
Pharmacogenomics in Solid Tumors: A Comprehensive Review of Genetic Variability and Its Clinical Implications


Simple Summary
Abstract
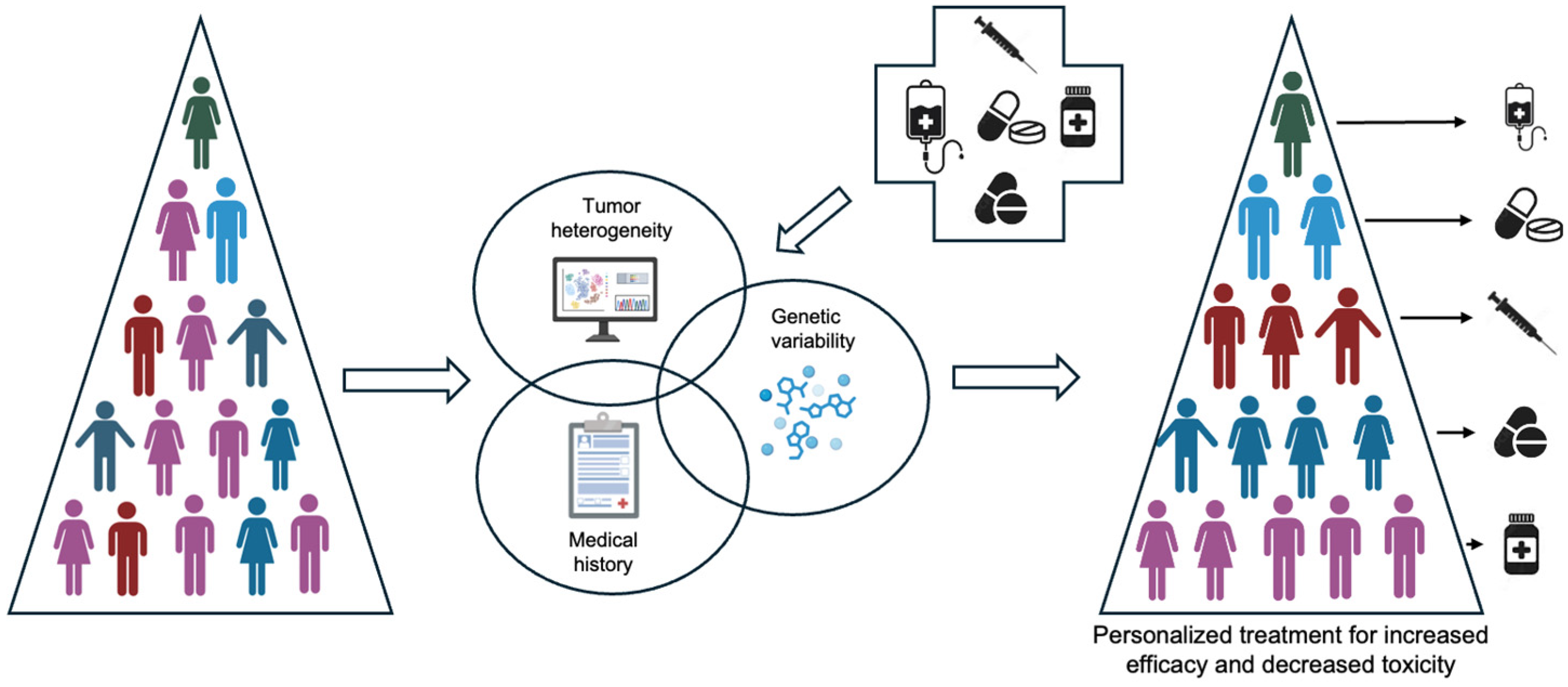
1. Introduction
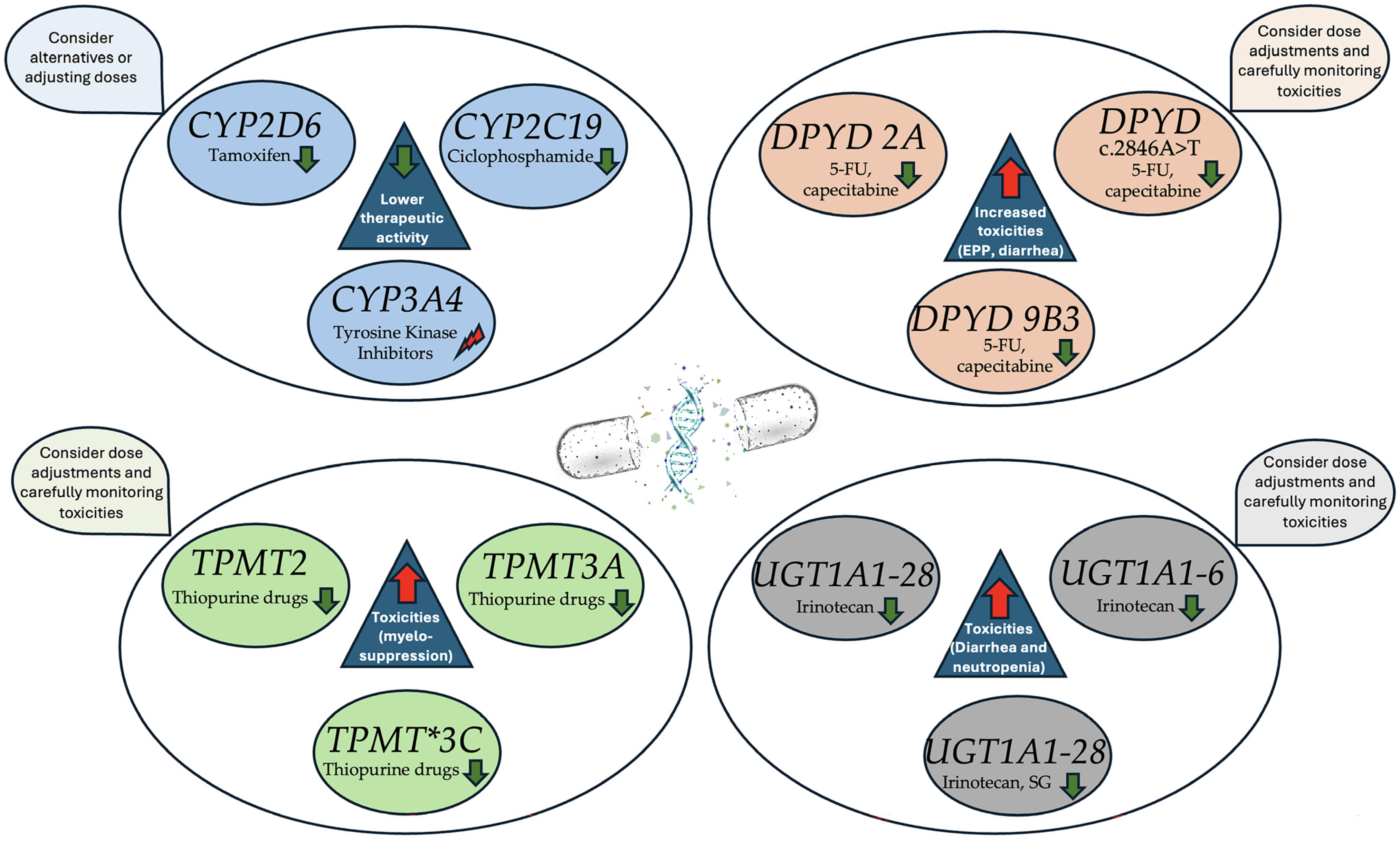
2. Key Pharmacogenomic Genes with Clinical Implications
2.1. Cytochrome P450 Enzymes
2.1.1. CYP2D6 and Tamoxifen Metabolism
2.1.2. CYP2C19 and Cyclophosphamide Activation
2.1.3. CYP3A4 and Tyrosine Kinase Inhibitors (TKIs)
2.2. DPYD (Dihydropyrimidine Dehydrogenase)
2.2.1. DPYD Function and the Importance of Metabolism in Fluoropyrimidines
2.2.2. DPYD Polymorphisms and Their Clinical Implications
2.3. Thiopurine Methyl Transferase (TPMT)
2.3.1. TPMT Genetic Variability and Clinical Implications
2.3.2. TPMT Testing in Clinical Practice
2.3.3. Beyond Thiopurines: TPMT and Other Therapeutics
2.4. UGT1A1 (Uridine Diphosphate Glucuronosyltransferase 1A1)
2.4.1. UGT1A1 and Irinotecan Metabolism
2.4.2. Clinical Implications of UGT1A1 Polymorphisms in Irinotecan Therapy
2.4.3. Beyond Irinotecan: UGT1A1 in Other Cancer Treatments
3. Ongoing Research and Future Directions
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Marcu, L.G.; Marcu, D.C. Pharmacogenomics and Big Data in medical oncology: Developments and challenges. Ther. Adv. Med. Oncol. 2024, 16, 17588359241287658. [Google Scholar] [PubMed]
- Rashid, N.; Koh, H.A.; Baca, H.C.; Lin, K.J.; Malecha, S.E.; Masaquel, A. Breast Cancer-Targets and Therapy Dovepress Economic burden related to chemotherapy-related adverse events in patients with metastatic breast cancer in an integrated health care system. Breast Cancer Targets Ther. 2016, 8, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Saxena, A.; Rubens, M.; Ramamoorthy, V.; Tonse, R.; Veledar, E.; McGranaghan, P.; Sundil, S.; Chuong, M.D.; Hall, M.D.; Odia, Y.; et al. Hospitalization rates for complications due to systemic therapy in the United States. Sci. Rep. 2021, 11, 7385. [Google Scholar] [CrossRef]
- Gilani, B.; Cassagnol, M. StatPearls. StatPearls Publishing; 2023. Biochemistry, Cytochrome P450. Available online: https://www.ncbi.nlm.nih.gov/books/NBK557698/ (accessed on 14 February 2025).
- Weng, L.; Zhang, L.; Peng, Y.; Huang, R.S. Pharmacogenetics and pharmacogenomics: A bridge to individualized cancer therapy. Pharmacogenomics 2013, 14, 315–324. [Google Scholar] [CrossRef]
- Lamb, D.C.; Waterman, M.R.; Kelly, S.L.; Guengerich, F.P. Cytochromes P450 and drug discovery. Curr. Opin. Biotechnol. 2007, 18, 504–512. [Google Scholar] [CrossRef]
- Taylor, C.; Crosby, I.; Yip, V.; Maguire, P.; Pirmohamed, M.; Turner, R.M. A Review of the Important Role of CYP2D6 in Pharmacogenomics. Genes 2020, 11, 1295. [Google Scholar] [CrossRef]
- Gaedigk, A. Complexities of CYP2D6 gene analysis and interpretation. Int. Rev. Psychiatry 2013, 25, 534–553. [Google Scholar] [CrossRef]
- Hicks, J.K.; Bishop, J.R.; Sangkuhl, K.; Müller, D.J.; Ji, Y.; Leckband, S.G.; Leeder, J.S.; Graham, R.L.; Chiulli, D.L.; Llerena, A.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2D6 and CYP2C19 Genotypes and Dosing of Selective Serotonin Reuptake Inhibitors. Clin. Pharmacol. Ther. 2015, 98, 127–134. [Google Scholar] [CrossRef]
- Beoris, M.; Wilson, J.A.; Garces, J.A.; Lukowiak, A.A. CYP2D6 copy number distribution in the US population. Pharmacogenet. Genom. 2016, 26, 96–99. [Google Scholar] [CrossRef]
- Fleeman, N.; Dundar, Y.; Dickson, R.; Jorgensen, A.; Pushpakom, S.; McLeod, C.; Pirmohamed, M.; Walley, T. Cytochrome P450 testing for prescribing antipsychotics in adults with schizophrenia: Systematic review and meta-analyses. Pharm. J. 2010, 11, 1–14. [Google Scholar] [CrossRef]
- Ferraldeschi, R.; Newman, W.G. The Impact of CYP2D6 Genotyping on Tamoxifen Treatment. Pharmaceuticals 2010, 3, 1122–1138. [Google Scholar] [CrossRef]
- Goetz, M.P.; Suman, V.J.; Hoskin, T.L.; Gnant, M.; Filipits, M.; Safgren, S.L.; Kuffel, M.; Jakesz, R.; Rudas, M.; Greil, R.; et al. CYP2D6 metabolism and patient outcome in the Austrian breast and colorectal cancer study group trial (ABCSG) 8. Clin. Cancer Res. 2013, 19, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Newman, W.G.; Hadfield, K.D.; Latif, A.; Roberts, S.A.; Shenton, A.; McHague, C.; Lalloo, F.; Howell, S.; Evans, D.G. Impaired tamoxifen metabolism reduces survival in familial breast cancer patients. Clin. Cancer Res. 2008, 14, 5913–5918. [Google Scholar] [CrossRef] [PubMed]
- E Abraham, J.; Maranian, M.J.; E Driver, K.; Platte, R.; Kalmyrzaev, B.; Baynes, C.; Luccarini, C.; Shah, M.; Ingle, S.; Greenberg, D.; et al. CYP2D6 gene variants: Association with breast cancer specific survival in a cohort of breast cancer patients from the United Kingdom treated with adjuvant tamoxifen. Breast Cancer Res. 2010, 12, R64. [Google Scholar] [CrossRef] [PubMed]
- Lyon, E.; Foster, J.G.; Palomaki, G.E.; Pratt, V.M.; Reynolds, K.; Sábato, M.F.; Scott, S.A.; Vitazka, P. Laboratory testing of CYP2D6 alleles in relation to tamoxifen therapy. Anesthesia Analg. 2012, 14, 990–1000. [Google Scholar] [CrossRef]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef]
- Pratt, V.M.; Cavallari, L.H.; Del Tredici, A.L.; Gaedigk, A.; Hachad, H.; Ji, Y.; Kalman, L.V.; Ly, R.C.; Moyer, A.M.; Scott, S.A.; et al. Recommendations for Clinical CYP2D6 Genotyping Allele Selection: A Joint Consensus Recommendation of the Association for Molecular Pathology, College of American Pathologists, Dutch Pharmacogenetics Working Group of the Royal Dutch Pharmacists Association, and the European Society for Pharmacogenomics and Personalized Therapy. J. Mol. Diagn. 2021, 23, 1047–1064. [Google Scholar]
- Goetz, M.P.; Sangkuhl, K.; Guchelaar, H.J.; Schwab, M.; Province, M.; Whirl-Carrillo, M.; Symmans, W.F.; McLeod, H.L.; Ratain, M.J.; Zembutsu, H.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2D6 and Tamoxifen Therapy. Clin. Pharmacol. Ther. 2018, 103, 770–777. [Google Scholar] [CrossRef]
- Helsby, N.A.; Yong, M.; Van Kan, M.; De Zoysa, J.R.; Burns, K.E. The importance of both CYP2C19 and CYP2B6 germline variations in cyclophosphamide pharmacokinetics and clinical outcomes. Br. J. Clin. Pharmacol. 2019, 85, 1925–1934. [Google Scholar] [CrossRef]
- Ekhart, C.; Doodeman, V.D.; Rodenhuis, S.; Smits, P.H.M.; Beijnen, J.H.; Huitema, A.D.R. Influence of polymorphisms of drug metabolizing enzymes (CYP2B6, CYP2C9, CYP2C19, CYP3A4, CYP3A5, GSTA1, GSTP1, ALDH1A1 and ALDH3A1) on the pharmacokinetics of cyclophosphamide and 4-hydroxycyclophosphamide. Pharmacogenet. Genom. 2008, 18, 515–523. [Google Scholar] [CrossRef]
- Teo, Y.L.; Ho, H.K.; Chan, A. Metabolism-related pharmacokinetic drug-drug interactions with tyrosine kinase inhibitors: Current understanding, challenges and recommendations. Br. J. Clin. Pharmacol. 2015, 79, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Mikus, G.; Foerster, K.I. Role of CYP3A4 in kinase inhibitor metabolism and assessment of CYP3A4 activity. Transl. Cancer Res. 2017, 6, S1592–S1599. [Google Scholar] [CrossRef]
- Lamba, J.K.; Lin, Y.S.; Schuetz, E.G.; Thummel, K.E. Genetic contribution to variable human CYP3A-mediated metabolism. Adv. Drug Deliv. Rev. 2002, 54, 1271–1294. [Google Scholar] [CrossRef] [PubMed]
- Verougstraete, N.; Stove, V.; Verstraete, A.G.; Stove, C.P. Therapeutic Drug Monitoring of Tyrosine Kinase Inhibitors Using Dried Blood Microsamples. Front. Oncol. 2022, 12, 821807. [Google Scholar] [CrossRef]
- Van Kuilenburg, A.B.P.; Meinsma, R.; Zoetekouw, L.; Van Gennip, A.H. High prevalence of the IVS14 + 1G>A mutation in the dihydropyrimidine dehydrogenase gene of patients with severe 5-fluorouracil-associated toxicity. Pharmacogenetics 2002, 12, 555–558. [Google Scholar] [CrossRef]
- Amstutz, U.; Henricks, L.M.; Offer, S.M.; Barbarino, J.; Schellens, J.H.M.; Swen, J.J.; Klein, T.E.; McLeod, H.L.; Caudle, K.E.; Diasio, R.B.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for Dihydropyrimidine Dehydrogenase Genotype and Fluoropyrimidine Dosing: 2017 Update HHS Public Access. Clin. Pharmacol. Ther. 2018, 103, 210–216. [Google Scholar] [CrossRef]
- Etienne-Grimaldi, M.-C.; Boyer, J.-C.; Beroud, C.; Mbatchi, L.; van Kuilenburg, A.; Bobin-Dubigeon, C.; Thomas, F.; Chatelut, E.; Merlin, J.-L.; Pinguet, F.; et al. New advances in DPYD genotype and risk of severe toxicity under capecitabine. PLoS ONE 2017, 12, e0175998. [Google Scholar] [CrossRef]
- Gross, E.; Busse, B.; Riemenschneider, M.; Neubauer, S.; Seck, K.; Klein, H.-G.; Kiechle, M.; Lordick, F.; Meindl, A. Strong Association of a Common Dihydropyrimidine Dehydrogenase Gene Polymorphism with Fluoropyrimidine-Related Toxicity in Cancer Patients. PLoS ONE 2008, 3, e4003. [Google Scholar] [CrossRef]
- Diasio, R.B.; Johnson, M.R. Dihydropyrimidine Dehydrogenase: Its Role in 5-Fluorouracil Clinical Toxicity and Tumor Resistance. Clin. Cancer Res. 1999, 5, 2672–2673. [Google Scholar]
- de With, M.; Sadlon, A.; Cecchin, E.; Haufroid, V.; Thomas, F.; Joerger, M.; van Schaik, R.; Mathijssen, R.; Largiadèr, C. Implementation of dihydropyrimidine dehydrogenase deficiency testing in Europe. ESMO Open 2023, 8, 101197. [Google Scholar] [CrossRef]
- Weinshilboum1, R.M.; Sladek, S.L. Mercaptopurine Pharmacogenetics: Monogenic Inheritance of Erythrocyte Thiopurine Methyltransferase Activity. Am. J. Hum. Genet. 1980, 32, 651–662. [Google Scholar]
- Relling, M.V.; Schwab, M.; Whirl-Carrillo, M.; Suarez-Kurtz, G.; Pui, C.H.; Stein, C.M.; Moyer, A.M.; Evans, W.E.; Klein, T.E.; Antillon-Klussmann, F.G.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for thiopurine dosing based on TPMT and NUDT15 genotypes: 2018 update. Clin. Pharmacol. Ther. 2019, 105, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- McLeod, H.L.; Siva, C. The thiopurine S-methyltransferase gene locus-implications for clinical pharmacogenomics. Pharmacogenomics 2002, 3, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Lennard, L. The clinical pharmacology of 6-mercaptopurine. Eur. J. Clin. Pharmacol. 1992, 43, 329–339. [Google Scholar] [CrossRef]
- Wang, L.; Weinshilboum, R. Thiopurine S-methyltransferase pharmacogenetics: Insights, challenges and future directions. Oncogene 2006, 25, 1629–1638. [Google Scholar] [CrossRef]
- Ando, Y.; Saka, H.; Ando, M.; Sawa, T.; Muro, K.; Ueoka, H.; Yokoyama, A.; Saitoh, S.; Shimokata, K.; Hasegawa, Y. Polymorphisms of UDP-glucuronosyltransferase gene and irinotecan toxicity: A pharmacogenetic analysis. Cancer Res. 2000, 60, 6921–6926. [Google Scholar]
- Marsh, S.; Hoskins, J.M. Irinotecan pharmacogenomics. Pharmacogenomics 2010, 11, 1003–1010. [Google Scholar] [CrossRef]
- Innocenti, F.; Kroetz, D.L.; Schuetz, E.; Dolan, M.E.; Ramírez, J.; Relling, M.; Chen, P.; Das, S.; Rosner, G.L.; Ratain, M.J. Comprehensive pharmacogenetic analysis of irinotecan neutropenia and pharmacokinetics. J. Clin. Oncol. 2009, 27, 2604–2614. [Google Scholar] [CrossRef]
- Hoskins, J.M.; McLeod, H.L. UGT1A and irinotecan toxicity: Keeping it in the family. J. Clin. Oncol. 2009, 27, 2419–2421. [Google Scholar] [CrossRef]
- Hoskins, J.M.; Marcuello, E.; Altes, A.; Marsh, S.; Maxwell, T.; Van Booven, D.J.; Pare, L.; Culverhouse, R.; McLeod, H.L.; Baiget, M. Irinotecan pharmacogenetics: Influence of pharmacodynamic genes. Clin. Cancer Res. 2008, 14, 1788–1796. [Google Scholar] [CrossRef]
- Czejka, M.; Johannes, S.; Katharina, H. Pharmacokinetics and Metabolism of Irinotecan Combined with Capecitabine in Patients with Advanced Colorectal Cancer. Anticancer Res. 2005, 25, 2985–2990. [Google Scholar]
- Klotz, U.; Schwab, M.; Treiber, G. CYP2C19 polymorphism and proton pump inhibitors. Basic Clin. Pharmacol. Toxicol. 2004, 95, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Guillemette, C. Pharmacogenomics of human UDP-glucuronosyltransferase enzymes. Pharmacogenom. J. 2003, 3, 136–158. [Google Scholar] [CrossRef] [PubMed]
- Rugo, H.S.; Tolaney, S.M.; Loirat, D.; Punie, K.; Bardia, A.; Hurvitz, S.A.; O’shaughnessy, J.; Cortés, J.; Diéras, V.; Carey, L.A.; et al. Safety analyses from the phase 3 ASCENT trial of sacituzumab govitecan in metastatic triple-negative breast cancer. NPJ Breast Cancer 2022, 8, 98. [Google Scholar] [CrossRef]
- Ando, M.; Ando, Y.; Sekido, Y.; Ando, M.; Shimokata, K.; Hasegawa, Y. Genetic Polymorphisms of the UDP-Glucuronosyltransferase 1A7 Gene and Irinotecan Toxicity in Japanese Cancer Patients. Jpn. J. Cancer Res. 2002, 93, 591–597. [Google Scholar] [CrossRef]
- Benson, A.B.; Venook, A.P.; Adam, M.; Chang, G.J.; Chen, Y.J.; Ciombor, K.K.; Cohen, S.A.; Cooper, H.S.; Deming, D.; Garrido-Laguna, I.; et al. NCCN Guidelines Version 1.2025 Colon Cancer Continue NCCN Guidelines Panel Disclosures. Available online: https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1428 (accessed on 12 February 2025).
- Mathijssen, R.H.J.; Marsh, S.; O Karlsson, M.; Xie, R.; Baker, S.D.; Verweij, J.; Sparreboom, A.; McLeod, H.L. Irinotecan Pathway Genotype Analysis to Predict Pharmacokinetics 1. Clin. Cancer Res. 2003, 9, 3246–3253. [Google Scholar]
- Ruzzo, A.; Graziano, F.; Kawakami, K.; Watanabe, G.; Santini, D.; Catalano, V.; Bisonni, R.; Canestrari, E.; Ficarelli, R.; Menichetti, E.T.; et al. Pharmacogenetic profiling and clinical outcome of patients with advanced gastric cancer treated with palliative chemotherapy. J. Clin. Oncol. 2006, 24, 1883–1891. [Google Scholar] [CrossRef]
- Rabaan, A.A.; AlSaihati, H.; Bukhamsin, R.; Bakhrebah, M.A.; Nassar, M.S.; Alsaleh, A.A.; Alhashem, Y.N.; Bukhamseen, A.Y.; Al-Ruhimy, K.; Alotaibi, M.; et al. Application of CRISPR/Cas9 Technology in Cancer Treatment: A Future Direction. Curr. Oncol. 2023, 30, 1954–1976. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Gene (Enzyme) | Drug Affected | Cancer Type | Polymorphism | Clinical Implications | Recommendations |
|---|---|---|---|---|---|
| CYP2D6 | Tamoxifen | Breast cancer | Poor metabolizers | Reduced conversion of tamoxifen to active metabolite (endoxifen), leading to decreased efficacy | Consider alternative therapy or increased dose of tamoxifen. |
| CYP2C19 | Cyclophosphamide | Sarcoma, lymphoma, breast cancer | Poor metabolizers | Reduced activation of cyclophosphamide, potentially leading to lower therapeutic efficacy. | Adjust dosing or use alternative chemotherapeutic agents. |
| CYP3A4 | Tyrosine kinase inhibitors (Imatinib, gefitinib) | GIST, lung cancer, sarcoma | Polymorphisms affecting activity | Altered drug metabolism (either enhanced clearance or toxicity due to poor metabolism). | Monitor drug levels closely; consider dose adjustments. |
| Drug Affected | Polymorphism | Clinical Implications | Recommendations |
|---|---|---|---|
| 5-Fluorouracil (5-FU), Capecitabine | DPYD 2A (c.1905+1G>A) | Reduced enzyme activity, leading to accumulation of 5-FU and severe toxicities (myelosuppression, GI toxicity, neurotoxicity). | Reduce starting dose or consider alternative therapies. |
| 5-Fluorouracil (5-FU), Capecitabine | c.2846A>T | Moderate reduction in enzyme activity, associated with increased toxicity risk. | Adjust dose based on genotype or consider alternative treatments. |
| 5-Fluorouracil (5-FU), Capecitabine | DPYD 9B3 (c.1679T>G) | Partial deficiency in enzyme activity, resulting in increased risk of severe toxicity. | Dose reduction and careful monitoring for toxicity. |
| Drug Affected | Polymorphism | Clinical Implications | Recommendations |
|---|---|---|---|
| Thiopurine drugs | TPMT2, TPMT3A, TPMT*3C | Reduced TPMT enzymatic activity, leading to the accumulation of toxic thioguanine TGNs, increasing a the risk of developing severe myelosuppression | Dose reductions of up to 90% for poor metabolizers, and moderate dose reduction and careful monitoring for intermediate metabolizers |
| Drug Affected | Polymorphism | Clinical Implications | Recommendations |
|---|---|---|---|
| Irinotecan | UGT1A1 28 (7/7) | Reduced enzyme activity, leading to impaired SN-38 glucuronidation, increased risk of severe neutropenia, and diarrhea. | Reduce irinotecan dose by 30–50%; monitor closely for toxicity. |
| Irinotecan | UGT1A1 28 (6/7) | Intermediate enzyme activity, moderate risk of toxicity (neutropenia, diarrhea). | Consider moderate dose reduction or close monitoring. |
| Irinotecan | UGT1A1 6 (Asian populations) | Similar to UGT1A1 28, leading to reduced metabolism and increased toxicity. | Adjust dose based on genotype, particularly in Asian populations. |
| Sacituzumab govitecan | UGT1A1 28 | Increased risk of neutropenia and diarrhea. | Monitor closely and consider dose reduction. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Bayona, R.; Catalán, C.; Cobos, M.A.; Bergamino, M. Pharmacogenomics in Solid Tumors: A Comprehensive Review of Genetic Variability and Its Clinical Implications. Cancers 2025, 17, 913. https://doi.org/10.3390/cancers17060913
Sánchez-Bayona R, Catalán C, Cobos MA, Bergamino M. Pharmacogenomics in Solid Tumors: A Comprehensive Review of Genetic Variability and Its Clinical Implications. Cancers. 2025; 17(6):913. https://doi.org/10.3390/cancers17060913
Chicago/Turabian StyleSánchez-Bayona, Rodrigo, Camila Catalán, Maria Angeles Cobos, and Milana Bergamino. 2025. "Pharmacogenomics in Solid Tumors: A Comprehensive Review of Genetic Variability and Its Clinical Implications" Cancers 17, no. 6: 913. https://doi.org/10.3390/cancers17060913
APA StyleSánchez-Bayona, R., Catalán, C., Cobos, M. A., & Bergamino, M. (2025). Pharmacogenomics in Solid Tumors: A Comprehensive Review of Genetic Variability and Its Clinical Implications. Cancers, 17(6), 913. https://doi.org/10.3390/cancers17060913