407–1414. [Google Scholar] [CrossRef]
- Mitsuhashi, A.; Goto, H.; Saijo, A.; Trung, V.T.; Aono, Y.; Ogino, H.; Kuramoto, T.; Tabata, S.; Uehara, H.; Izumi, K.; et al. Fibrocyte-like cells mediate acquired resistance to anti-angiogenic therapy with bevacizumab. Nat. Commun. 2015, 6, 8792. [Google Scholar] [CrossRef] [PubMed]
- Goto, H.; Nishioka, Y. Fibrocytes: A novel stromal cell to regulate resistance to anti-angiogenic therapy and cancer progression. Int. J. Mol. Sci. 2017, 19, 98. [Google Scholar] [CrossRef] [PubMed]
- Semrad, T.J.; Kim, E.J.; Tanaka, M.S.; Sands, J.; Roberts, C.; Burich, R.A.; Li, Y.; Gandara, D.R.; Lara, P., Jr.; Mack, P.C.; et al. Phase II Study of Dovitinib in Patients Progressing on Anti-Vascular Endothelial Growth Factor Therapy. Cancer Treat. Res. Commun. 2017, 10, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Jahangiri, A.; De Lay, M.; Miller, L.M.; Carbonell, W.S.; Hu, Y.L.; Lu, K.; Tom, M.W.; Paquette, J.; Tokuyasu, T.A.; Tsao, S.; et al. Gene expression profile identifies tyrosine kinase c-Met as a targetable mediator of antiangiogenic therapy resistance. Clin. Cancer Res. 2013, 19, 1773–1783. [Google Scholar] [CrossRef]
- Zhao, Y.; Guo, S.; Deng, J.; Shen, J.; Du, F.; Wu, X.; Chen, Y.; Li, M.; Chen, M.; Li, X.; et al. VEGF/VEGFR-targeted therapy and immunotherapy in non-small cell lung cancer: Targeting the tumor microenvironment. Int. J. Biol. Sci. 2022, 18, 3845–3858. [Google Scholar] [CrossRef]
- Carbone, C.; Tamburrino, A.; Piro, G.; Boschi, F.; Cataldo, I.; Zanotto, M.; Mina, M.M.; Zanini, S.; Sbarbati, A.; Scarpa, A.; et al. Combined inhibition of IL1, CXCR1/2, and TGFβ signaling pathways modulates in-vivo resistance to anti-VEGF treatment. Anti-Cancer Drugs 2016, 27, 29–40. [Google Scholar] [CrossRef]
- Castro, B.A.; Flanigan, P.; Jahangiri, A.; Hoffman, D.; Chen, W.; Kuang, R.; De Lay, M.; Yagnik, G.; Wagner, J.R.; Mascharak, S.; et al. Macrophage migration inhibitory factor downregulation: A novel mechanism of resistance to anti-angiogenic therapy. Oncogene 2017, 36, 3749–3759. [Google Scholar] [CrossRef]
- Liu, T.; Ma, W.; Xu, H.; Huang, M.; Zhang, D.; He, Z.; Zhang, L.; Brem, S.; O’Rourke, D.M.; Gong, Y.; et al. PDGF-mediated mesenchymal transformation renders endothelial resistance to anti-VEGF treatment in glioblastoma. Nat. Commun. 2018, 9, 3439. [Google Scholar] [CrossRef]
- Lièvre, A.; Bachet, J.B.; Le Corre, D.; Boige, V.; Landi, B.; Emile, J.F.; Côté, J.F.; Tomasic, G.; Penna, C.; Ducreux, M.; et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006, 66, 3992–3995. [Google Scholar] [CrossRef]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 2008, 359, 1757–1765. [Google Scholar] [CrossRef]
- De Roock, W.; Claes, B.; Bernasconi, D.; De Schutter, J.; Biesmans, B.; Fountzilas, G.; Kalogeras, K.T.; Kotoula, V.; Papamichael, D.; Laurent-Puig, P.; et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: A retrospective consortium analysis. Lancet Oncol. 2010, 11, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.; Ciombor, K.; Wu, C.; Bekaii-Saab, T.; Strickler, J. Addressing Resistance to Targeted Therapies in Metastatic Colorectal Cancer. Oncology 2021, 35, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Di Nicolantonio, F.; Martini, M.; Molinari, F.; Sartore-Bianchi, A.; Arena, S.; Saletti, P.; De Dosso, S.; Mazzucchelli, L.; Frattini, M.; Siena, S.; et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 5705–5712. [Google Scholar] [CrossRef] [PubMed]
- Martin, V.; Landi, L.; Molinari, F.; Fountzilas, G.; Geva, R.; Riva, A.; Saletti, P.; De Dosso, S.; Spitale, A.; Tejpar, S.; et al. HER2 gene copy number status may influence clinical efficacy to anti-EGFR monoclonal antibodies in metastatic colorectal cancer patients. Br. J. Cancer 2013, 108, 668–675. [Google Scholar] [CrossRef]
- Arena, S.; Bellosillo, B.; Siravegna, G.; Martínez, A.; Cañadas, I.; Lazzari, L.; Ferruz, N.; Russo, M.; Misale, S.; González, I.; et al. Emergence of multiple EGFR extracellular mutations during cetuximab treatment in colorectal cancer. Clin. Cancer Res. 2015, 21, 2157–2166. [Google Scholar] [CrossRef]
- Pietrantonio, F.; Vernieri, C.; Siravegna, G.; Mennitto, A.; Berenato, R.; Perrone, F.; Gloghini, A.; Tamborini, E.; Lonardi, S.; Morano, F.; et al. Heterogeneity of acquired resistance to anti-EGFR monoclonal antibodies in patients with metastatic colorectal cancer. Clin. Cancer Res. 2017, 23, 2414–2422. [Google Scholar] [CrossRef]
- Siravegna, G.; Mussolin, B.; Buscarino, M.; Corti, G.; Cassingena, A.; Crisafulli, G.; Ponzetti, A.; Cremolini, C.; Amatu, A.; Lauricella, C.; et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat. Med. 2015, 21, 795–801. [Google Scholar] [CrossRef]
- Parseghian, C.M.; Loree, J.M.; Morris, V.K.; Liu, X.; Clifton, K.K.; Napolitano, S.; Henry, J.T.; Pereira, A.A.; Vilar, E.; Johnson, B.; et al. Anti-EGFR-resistant clones decay exponentially after progression: Implications for anti-EGFR re-challenge. Ann. Oncol. 2019, 30, 243–249. [Google Scholar] [CrossRef]
- Oddo, D.; Sennott, E.M.; Barault, L.; Valtorta, E.; Arena, S.; Cassingena, A.; Filiciotto, G.; Marzolla, G.; Elez, E.; van Geel, R.M.; et al. Molecular landscape of acquired resistance to targeted therapy combinations in BRAF-mutant colorectal cancer. Cancer Res. 2016, 76, 4504–4515. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Pietrantonio, F.; Lonardi, S.; Mussolin, B.; Rua, F.; Fenocchio, E.; Amatu, A.; Corallo, S.; Manai, C.; Tosi, F.; et al. Phase II study of anti-EGFR rechallenge therapy with panitumumab driven by circulating tumor DNA molecular selection in metastatic colorectal cancer: The CHRONOS trial. J. Clin. Oncol. 2021, 39, 3506. [Google Scholar] [CrossRef]
- Cremolini, C.; Rossini, D.; Dell’Aquila, E.; Lonardi, S.; Conca, E.; Del Re, M.; Busico, A.; Pietrantonio, F.; Danesi, R.; Aprile, G.; et al. Rechallenge for patients with RAS and BRAF wild-type metastatic colorectal cancer with acquired resistance to first-line cetuximab and irinotecan: A phase 2 single-arm clinical trial. JAMA Oncol. 2019, 5, 343–350. [Google Scholar] [CrossRef]
- Strickler, J.; Ou, F.S.; Bekaii-Saab, T.; Parseghian, C.; Cercek, A.; Ng, K.; Sanchez, F.; Bruggeman, S.; Larson, J.; Finley, G.; et al. PULSE: A randomized phase II open-label study of panitumumab rechallenge versus standard therapy after progression on anti-EGFR therapy in patients with RAS wild-type metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2021, 39 (Suppl. 3), TPS143. [Google Scholar] [CrossRef]
- Loupakis, F.; Cremolini, C.; Masi, G.; Lonardi, S.; Zagonel, V.; Salvatore, L.; Cortesi, E.; Tomasello, G.; Ronzoni, M.; Spadi, R.; et al. Initial therapy with FOLFOXIRI and bevacizumab for metastatic colorectal cancer. N. Engl. J. Med. 2014, 371, 1609–1618. [Google Scholar] [CrossRef]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical sequencing defines the genomic landscape of metastatic colorectal cancer. Cancer Cell 2018, 33, 125–136.e3. [Google Scholar] [CrossRef]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, R.B.; Ebi, H.; Turke, A.B.; Coffee, E.M.; Nishino, M.; Cogdill, A.P.; Brown, R.D.; Della Pelle, P.; Dias-Santagata, D.; Hung, K.E.; et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012, 2, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Yao, Z.; Hyman, D.M.; Hechtman, J.F.; Vakiani, E.; Zhao, H.; Su, W.; Wang, L.; Joelson, A.; Cercek, A.; et al. Mechanisms of acquired resistance to BRAF V600E inhibition in colon cancers converge on RAF dimerization and are sensitive to its inhibition. Cancer Res. 2017, 77, 6513–6523. [Google Scholar] [CrossRef]
- Ji, J.; Wang, C.; Fakih, M. Rechallenge with BRAF and anti-EGFR inhibitors in patients with metastatic colorectal cancer harboring BRAFV600E mutation who progressed on cetuximab and encorafenib with or without binimetinib: A case series. Clin. Color. Cancer 2022, 21, 267–271. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Chitkara, A.; Bakhtiar, M.; Sahin, I.H.; Hsu, D.; Zhang, J.; Anamika, F.; Mahnoor, M.; Ahmed, R.; Gholami, S.; Saeed, A. A Meta-Analysis to Assess the Efficacy of HER2-Targeted Treatment Regimens in HER2-Positive Metastatic Colorectal Cancer (mCRC). Curr. Oncol. 2023, 30, 8266–8277. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Siena, S.; Di Bartolomeo, M.; Raghav, K.; Masuishi, T.; Loupakis, F.; Kawakami, H.; Yamaguchi, K.; Nishina, T.; Fakih, M.; Elez, E.; et al. Trastuzumab deruxtecan (DS-8201) in patients with HER2-expressing metastatic colorectal cancer (DESTINY-CRC01): A multicentre, open-label, phase 2 trial. Lancet Oncol. 2021, 22, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Sartore-Bianchi, A.; Trusolino, L.; Martino, C.; Bencardino, K.; Lonardi, S.; Bergamo, F.; Zagonel, V.; Leone, F.; Depetris, I.; Martinelli, E.; et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): A proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 738–746, Erratum in Lancet Oncol. 2016, 17, e420. https://doi.org/10.1016/S1470-2045(16)30463-6. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.; Borad, M.J.; Azad, N.S.; Kurzrock, R.; Abou-Alfa, G.K.; George, B.; Hainsworth, J.; Meric-Bernstam, F.; Swanton, C.; Sweeney, C.J.; et al. Pertuzumab and trastuzumab for HER2-positive, metastatic biliary tract cancer (MyPathway): A multicentre, open-label, phase 2a, multiple basket study. Lancet Oncol. 2021, 22, 1290–1300. [Google Scholar] [CrossRef]
- Passardi, A.; Canale, M.; Valgiusti, M.; Ulivi, P. Immune checkpoints as a target for colorectal cancer treatment. Int. J. Mol. Sci. 2017, 18, 1324. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef]
- Giannakis, M.; Mu, X.J.; Shukla, S.A.; Qian, Z.R.; Cohen, O.; Nishihara, R.; Bahl, S.; Cao, Y.; Amin-Mansour, A.; Yamauchi, M.; et al. Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Rep. 2016, 15, 857–865, Erratum in Cell Rep. 2016, 17, 1206. https://doi.org/10.1016/j.celrep.2016.10.009. [Google Scholar] [CrossRef]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S.; et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015, 5, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Lee, J.J.; Chu, E. Recent advances in the clinical development of immune checkpoint blockade therapy for mismatch repair proficient (pMMR)/non-MSI-H metastatic colorectal cancer. Clin. Color. Cancer 2018, 17, 258–273. [Google Scholar] [CrossRef]
- Ganesh, K.; Stadler, Z.K.; Cercek, A.; Mendelsohn, R.B.; Shia, J.; Segal, N.H.; Diaz, L.A., Jr. Immunotherapy in colorectal cancer: Rationale, challenges and potential. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 361–375. [Google Scholar] [CrossRef]
- Liu, B.; Zhou, H.; Tan, L.; Siu, K.T.H.; Guan, X.Y. Exploring treatment options in cancer: Tumor treatment strategies. Signal Transduct. Target. Ther. 2024, 9, 175. [Google Scholar] [CrossRef] [PubMed]
- van Westrienen, P.E.; de Wit, N.; Toonders, S.; Veenhof, C.; Gerrits, M.; Pisters, M. Effectiveness of a blended multidisciplinary intervention for patients with moderate medically unexplained physical symptoms (PARASOL): A cluster randomized clinical trial. PLoS ONE 2023, 18, e0283162. [Google Scholar] [CrossRef]
- Jardim, D.L.; De Melo Gagliato, D.; Nikanjam, M.; Barkauskas, D.A.; Kurzrock, R. Efficacy and safety of anticancer drug combinations: A meta-analysis of randomized trials with a focus on immunotherapeutics and gene-targeted compounds. OncoImmunology 2020, 9, 1710052. [Google Scholar] [CrossRef] [PubMed]
- Parikh, A.R.; Mojtahed, A.; Schneider, J.L.; Kanter, K.; Van Seventer, E.E.; Fetter, I.J.; Thabet, A.; Fish, M.G.; Teshome, B.; Fosbenner, K.; et al. Serial ctDNA monitoring to predict response to systemic therapy in metastatic gastrointestinal cancers. Clin. Cancer Res. 2020, 26, 1877–1885. [Google Scholar] [CrossRef]
- Azad, N.S.; Posadas, E.M.; Kwitkowski, V.E.; Steinberg, S.M.; Jain, L.; Annunziata, C.M.; Minasian, L.; Sarosy, G.; Kotz, H.L.; Premkumar, A.; et al. Combination targeted therapy with sorafenib and bevacizumab results in enhanced toxicity and antitumor activity. J. Clin. Oncol. 2008, 26, 3709–3714, Erratum in J. Clin. Oncol. 2008, 26, 4363. [Google Scholar] [CrossRef]
- Khan, K.H.; Wong, M.; Rihawi, K.; Bodla, S.; Morganstein, D.; Banerji, U.; Molife, L.R. Hyperglycemia and phosphatidylinositol 3-kinase/protein kinase B/mammalian target of rapamycin (PI3K/AKT/mTOR) inhibitors in phase I trials: Incidence, predictive factors, and management. Oncologist 2016, 21, 855–860. [Google Scholar] [CrossRef]
- Robinson, E.S.; Khankin, E.V.; Karumanchi, S.A.; Humphreys, B.D. Hypertension induced by vascular endothelial growth factor signaling pathway inhibition: Mechanisms and potential use as a biomarker. Semin. Nephrol. 2010, 30, 591–601. [Google Scholar] [CrossRef]
- Perez-Soler, R. Rash as a surrogate marker for efficacy of epidermal growth factor receptor inhibitors in lung cancer. Clin. Lung Cancer 2006, 8 (Suppl. 1), S7–S14. [Google Scholar] [CrossRef]
- Uprety, D.; Mansfield, A.S. Targeting the cardiotoxicity of epidermal growth factor receptor inhibitors. JACC CardioOncology 2020, 2, 11–12. [Google Scholar] [CrossRef]
- Hamilton, M.; Wolf, J.L.; Rusk, J.; Beard, S.E.; Clark, G.M.; Witt, K.; Cagnoni, P.J. Effects of smoking on the pharmacokinetics of erlotinib. Clin. Cancer Res. 2006, 12, 2166–2171. [Google Scholar] [CrossRef]
- Desai, A.; Scheckel, C.; Jensen, C.J.; Orme, J.; Williams, C.; Shah, N.; Leventakos, K.; Adjei, A.A. Trends in prices of drugs used to treat metastatic non-small cell lung cancer in the US from 2015 to 2020. JAMA Netw. Open 2022, 5, e2144923. [Google Scholar] [CrossRef] [PubMed]
- Shih, Y.C.T.; Smieliauskas, F.; Geynisman, D.M.; Kelly, R.J.; Smith, T.J. Trends in the cost and use of targeted cancer therapies for the privately insured nonelderly: 2001 to 2011. J. Clin. Oncol. 2015, 33, 2190–2196. [Google Scholar] [CrossRef]
- Dos-Santos-Silva, I.; Gupta, S.; Orem, J.; Shulman, L.N. Global disparities in access to cancer care. Commun. Med. 2022, 2, 31. [Google Scholar] [CrossRef] [PubMed]
- Meillon-Garcia, L.A.; Demichelis-Gómez, R. Access to therapy for acute myeloid leukemia in the developing world: Barriers and solutions. Curr. Oncol. Rep. 2020, 22, 125. [Google Scholar] [CrossRef] [PubMed]
- Colomer, R.; Mondejar, R.; Romero-Laorden, N.; Alfranca, A.; Sanchez-Madrid, F.; Quintela-Fandino, M. When should we order a next generation sequencing test in a patient with cancer? eClinicalMedicine 2020, 25, 100487. [Google Scholar] [CrossRef]
- Mosele, F.; Remon, J.; Mateo, J.; Westphalen, C.B.; Barlesi, F.; Lolkema, M.P.; Normanno, N.; Scarpa, A.; Robson, M.; Meric-Bernstam, F.; et al. Recommendations for the use of next-generation sequencing (NGS) for patients with metastatic cancers: A report from the ESMO Precision Medicine Working Group. Ann. Oncol. 2020, 31, 1491–1505. [Google Scholar] [CrossRef]
- Nakamura, Y.; Taniguchi, H.; Ikeda, M.; Bando, H.; Kato, K.; Morizane, C.; Esaki, T.; Komatsu, Y.; Kawamoto, Y.; Takahashi, N.; et al. Clinical utility of circulating tumor DNA sequencing in advanced gastrointestinal cancer: SCRUM-Japan GI-SCREEN and GOZILA studies. Nat. Med. 2020, 26, 1859–1864. [Google Scholar] [CrossRef]
- D’Haene, N.; Fontanges, Q.; De Nève, N.; Blanchard, O.; Melendez, B.; Delos, M.; Dehou, M.F.; Maris, C.; Nagy, N.; Rousseau, E.; et al. Clinical application of targeted next-generation sequencing for colorectal cancer patients: A multicentric Belgian experience. Oncotarget 2018, 9, 20761–20768. [Google Scholar] [CrossRef]
- Suzuki, Y.; Ng, S.B.; Chua, C.; Leow, W.Q.; Chng, J.; Liu, S.Y.; Ramnarayanan, K.; Gan, A.; Ho, D.L.; Ten, R.; et al. Multiregion ultra-deep sequencing reveals early intermixing and variable levels of intratumoral heterogeneity in colorectal cancer. Mol. Oncol. 2017, 11, 124–139. [Google Scholar] [CrossRef]
- Giardina, T.; Robinson, C.; Grieu-Iacopetta, F.; Millward, M.; Iacopetta, B.; Spagnolo, D.; Amanuel, B. Implementation of next generation sequencing technology for somatic mutation detection in routine laboratory practice. Pathology 2018, 50, 389–401. [Google Scholar] [CrossRef]
- Lam, M.; Lum, C.; Latham, S.; Tipping Smith, S.; Prenen, H.; Segelov, E. Refractory metastatic colorectal cancer: Current challenges and future prospects. Cancer Manag. Res. 2020, 12, 5819–5830. [Google Scholar] [CrossRef] [PubMed]
- Joshi, D.C.; Sharma, A.; Prasad, S.; Singh, K.; Kumar, M.; Sherawat, K.; Tuli, H.S.; Gupta, M. Novel therapeutic agents in clinical trials: Emerging approaches in cancer therapy. Discov. Oncol. 2024, 15, 342. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Drug(s) | Clinical Indication | Key Clinical Outcomes | Clinical Observations |
|---|---|---|---|---|
| EGFR | Cetuximab, Panitumumab | RAS wild-type; first-/second-line or post chemotherapy | ORR of ~40–50%; reduced effectiveness in right-sided tumors | Benefit is confined to RAS wild-type tumors; intrinsic resistance is common in right-sided cancers |
| BRAF V600E | Encorafenib (+Cetuximab ± Binimetinib) | BRAF V600E-mutant; typically second-line | Improved OS (9.0–9.3 months vs. 5.4 months with SOC); ORR of 20–26% versus 2% with SOC | Median survival around 12 months |
| HER2 | Trastuzumab ± Pertuzumab | HER2-positive; second-line | ORR 9.7% to 35%, with some reports up to 45% using trastuzumab; potential pulmonary toxicity | Dual HER2 blockade more effective than single |
| MEK | Trametinib, Cobimetinib | Later lines, typically in combination | Limited activity as monotherapy; enhanced outcomes in combination therapies | Not approved as monotherapy; benefits are mainly observed in combination regimens |
| Immune Checkpoints | Pembrolizumab, Nivolumab (± Ipilimumab) | MSI-H/dMMR | Marked improvements in response and survival in MSI-H/dMMR tumors; robust benefit in selected patient groups | Minimal benefit in microsatellite stable mCRC; efficacy is confined to MSI-H/dMMR subgroups |
| Mechanism of Action | Medication |
|---|---|
| VEGF Inhibitor | Bevacizumab (Avastin®) |
| Aflibercept (Eylea®, Zaltrap®) | |
| EGFR inhibitor | Cetuximab (Erbitux®) |
| Panitumumab (Vectibix®) | |
| BRAF Inhibitor | Vemurafenib (Zelboraf®) |
| Encorafenib (Braftovi®) | |
| HER2 Inhibitor | Pertuzumab (Perjeta®) |
| Trastuzumab (Herceptin®) | |
| MEK1, MEK2 Inhibitor | Trametinib (Mekinist®) |
| MEK 1 Inhibitor | Cobimetinib (Cotellic®) |
| PD-1 Inhibitor | Pembrolizumab (Keytruda®) |
| Volumab (Opdivo®) |
| Medication | Brand Name | Mechanism of Action | Mechanism of toxicity |
|---|---|---|---|
| Bevacizumab | Avastin® | VEGF inhibitor | 1. Cardiovascular Toxicity: VEGF inhibitors can affect the cardiovascular system by disrupting the normal balance of blood vessel growth and maintenance. By inhibiting VEGF, these medications can lead to hypertension and increase the risk of cardiovascular events such as heart attack or heart failure. |
| Aflibercept | Eylea®, Zaltrap® | VEGF Inhibitor | 2. Impaired Wound Healing: VEGF plays a crucial role in the process of wound healing, as it promotes angiogenesis and the growth of new blood vessels in injured tissues. By inhibiting VEGF, VEGF inhibitors can impair wound healing and increase the risk of delayed healing or complications following surgery. |
| 3. Hemorrhage: VEGF inhibitors can interfere with the formation and maintenance of blood vessels, leading to fragile blood vessels that are more prone to bleeding. This can result in an increased risk of hemorrhage, both externally and internally. | |||
| 4. Proteinuria: VEGF inhibitors can affect the filtration function of the kidneys, leading to an increased excretion of protein in the urine. This occurs due to the disruption of normal blood vessel development and maintenance in the kidneys. | |||
| 5. Gastrointestinal Perforation:. The inhibition of VEGF can weaken the integrity of the gastrointestinal tract, leading to the development of holes or tears in the stomach, intestines, or other parts of the gastrointestinal system. | |||
| Cetuximab | Erbitux® | Targets EGFR | 1. Skin Toxicity: EGFR inhibitors can cause skin-related toxicities, such as rash and dermatitis. This occurs because EGFR is also expressed in the skin, and inhibiting EGFR can disrupt normal skin cell growth and maintenance. |
| Panitumumab | Vectibix® | Targets EGFR | 2. Diarrhea: EGFR inhibitors can affect the gastrointestinal tract and lead to an increased frequency of bowel movements and diarrhea. The exact mechanism of EGFR inhibitor-induced diarrhea is not fully understood, but it is thought to be related to the effects of EGFR inhibition on the gut lining and water absorption. |
| 3. Nail Changes: EGFR inhibitors may cause changes in the nails, including nail discoloration, brittle nails, or nail inflammation (paronychia). These nail changes are generally reversible once the treatment is completed or the dose is adjusted. | |||
| 4. Mucositis: EGFR inhibitors can result in inflammation and ulceration of the mucous membranes, leading to mucositis. This can affect the lining of the mouth, throat, and gastrointestinal tract, causing pain, difficulty swallowing, and mouth sores. | |||
| 5. Ocular Toxicity: EGFR inhibitors may cause ocular toxicities such as dry eyes, conjunctivitis (inflammation of the conjunctiva), and corneal erosion. | |||
| Vemurafenib | Zelboraf® | BRAF Inhibitor | 1. Cutaneous Toxicity: BRAF inhibitors can cause skin-related toxicities, including rash, photosensitivity, and hyperkeratosis (thickening of the outer layer of the skin). These skin toxicities can manifest as dryness, redness, itching, or the development of acneiform eruptions. Regular monitoring of the skin and appropriate skincare measures are important to manage these toxicities. |
| Encorafenib | Braftovi® | BRAF Inhibitor | 2. Pyrexia (Fever): Fever is a common side effect associated with BRAF inhibitors. It is usually low-grade and self-limiting, but occasionally it can be severe and require medical attention. |
| 3. Gastrointestinal Toxicity: BRAF inhibitors can cause gastrointestinal toxicities, such as diarrhea and nausea. Diarrhea can range from mild to severe and may require supportive care and management to prevent dehydration. Nausea and vomiting can also occur. | |||
| 4. Hepatotoxicity: There have been reports of liver toxicity associated with BRAF inhibitors, including elevation of liver enzymes. | |||
| 5. Cardiotoxicity: In some cases, BRAF inhibitors have been associated with cardiotoxic effects, including arrhythmias, left ventricular dysfunction, and cardiomyopathy. | |||
| 6. Photosensitivity: BRAF inhibitors can make the skin more sensitive to sunlight, leading to an increased risk of sunburn. | |||
| Trastuzumab | Herceptin® | HER2 Inhibitor | 1. Cardiotoxicity: HER2 inhibitors can have cardiotoxic effects, including a risk of decreased heart function and heart failure. This occurs because HER2 plays a role in the normal functioning and maintenance of heart cells. |
| Pertuzumab | Perjeta® | HER2 Inhibitor | 2. Infusion Reactions: HER2 inhibitors are typically administered intravenously, and infusion reactions may occur during or shortly after administration. These reactions can include symptoms such as fever, chills, skin rash, itching, shortness of breath, or low blood pressure. |
| 3. Diarrhea: HER2 inhibitors can cause gastrointestinal toxicities, with diarrhea being a common side effect. The severity of diarrhea can range from mild to severe. | |||
| 4. Fatigue: Fatigue or excessive tiredness is a frequent side effect associated with HER2 inhibitors. It can affect a patient’s daily activities and quality of life. | |||
| 5. Hepatotoxicity: In rare cases, HER2 inhibitors may cause liver toxicities, such as elevated liver enzymes. | |||
| 6. Skin and Nail Toxicities: HER2 inhibitors can lead to skin-related toxicities, including rash, dry skin, and changes in the nails. Some patients may experience skin redness, itching, or skin peeling. Nail changes, such as discoloration or brittleness, can also occur. | |||
| Trametinib | Mekinist® | MEK1, MEK2 inhibitor | 1. Dermatological Toxicity: MEK1 inhibitors can cause various dermatological toxicities, including rash, acneiform eruptions, dry skin, and pruritus (itching). These skin-related toxicities are commonly observed and can vary in severity. |
| Cobimetinib | Cotellic® | MEK1 inhibitor | 2. Gastrointestinal Toxicity: MEK1 inhibitors can cause gastrointestinal toxicities, such as diarrhea, nausea, vomiting, and abdominal pain. Diarrhea is a common side effect and can range from mild to severe. |
| 3. Hepatotoxicity: MEK1 inhibitors have been associated with hepatotoxic effects, including elevation of liver enzymes (transaminases) and hepatocellular injury. | |||
| 4. Ocular Toxicity: MEK1 inhibitors can cause ocular toxicities, including dry eyes, blurred vision, and ocular inflammation. | |||
| 5. Cardiovascular Toxicity: In some cases, MEK1 inhibitors may lead to cardiovascular toxicities, including cardiomyopathy and prolongation of the QT interval. | |||
| 6. Interstitial Lung Disease: Rarely, MEK1 inhibitors have been associated with interstitial lung disease, which is characterized by inflammation and scarring of lung tissue. Symptoms may include shortness of breath, cough, and fever. | |||
| Pembrolizumab | Keytruda® | PD-1 Inhibitor | 1. Immune-Related Adverse Events: Anti-PD-1 inhibitors can cause immune-related adverse events, which occur due to the activation of the immune system. These adverse events can affect various organs and systems in the body. Common irAEs are listed subsequently. |
| Nivolumab | Opdivo® | PD-1 Inhibitor | 2. Skin Toxicity: Skin toxicities can include rash, itching, and blistering. More severe reactions such as Stevens-Johnson syndrome or toxic epidermal necrolysis can occur but are rare. |
| 3. Gastrointestinal Toxicity: Gastrointestinal toxicities can manifest as diarrhea, colitis, or hepatitis. Symptoms may include abdominal pain, diarrhea with or without blood, or jaundice. | |||
| 4. Endocrine Toxicity: Endocrine toxicities can result in the dysfunction of various glands in the body, such as the thyroid, pituitary, or adrenal glands. This can lead to conditions like hypothyroidism, hyperthyroidism, adrenal insufficiency, or hypophysitis. | |||
| 5. Pneumonitis: Pneumonitis is inflammation of the lungs, which can cause symptoms such as cough, shortness of breath, and chest pain. | |||
| 6. Nephritis: Nephritis refers to inflammation of the kidneys, which can cause kidney dysfunction and abnormal urine tests. | |||
| 7. Fatigue: Fatigue or excessive tiredness is a common side effect associated with anti-PD-1 inhibitors. It can impact a patient’s daily activities and quality of life. | |||
| 8. Infusion Reactions: Infusion reactions may occur during or shortly after the administration of anti-PD-1 inhibitors. These reactions can include symptoms such as fever, chills, itching, rash, or low blood pressure. | |||
| 9. Autoimmune Disorders: Anti-PD-1 inhibitors can trigger or exacerbate pre-existing autoimmune disorders or lead to the development of new autoimmune conditions. These can include conditions like rheumatoid arthritis, autoimmune thyroiditis, or type 1 diabetes. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Hage, M.; Su, Z.; Linnebacher, M. Addressing Challenges in Targeted Therapy for Metastatic Colorectal Cancer. Cancers 2025, 17, 1098. https://doi.org/10.3390/cancers17071098
El Hage M, Su Z, Linnebacher M. Addressing Challenges in Targeted Therapy for Metastatic Colorectal Cancer. Cancers. 2025; 17(7):1098. https://doi.org/10.3390/cancers17071098
Chicago/Turabian StyleEl Hage, Maria, Zhaoran Su, and Michael Linnebacher. 2025. "Addressing Challenges in Targeted Therapy for Metastatic Colorectal Cancer" Cancers 17, no. 7: 1098. https://doi.org/10.3390/cancers17071098
APA StyleEl Hage, M., Su, Z., & Linnebacher, M. (2025). Addressing Challenges in Targeted Therapy for Metastatic Colorectal Cancer. Cancers, 17(7), 1098. https://doi.org/10.3390/cancers17071098