
IL-10-Directed Cancer Immunotherapy: Preclinical Advances, Clinical Insights, and Future Perspectives

 , ,
, ,  and
and 
Simple Summary
Abstract

1. Introduction
2. Search Strategy
3. IL-10: A Historical Journey as a Tumor-Suppressor Cytokine
4. Preclinical Strategies for Bioengineered IL-10 and Innovative Delivery Systems in Cancer Therapy

{kind=link}
| Strategy | Design and Therapeutic Agent(s) | Key Findings | Ref. | |
|---|---|---|---|---|
| IL-10/Fc Fusion protein | 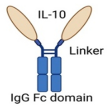 | Fusion of IL-10 with IgG Fc domain |
| [65] |
| Bispecific CmAb(IL-10)2 Fusion Protein | 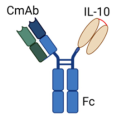 | Fusion of Anti-EGFR antibody (Cetuximab “CmAb”) with IL-10 dimer to engage EGFR and IL-10R in EGFR+ tumors |
| [66] |
| BF10 Fusion Protein | 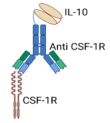 | Bispecific Anti-CSF-IR/IL-10 Fusion Protein, combining IL-10 with anti-CSF-1R- antibody |
| [67] |
| IL-10 variants for IL-10Rβ | 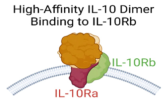 | Bioengineered IL-10 variant with increased binding affinity for IL-10Rβ |
| [68] |
| Oncolytic Viruses (OVs) Based Strategy |  | An oncolytic vaccina virus (OVV) armed with armed with IL-10 gene (OVV-IL10) | In pancreatic cancer models, OVV-IL-10 showed
| [69] |
| Nanoparticles & Scaffolds Based Systems |  | Conjugation of IL-10 into nanoparticles or scaffolds Delivery systems |
| [70,71] |
4.1. Fusion Proteins and Immunocytokine-Based Strategies
4.1.1. Il-10/Fc Fusion Protein Approach
4.1.2. Bispecific CmAb-(IL-10)2 Fusion Protein: Targeting EGFR+ Tumors
4.1.3. Bifunctional Anti-CSF-1R/IL-10 Fusion Protein: Targeting TAMs-Enriched Tumors
4.2. Bioengineering of IL-10 Variants with Enhanced Affinity for IL-10Rβ
4.3. Oncolytic Viruses and Nanoparticle-Based Carriers for Il-10 and Other Biologically Active Il-10 Isoforms
5. Clinical Trials of Il-10 Directed in Cancer Immunotherapy
| Trial (NCT) and [Ref.] | Phase, Patients, and Design | Therapeutic Agent(s) | Key Outcomes | Interpretation |
|---|---|---|---|---|
| IVY (NCT02009449) [115] | Phase 1; patients with various advanced solid tumors | PEG alone | An acceptable safety and early antitumor activity | Warranting further evaluation |
| IVY (NCT02009449) [116] | Phase 1b; patients with various solid tumors, including patients with treatment-refractory NSCLC and RCC | PEG + Anti-PD1 therapy (PEMB or NIVO) | ORR: 43% (NSCLC) and 40% (RCC) | Favorable anti-tumor activity between PEG and Anti-PD1 therapy |
| IVY (NCT02009449) [117] | Phase 1b; mRCC patients | PEG + NIVO vs. PEG + Pazopanib | ORR: 43% (PEG/NIVO) vs. 33% (PEG/Pazopanib) | PEG/NIVO showed better activity and tolerable safety profile |
| Ovarian Cancer [118] | Phase II; patients with metastatic Ovarian cancer | PEG alone vs. PEG + platinum-taxane | PFS:2.4 months (PEG) vs. 5.2 months (co-therapy) | Warranting further evaluation |
| CYPRESS 1 and 2 (NCT03382899 and (NCT03382912) [119] | Phase II; mNSCLC patients divided into: CYPRESS 1 (PD-L1 TPS ≥ 50%) and CYPRESS 2 (PD-L1 TPS 0–49%) | PEG + PEMB (or NIVO) vs. PEMB (or NIVO) alone | CYPRESS 1: ORR (47% vs. 44%) and PFS (6.3 vs. 6.1 months) and CYPRESS 2: ORR (15% vs. 12%), PFS (1.9 vs. 1.9 months), and OS (6.7 vs. 10.7 months) | PEG + Anti-PD1 therapy (PEMB or NIVO) did not offer benefits over Anti-PD1 alone |
| SEQUOIA (NCT02923921) [120,121] | Phase III; gemcitabine-refractory PDAC patients | PEG + FOLFOX vs. FOLFOX alone | ORR: 4.6% vs. 5.6%; PFS: 2.1 vs. 2.1 months; and OS: 5.8 vs. 6.3 months | PEG did not improve FOLFOX efficacy in advanced gemcitabine-refractory PDAC patients |
5.1. Early-Phase Dose Escalation Study of Pegilodecakin Monotherapy
5.2. Evaluation of Pegilodecakin in Combination with Anti-PD-1 Therapy
5.3. Evaluation of Pegilodecakin as an Add-On Therapy in CYPRESS and SEQUOIA Clinical Trials
6. Conclusion
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lim, Y.Y.; Chin, Y.M.; Tai, M.C.; Fani, S.; Chang, K.M.; Ong, T.C.; Bee, P.C.; Gan, G.G.; Ng, C.C. Analysis of interleukin-10 promoter single nucleotide polymorphisms and risk of non-Hodgkin lymphoma in a Malaysian population. Leuk. Lymphoma 2015, 56, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; Cheng, G. Role of interleukin 10 transcriptional regulation in inflammation and autoimmune disease. Crit. Rev. Immunol. 2012, 32, 23–63. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Wang, X.; Xiao, F.; Ma, K.; Liu, L.; Wang, X.; Xu, D.; Wang, F.; Shi, X.; Liu, D.; et al. IL-10-producing regulatory B cells restrain the T follicular helper cell response in primary Sjogren’s syndrome. Cell Mol. Immunol. 2019, 16, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Li, B.; Sun, A.; Guo, F. Interleukin-10 Family Cytokines Immunobiology and Structure. Adv. Exp. Med. Biol. 2019, 1172, 79–96. [Google Scholar] [CrossRef]
- Itakura, E.; Huang, R.R.; Wen, D.R.; Paul, E.; Wunsch, P.H.; Cochran, A.J. IL-10 expression by primary tumor cells correlates with melanoma progression from radial to vertical growth phase and development of metastatic competence. Mod. Pathol. 2011, 24, 801–809. [Google Scholar] [CrossRef]
- Ozaki, K.; Leonard, W.J. Cytokine and cytokine receptor pleiotropy and redundancy. J. Biol. Chem. 2002, 277, 29355–29358. [Google Scholar] [CrossRef]
- Pestka, S.; Krause, C.D.; Sarkar, D.; Walter, M.R.; Shi, Y.; Fisher, P.B. Interleukin-10 and related cytokines and receptors. Annu. Rev. Immunol. 2004, 22, 929–979. [Google Scholar] [CrossRef]
- Yoon, S.I.; Logsdon, N.J.; Sheikh, F.; Donnelly, R.P.; Walter, M.R. Conformational changes mediate interleukin-10 receptor 2 (IL-10R2) binding to IL-10 and assembly of the signaling complex. J. Biol. Chem. 2006, 281, 35088–35096. [Google Scholar] [CrossRef]
- Murray, P.J. Understanding and exploiting the endogenous interleukin-10/STAT3-mediated anti-inflammatory response. Curr. Opin. Pharmacol. 2006, 6, 379–386. [Google Scholar] [CrossRef]
- Murray, P.J. The JAK-STAT signaling pathway: Input and output integration. J. Immunol. 2007, 178, 2623–2629. [Google Scholar] [CrossRef]
- Hutchins, A.P.; Diez, D.; Miranda-Saavedra, D. The IL-10/STAT3-mediated anti-inflammatory response: Recent developments and future challenges. Brief. Funct. Genom. 2013, 12, 489–498. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Schwartz, D.M.; Villarino, A.V.; Gadina, M.; McInnes, I.B.; Laurence, A. The JAK-STAT pathway: Impact on human disease and therapeutic intervention. Annu. Rev. Med. 2015, 66, 311–328. [Google Scholar] [CrossRef]
- Shouval, D.S.; Ouahed, J.; Biswas, A.; Goettel, J.A.; Horwitz, B.H.; Klein, C.; Muise, A.M.; Snapper, S.B. Interleukin 10 receptor signaling: Master regulator of intestinal mucosal homeostasis in mice and humans. Adv. Immunol. 2014, 122, 177–210. [Google Scholar] [CrossRef]
- Walter, M.R. The molecular basis of IL-10 function: From receptor structure to the onset of signaling. In Interleukin-10 in Health and Disease; Springer: Berlin/Heidelberg, Germany, 2014; Volume 380, pp. 191–212. [Google Scholar] [CrossRef]
- Fiorentino, D.F.; Zlotnik, A.; Mosmann, T.R.; Howard, M.; O’Garra, A. IL-10 inhibits cytokine production by activated macrophages. J. Immunol. 1991, 147, 3815–3822. [Google Scholar] [CrossRef]
- Oft, M. IL-10: Master switch from tumor-promoting inflammation to antitumor immunity. Cancer Immunol. Res. 2014, 2, 194–199. [Google Scholar] [CrossRef]
- Bedke, T.; Muscate, F.; Soukou, S.; Gagliani, N.; Huber, S. IL-10-producing T cells and their dual functions. Semin. Immunol. 2019, 44, 101335. [Google Scholar] [CrossRef]
- Saraiva, M.; Vieira, P.; O’Garra, A. Biology and therapeutic potential of interleukin-10. J. Exp. Med. 2020, 217, e20190418. [Google Scholar] [CrossRef]
- Wang, X.; Wang, X.; Wang, D.; Zhou, C.; Lv, K.; Ma, Y.; Chang, W.; Wang, B.; Hu, J.; Ji, Y.; et al. Interleukin-10 overexpression in 4T1 cells: A gateway to suppressing mammary carcinoma growth. Int. Immunopharmacol. 2024, 142, 113089. [Google Scholar] [CrossRef]
- Levin, G.; Gotlieb, W.H. Interleukine-10 in ovarian cancer. Chin. Clin. Oncol. 2024, 13, 48. [Google Scholar] [CrossRef]
- Mannino, M.H.; Zhu, Z.; Xiao, H.; Bai, Q.; Wakefield, M.R.; Fang, Y. The paradoxical role of IL-10 in immunity and cancer. Cancer Lett. 2015, 367, 103–107. [Google Scholar] [CrossRef]
- Rallis, K.S.; Corrigan, A.E.; Dadah, H.; Stanislovas, J.; Zamani, P.; Makker, S.; Szabados, B.; Sideris, M. IL-10 in cancer: An essential thermostatic regulator between homeostatic immunity and inflammation—A comprehensive review. Future Oncol. 2022, 18, 3349–3365. [Google Scholar] [CrossRef] [PubMed]
- Guan, Q.; Han, M.; Guo, Q.; Yan, F.; Wang, M.; Ning, Q.; Xi, D. Strategies to reinvigorate exhausted CD8(+) T cells in tumor microenvironment. Front. Immunol. 2023, 14, 1204363. [Google Scholar] [CrossRef] [PubMed]
- Carlini, V.; Noonan, D.M.; Abdalalem, E.; Goletti, D.; Sansone, C.; Calabrone, L.; Albini, A. The multifaceted nature of IL-10: Regulation, role in immunological homeostasis and its relevance to cancer, COVID-19 and post-COVID conditions. Front. Immunol. 2023, 14, 1161067. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Chen, S.; He, X.; Yuan, Y.; Wei, X. Targeting inflammation as cancer therapy. J. Hematol. Oncol. 2024, 17, 13. [Google Scholar] [CrossRef]
- Huang, Y.; Zou, K.; Jiang, H.; Li, Z. The complex role of IL-10 in malignant ascites: A review. Cancer Immunol. Immunother. 2024, 73, 32. [Google Scholar] [CrossRef]
- Ni, G.; Zhang, L.; Yang, X.; Li, H.; Ma, B.; Walton, S.; Wu, X.; Yuan, J.; Wang, T.; Liu, X. Targeting interleukin-10 signalling for cancer immunotherapy, a promising and complicated task. Hum. Vaccin. Immunother. 2020, 16, 2328–2332. [Google Scholar] [CrossRef]
- Gabryšová, L.; Howes, A.; Saraiva, M.; O’Garra, A. The regulation of IL-10 expression. In Interleukin-10 in Health and Disease; Springer: Berlin/Heidelberg, Germany, 2014; Volume 380, pp. 157–190. [Google Scholar] [CrossRef]
- Rutz, S.; Ouyang, W. Regulation of Interleukin-10 Expression. Adv. Exp. Med. Biol. 2016, 941, 89–116. [Google Scholar] [CrossRef]
- Fan, L.; Qiu, D.; Huang, G.; Chen, J.; Wu, Q.; Xiong, S.; Wu, C.; Peng, Y.; Zhang, Q. Wogonin Suppresses IL-10 Production in B Cells via STAT3 and ERK Signaling Pathway. J. Immunol. Res. 2020, 2020, 3032425. [Google Scholar] [CrossRef]
- Saraiva, M.; O’Garra, A. The regulation of IL-10 production by immune cells. Nat. Rev. Immunol. 2010, 10, 170–181. [Google Scholar] [CrossRef]
- Zhu, J.; Yamane, H.; Paul, W.E. Differentiation of effector CD4 T cell populations. Annu. Rev. Immunol. 2010, 28, 445–489. [Google Scholar] [CrossRef]
- Yi, A.K.; Yoon, J.G.; Yeo, S.J.; Hong, S.C.; English, B.K.; Krieg, A.M. Role of mitogen-activated protein kinases in CpG DNA-mediated IL-10 and IL-12 production: Central role of extracellular signal-regulated kinase in the negative feedback loop of the CpG DNA-mediated Th1 response. J. Immunol. 2002, 168, 4711–4720. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, F.; Cook, D.; Papoutsopoulou, S.; Rajsbaum, R.; Wu, X.; Yang, H.T.; Grant, S.; Ricciardi-Castagnoli, P.; Tsichlis, P.N.; Ley, S.C.; et al. TPL-2 negatively regulates interferon-beta production in macrophages and myeloid dendritic cells. J. Exp. Med. 2009, 206, 1863–1871. [Google Scholar] [CrossRef]
- Martin, M.; Schifferle, R.E.; Cuesta, N.; Vogel, S.N.; Katz, J.; Michalek, S.M. Role of the phosphatidylinositol 3 kinase-Akt pathway in the regulation of IL-10 and IL-12 by Porphyromonas gingivalis lipopolysaccharide. J. Immunol. 2003, 171, 717–725. [Google Scholar] [CrossRef]
- Kanters, E.; Pasparakis, M.; Gijbels, M.J.; Vergouwe, M.N.; Partouns-Hendriks, I.; Fijneman, R.J.; Clausen, B.E.; Förster, I.; Kockx, M.M.; Rajewsky, K.; et al. Inhibition of NF-kappaB activation in macrophages increases atherosclerosis in LDL receptor-deficient mice. J. Clin. Invest. 2003, 112, 1176–1185. [Google Scholar] [CrossRef]
- Weichhart, T.; Costantino, G.; Poglitsch, M.; Rosner, M.; Zeyda, M.; Stuhlmeier, K.M.; Kolbe, T.; Stulnig, T.M.; Hörl, W.H.; Hengstschläger, M.; et al. The TSC-mTOR signaling pathway regulates the innate inflammatory response. Immunity 2008, 29, 565–577. [Google Scholar] [CrossRef]
- Cao, S.; Liu, J.; Song, L.; Ma, X. The protooncogene c-Maf is an essential transcription factor for IL-10 gene expression in macrophages. J. Immunol. 2005, 174, 3484–3492. [Google Scholar] [CrossRef]
- Wang, Z.Y.; Sato, H.; Kusam, S.; Sehra, S.; Toney, L.M.; Dent, A.L. Regulation of IL-10 gene expression in Th2 cells by Jun proteins. J. Immunol. 2005, 174, 2098–2105. [Google Scholar] [CrossRef]
- Jones, M.B.; Alvarez, C.A.; Johnson, J.L.; Zhou, J.Y.; Morris, N.; Cobb, B.A. CD45Rb-low effector T cells require IL-4 to induce IL-10 in Foxp3 Tregs and to protect mice from inflammation. PLoS ONE 2019, 14, e0216893. [Google Scholar] [CrossRef]
- Zhu, J.; Luo, L.; Tian, L.; Yin, S.; Ma, X.; Cheng, S.; Tang, W.; Yu, J.; Ma, W.; Zhou, X.; et al. Aryl hydrocarbon receptor promotes IL-10 expression in inflammatory macrophages through Src-STAT3 signaling pathway. Front. Immunol. 2018, 9, 2033. [Google Scholar] [CrossRef]
- Meng, X.; Grötsch, B.; Luo, Y.; Knaup, K.X.; Wiesener, M.S.; Chen, X.X.; Jantsch, J.; Fillatreau, S.; Schett, G.; Bozec, A. Hypoxia-inducible factor-1α is a critical transcription factor for IL-10-producing B cells in autoimmune disease. Nat. Commun. 2018, 9, 251. [Google Scholar] [CrossRef]
- Selig, M.; Poehlman, L.; Lang, N.C.; Völker, M.; Rolauffs, B.; Hart, M.L. Prediction of six macrophage phenotypes and their IL-10 content based on single-cell morphology using artificial intelligence. Front. Immunol. 2024, 14, 1336393. [Google Scholar] [CrossRef] [PubMed]
- Rallis, K.S.; Corrigan, A.E.; Dadah, H.; George, A.M.; Keshwara, S.M.; Sideris, M.; Szabados, B. Cytokine-based cancer immunotherapy: Challenges and opportunities for IL-10. Anticancer Res. 2021, 41, 3247–3252. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Li, T.; Niu, M.; Zhang, H.; Wu, Y.; Wu, K.; Dai, Z. Targeting cytokine and chemokine signaling pathways for cancer therapy. Signal Transduct. Target. Ther. 2024, 9, 176. [Google Scholar] [CrossRef]
- Han, J.; Wang, H. Cytokine-overexpressing dendritic cells for cancer immunotherapy. Exp. Mol. Med. 2024, 56, 2559–2568. [Google Scholar] [CrossRef]
- Berg, D.J.; Davidson, N.; Kuhn, R.; Muller, W.; Menon, S.; Holland, G.; Thompson-Snipes, L.; Leach, M.W.; Rennick, D. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) TH1-like responses. J. Clin. Invest. 1996, 98, 1010–1020. [Google Scholar] [CrossRef]
- Neven, B.; Mamessier, E.; Bruneau, J.; Kaltenbach, S.; Kotlarz, D.; Suarez, F.; Masliah-Planchon, J.; Billot, K.; Canioni, D.; Frange, P.; et al. A Mendelian predisposition to B-cell lymphoma caused by IL-10R deficiency. Blood 2013, 122, 3713–3722. [Google Scholar] [CrossRef]
- Gerard, C.M.; Bruyns, C.; Delvaux, A.; Baudson, N.; Dargent, J.L.; Goldman, M.; Velu, T. Loss of tumorigenicity and increased immunogenicity induced by interleukin-10 gene transfer in B16 melanoma cells. Hum. Gene Ther. 1996, 7, 23–31. [Google Scholar] [CrossRef]
- Groux, H.; Cottrez, F.; Rouleau, M.; Mauze, S.; Antonenko, S.; Hurst, S.; McNeil, T.; Bigler, M.; Roncarolo, M.G.; Coffman, R.L. A transgenic model to analyze the immunoregulatory role of IL-10 secreted by antigen-presenting cells. J. Immunol. 1999, 162, 1723–1729. [Google Scholar] [CrossRef]
- Mumm, J.B.; Emmerich, J.; Zhang, X.; Chan, I.; Wu, L.; Mauze, S.; Blaisdell, S.; Basham, B.; Dai, J.; Grein, J.; et al. IL-10 elicits IFNgamma-dependent tumor immune surveillance. Cancer Cell 2011, 20, 781–796. [Google Scholar] [CrossRef]
- Emmerich, J.; Mumm, J.B.; Chan, I.H.; LaFace, D.; Truong, H.; McClanahan, T.; Gorman, D.M.; Oft, M. IL-10 directly activates and expands tumor-resident CD8(+) T cells without de novo infiltration from secondary lymphoid organs. Cancer Res. 2012, 72, 3570–3581. [Google Scholar] [CrossRef]
- Qiao, J.; Fu, Y.X. Cytokines that target immune killer cells against tumors. Cell Mol. Immunol. 2020, 17, 722–727. [Google Scholar] [CrossRef] [PubMed]
- Oft, M. Immune regulation and cytotoxic T cell activation of IL-10 agonists—Preclinical and clinical experience. Semin. Immunol. 2019, 44, 101325. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Hu, X.; Gao, L.; Chen, L.; Chen, J.; Yuan, J.; Huang, C.; Xu, X.; Yang, J. Interleukin 10 enhanced CD8+ T cell activity and reduced CD8(+) T cell apoptosis in patients with diffuse large B cell lymphoma. Exp. Cell Res. 2017, 360, 146–152. [Google Scholar] [CrossRef]
- Dennis, K.L.; Wang, Y.; Blatner, N.R.; Wang, S.; Saadalla, A.; Trudeau, E.; Roers, A.; Weaver, C.T.; Lee, J.J.; Gilbert, J.A.; et al. Adenomatous polyps are driven by microbe-instigated focal inflammation and are controlled by IL-10-producing T cells. Cancer Res. 2013, 73, 5905–5913. [Google Scholar] [CrossRef]
- Naing, A.; Infante, J.R.; Papadopoulos, K.P.; Chan, I.H.; Shen, C.; Ratti, N.P.; Rojo, B.; Autio, K.A.; Wong, D.J.; Patel, M.R.; et al. PEGylated IL-10 (Pegilodecakin) Induces Systemic Immune Activation, CD8(+) T Cell Invigoration and Polyclonal T Cell Expansion in Cancer Patients. Cancer Cell 2018, 34, 775-791.e3. [Google Scholar] [CrossRef]
- Cavallazzi Sebold, B.; Ni, G.; Li, J.; Li, H.; Liu, X.; Wang, T. PEGylated IL-10: Clinical Development in Cancer Immunotherapy, Where to Go? Curr. Oncol. Rep. 2023, 25, 115–122. [Google Scholar] [CrossRef]
- Veronese, F.M.; Mero, A. The impact of PEGylation on biological therapies. BioDrugs 2008, 22, 315–329. [Google Scholar] [CrossRef]
- Kontermann, R.E. Recombinant bispecific antibodies for cancer therapy. Acta Pharmacol. Sin. 2005, 26, 1–9. [Google Scholar] [CrossRef]
- Marradi, M.; Chiodo, F.; Garcia, I.; Penades, S. Glyconanoparticles as multifunctional and multimodal carbohydrate systems. Chem. Soc. Rev. 2013, 42, 4728–4745. [Google Scholar] [CrossRef]
- Kay, M.A.; Glorioso, J.C.; Naldini, L. Viral vectors for gene therapy: The art of turning infectious agents into vehicles of therapeutics. Nat. Med. 2001, 7, 33–40. [Google Scholar] [CrossRef]
- Minshawi, F.; Lanvermann, S.; McKenzie, E.; Jeffery, R.; Couper, K.; Papoutsopoulou, S.; Roers, A.; Muller, W. The generation of an engineered interleukin-10 protein with improved stability and biological function. Front. Immunol. 2020, 11, 1794. [Google Scholar] [CrossRef] [PubMed]
- Deckers, J.; Anbergen, T.; Hokke, A.M.; de Dreu, A.; Schrijver, D.P.; de Bruin, K.; Toner, Y.C.; Beldman, T.J.; Spangler, J.B.; de Greef, T.F.A.; et al. Engineering cytokine therapeutics. Nat. Rev. Bioeng. 2023, 1, 286–303. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Xie, Y.Q.; Gao, M.; Zhao, Y.; Franco, F.; Wenes, M.; Siddiqui, I.; Bevilacqua, A.; Wang, H.; Yang, H.; et al. Metabolic reprogramming of terminally exhausted CD8(+) T cells by IL-10 enhances anti-tumor immunity. Nat. Immunol. 2021, 22, 746–756. [Google Scholar] [CrossRef]
- Qiao, J.; Liu, Z.; Dong, C.; Luan, Y.; Zhang, A.; Moore, C.; Fu, K.; Peng, J.; Wang, Y.; Ren, Z.; et al. Targeting Tumors with IL-10 Prevents Dendritic Cell-Mediated CD8(+) T Cell Apoptosis. Cancer Cell 2019, 35, 901-915.e4. [Google Scholar] [CrossRef]
- Chang, Y.W.; Hsiao, H.W.; Chen, J.P.; Tzeng, S.F.; Tsai, C.H.; Wu, C.Y.; Hsieh, H.H.; Carmona, S.J.; Andreatta, M.; Di Conza, G.; et al. A CSF-1R-blocking antibody/IL-10 fusion protein increases anti-tumor immunity by effectuating tumor-resident CD8(+) T cells. Cell Rep. Med. 2023, 4, 101154. [Google Scholar] [CrossRef]
- Gorby, C.; Sotolongo Bellon, J.; Wilmes, S.; Warda, W.; Pohler, E.; Fyfe, P.K.; Cozzani, A.; Ferrand, C.; Walter, M.R.; Mitra, S.; et al. Engineered IL-10 variants elicit potent immunomodulatory effects at low ligand doses. Sci. Signal 2020, 13, eabc0653. [Google Scholar] [CrossRef]
- Chard, L.S.; Maniati, E.; Wang, P.; Zhang, Z.; Gao, D.; Wang, J.; Cao, F.; Ahmed, J.; El Khouri, M.; Hughes, J.; et al. A vaccinia virus armed with interleukin-10 is a promising therapeutic agent for treatment of murine pancreatic cancer. Clin. Cancer Res. 2015, 21, 405–416. [Google Scholar] [CrossRef]
- Baganizi, D.R.; Nyairo, E.; Duncan, S.A.; Singh, S.R.; Dennis, V.A. Interleukin-10 Conjugation to Carboxylated PVP-Coated Silver Nanoparticles for Improved Stability and Therapeutic Efficacy. Nanomaterials 2017, 7, 165. [Google Scholar] [CrossRef]
- Xue, Y.; Che, J.; Ji, X.; Li, Y.; Xie, J.; Chen, X. Recent advances in biomaterial-boosted adoptive cell therapy. Chem. Soc. Rev. 2022, 51, 1766–1794. [Google Scholar] [CrossRef]
- Murer, P.; Neri, D. Antibody-cytokine fusion proteins: A novel class of biopharmaceuticals for the therapy of cancer and of chronic inflammation. N. Biotechnol. 2019, 52, 42–53. [Google Scholar] [CrossRef]
- Williams, L.; Li, L.; Yazaki, P.J.; Wong, P.; Hong, T.; Poku, E.K.; Hui, S.; Ghimire, H.; Shively, J.E.; Kujawski, M. Comparison of IL-2-antibody to IL-2-Fc with or without stereotactic radiation therapy in CEA immunocompetent mice with CEA positive tumors. Cancer Med. 2024, 13, e6909. [Google Scholar] [CrossRef] [PubMed]
- Boersma, B.; Poinot, H.; Pommier, A. Stimulating the Antitumor Immune Response Using Immunocytokines: A Preclinical and Clinical Overview. Pharmaceutics 2024, 16, 974. [Google Scholar] [CrossRef] [PubMed]
- Kurtulus, S.; Madi, A.; Escobar, G.; Klapholz, M.; Nyman, J.; Christian, E.; Pawlak, M.; Dionne, D.; Xia, J.; Rozenblatt-Rosen, O.; et al. Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1(-)CD8(+) Tumor-Infiltrating T Cells. Immunity 2019, 50, 181-194.e6. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.C.; Sen, D.R.; Al Abosy, R.; Bi, K.; Virkud, Y.V.; LaFleur, M.W.; Yates, K.B.; Lako, A.; Felt, K.; Naik, G.S.; et al. Author Correction: Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 2019, 20, 1556. [Google Scholar] [CrossRef]
- Siddiqui, I.; Schaeuble, K.; Chennupati, V.; Fuertes Marraco, S.A.; Calderon-Copete, S.; Pais Ferreira, D.; Carmona, S.J.; Scarpellino, L.; Gfeller, D.; Pradervand, S.; et al. Intratumoral Tcf1(+)PD-1(+)CD8(+) T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 2019, 50, 195-211.e10. [Google Scholar] [CrossRef]
- Im, S.J.; Hashimoto, M.; Gerner, M.Y.; Lee, J.; Kissick, H.T.; Burger, M.C.; Shan, Q.; Hale, J.S.; Lee, J.; Nasti, T.H.; et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 2016, 537, 417–421. [Google Scholar] [CrossRef]
- Franco, F.; Jaccard, A.; Romero, P.; Yu, Y.R.; Ho, P.C. Metabolic and epigenetic regulation of T-cell exhaustion. Nat. Metab. 2020, 2, 1001–1012. [Google Scholar] [CrossRef]
- Vincenzi, B.; Zoccoli, A.; Pantano, F.; Venditti, O.; Galluzzo, S. Cetuximab: From bench to bedside. Curr. Cancer Drug Targets 2010, 10, 80–95. [Google Scholar] [CrossRef]
- Rajaram, P.; Chandra, P.; Ticku, S.; Pallavi, B.K.; Rudresh, K.B.; Mansabdar, P. Epidermal growth factor receptor: Role in human cancer. Indian J. Dent. Res. 2017, 28, 687–694. [Google Scholar] [CrossRef]
- Cheng, W.L.; Feng, P.H.; Lee, K.Y.; Chen, K.Y.; Sun, W.L.; Van Hiep, N.; Luo, C.S.; Wu, S.M. The Role of EREG/EGFR Pathway in Tumor Progression. Int. J. Mol. Sci. 2021, 22, 12828. [Google Scholar] [CrossRef]
- Yuan, X.; Zhang, J.; Li, D.; Mao, Y.; Mo, F.; Du, W.; Ma, X. Prognostic significance of tumor-associated macrophages in ovarian cancer: A meta-analysis. Gynecol. Oncol. 2017, 147, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.W.; Ge, X.X.; Xu, M.D.; Qin, H.; Wu, M.Y.; Shen, M.; Zhang, Y.; Liu, X.M.; Chen, K.; Li, W.; et al. Tumor-associated macrophages promote the metastasis and growth of non-small-cell lung cancer cells through NF-kappaB/PP2Ac-positive feedback loop. Cancer Sci. 2021, 112, 2140–2157. [Google Scholar] [CrossRef]
- Kersten, K.; Hu, K.H.; Combes, A.J.; Samad, B.; Harwin, T.; Ray, A.; Rao, A.A.; Cai, E.; Marchuk, K.; Artichoker, J.; et al. Spatiotemporal co-dependency between macrophages and exhausted CD8(+) T cells in cancer. Cancer Cell 2022, 40, 624-638.e9. [Google Scholar] [CrossRef]
- Chen, Y.; Jin, H.; Song, Y.; Huang, T.; Cao, J.; Tang, Q.; Zou, Z. Targeting tumor-associated macrophages: A potential treatment for solid tumors. J. Cell Physiol. 2021, 236, 3445–3465. [Google Scholar] [CrossRef]
- Sun, Y.; Cronin, M.F.; Mendonca, M.C.P.; Guo, J.; O’Driscoll, C.M. Sialic acid-targeted cyclodextrin-based nanoparticles deliver CSF-1R siRNA and reprogram tumour-associated macrophages for immunotherapy of prostate cancer. Eur. J. Pharm. Sci. 2023, 185, 106427. [Google Scholar] [CrossRef]
- Tomassetti, C.; Insinga, G.; Gimigliano, F.; Morrione, A.; Giordano, A.; Giurisato, E. Insights into CSF-1R Expression in the Tumor Microenvironment. Biomedicines 2024, 12, 2381. [Google Scholar] [CrossRef]
- Cersosimo, F.; Lonardi, S.; Ulivieri, C.; Martini, P.; Morrione, A.; Vermi, W.; Giordano, A.; Giurisato, E. CSF-1R in Cancer: More than a Myeloid Cell Receptor. Cancers 2024, 16, 282. [Google Scholar] [CrossRef]
- Xiang, C.; Li, H.; Tang, W. Targeting CSF-1R represents an effective strategy in modulating inflammatory diseases. Pharmacol. Res. 2023, 187, 106566. [Google Scholar] [CrossRef]
- Josephson, K.; Logsdon, N.J.; Walter, M.R. Crystal structure of the IL-10/IL-10R1 complex reveals a shared receptor binding site. Immunity 2001, 15, 35–46. [Google Scholar] [CrossRef]
- Logsdon, N.J.; Jones, B.C.; Josephson, K.; Cook, J.; Walter, M.R. Comparison of interleukin-22 and interleukin-10 soluble receptor complexes. J. Interferon Cytokine Res. 2002, 22, 1099–1112. [Google Scholar] [CrossRef]
- Ouyang, W.; Rutz, S.; Crellin, N.K.; Valdez, P.A.; Hymowitz, S.G. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu. Rev. Immunol. 2011, 29, 71–109. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Tsutsumi, N.; Su, L.L.; Abhiraman, G.C.; Mohan, K.; Henneberg, L.T.; Aduri, N.G.; Gati, C.; Garcia, K.C. Structure-based decoupling of the pro- and anti-inflammatory functions of interleukin-10. Science 2021, 371, eabc8433. [Google Scholar] [CrossRef]
- Saxton, R.A.; Garcia, K.C. Cryo-EM structure of the IL-10 receptor complex provides a blueprint for ligand engineering. FEBS J. 2022, 289, 8032–8036. [Google Scholar] [CrossRef]
- Li, M.; Zhang, M.; Ye, Q.; Liu, Y.; Qian, W. Preclinical and clinical trials of oncolytic vaccinia virus in cancer immunotherapy: A comprehensive review. Cancer Biol. Med. 2023, 20, 646–661. [Google Scholar] [CrossRef] [PubMed]
- El-Shemi, A.G.; Ashshi, A.M.; Na, Y.; Li, Y.; Basalamah, M.; Al-Allaf, F.A.; Oh, E.; Jung, B.K.; Yun, C.O. Combined therapy with oncolytic adenoviruses encoding TRAIL and IL-12 genes markedly suppressed human hepatocellular carcinoma both in vitro and in an orthotopic transplanted mouse model. J. Exp. Clin. Cancer Res. CR 2016, 35, 74. [Google Scholar] [CrossRef]
- Galal El-Shemi, A.; Mohammed Ashshi, A.; Oh, E.; Jung, B.K.; Basalamah, M.; Alsaegh, A.; Yun, C.O. Efficacy of combining ING4 and TRAIL genes in cancer-targeting gene virotherapy strategy: First evidence in preclinical hepatocellular carcinoma. Gene Ther. 2018, 25, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Oh, C.M.; Chon, H.J.; Kim, C. Combination Immunotherapy Using Oncolytic Virus for the Treatment of Advanced Solid Tumors. Int. J. Mol. Sci. 2020, 21, 7743. [Google Scholar] [CrossRef]
- Shi, T.; Song, X.; Wang, Y.; Liu, F.; Wei, J. Combining Oncolytic Viruses with Cancer Immunotherapy: Establishing a New Generation of Cancer Treatment. Front. Immunol. 2020, 11, 683. [Google Scholar] [CrossRef]
- Li, J.; Mooney, D.J. Designing hydrogels for controlled drug delivery. Nat. Rev. Mater. 2016, 1, 16071. [Google Scholar] [CrossRef]
- Nguyen, K.G.; Vrabel, M.R.; Mantooth, S.M.; Hopkins, J.J.; Wagner, E.S.; Gabaldon, T.A.; Zaharoff, D.A. Localized Interleukin-12 for Cancer Immunotherapy. Front. Immunol. 2020, 11, 575597. [Google Scholar] [CrossRef]
- Lv, Q.; He, C.; Quan, F.; Yu, S.; Chen, X. Corrigendum to “DOX/IL-2/IFN-gamma co-loaded thermo-sensitive polypeptide hydrogel for efficient melanoma treatment” [Bioact. Mater. 3 (2018) 118-128]. Bioact. Mater. 2019, 4, 167. [Google Scholar] [CrossRef]
- Ajona, D.; Ortiz-Espinosa, S.; Pio, R. Complement anaphylatoxins C3a and C5a: Emerging roles in cancer progression and treatment. Semin. Cell Dev. Biol. 2019, 85, 153–163. [Google Scholar] [CrossRef]
- Hameed, B.H.; Abdulsatar Al-Rayahi, I.; Muhsin, S.S. The preoperative serum levels of the anaphylatoxins C3a and C5a and their association with clinico-pathological factors in breast cancer patients. Arch. Razi Inst. 2022, 77, 1873–1879. [Google Scholar]
- Roumenina, L.T.; Daugan, M.V.; Petitprez, F.; Sautès-Fridman, C.; Fridman, W.H. Context-dependent roles of complement in cancer. Nat. Rev. Cancer 2019, 19, 698–715. [Google Scholar] [CrossRef]
- Afshar-Kharghan, V. The role of the complement system in cancer. J. Clin. Investig. 2017, 127, 780–789. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, S.N.; Liu, Q.; Yu, Y.Y.; Guo, J.; Wang, K.; Xing, B.C.; Zheng, Q.F.; Campa, M.J.; Patz, E.F., Jr.; et al. Autocrine complement inhibits IL-10-dependent T-cell-mediated antitumor immunity to promote tumor progression. Cancer Discov. 2016, 6, 1022–1035. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, H.; He, Y.W. The complement receptors C3aR and C5aR are a new class of immune checkpoint receptor in cancer immunotherapy. Front. Immunol. 2019, 10, 1574. [Google Scholar] [CrossRef]
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Perez-Gracia, J.L.; Rodriguez-Ruiz, M.E.; Ponz-Sarvise, M.; Castanon, E.; Melero, I. Cytokines in clinical cancer immunotherapy. Br. J. Cancer 2019, 120, 6–15. [Google Scholar] [CrossRef]
- Bohmer, M.; Xue, Y.; Jankovic, K.; Dong, Y. Advances in engineering and delivery strategies for cytokine immunotherapy. Expert. Opin. Drug Deliv. 2023, 20, 579–595. [Google Scholar] [CrossRef]
- Fedorak, R.N.; Gangl, A.; Elson, C.O.; Rutgeerts, P.; Schreiber, S.; Wild, G.; Hanauer, S.B.; Kilian, A.; Cohard, M.; LeBeaut, A.; et al. Recombinant human interleukin 10 in the treatment of patients with mild to moderately active Crohn’s disease. The Interleukin 10 Inflammatory Bowel Disease Cooperative Study Group. Gastroenterology 2000, 119, 1473–1482. [Google Scholar] [CrossRef]
- Schreiber, S.; Fedorak, R.N.; Nielsen, O.H.; Wild, G.; Williams, C.N.; Nikolaus, S.; Jacyna, M.; Lashner, B.A.; Gangl, A.; Rutgeerts, P.; et al. Safety and efficacy of recombinant human interleukin 10 in chronic active Crohn’s disease. Crohn’s Disease IL-10 Cooperative Study Group. Gastroenterology 2000, 119, 1461–1472. [Google Scholar] [CrossRef] [PubMed]
- Autio, K.; Oft, M. Pegylated Interleukin-10: Clinical Development of an Immunoregulatory Cytokine for Use in Cancer Therapeutics. Curr. Oncol. Rep. 2019, 21, 19. [Google Scholar] [CrossRef]
- Naing, A.; Papadopoulos, K.P.; Autio, K.A.; Ott, P.A.; Patel, M.R.; Wong, D.J.; Falchook, G.S.; Pant, S.; Whiteside, M.; Rasco, D.R.; et al. Safety, Antitumor Activity, and Immune Activation of Pegylated Recombinant Human Interleukin-10 (AM0010) in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2016, 34, 3562–3569. [Google Scholar] [CrossRef]
- Naing, A.; Wong, D.J.; Infante, J.R.; Korn, W.M.; Aljumaily, R.; Papadopoulos, K.P.; Autio, K.A.; Pant, S.; Bauer, T.M.; Drakaki, A.; et al. Pegilodecakin combined with pembrolizumab or nivolumab for patients with advanced solid tumours (IVY): A multicentre, multicohort, open-label, phase 1b trial. Lancet Oncol. 2019, 20, 1544–1555. [Google Scholar] [CrossRef]
- Tannir, N.M.; Papadopoulos, K.P.; Wong, D.J.; Aljumaily, R.; Hung, A.; Afable, M.; Kim, J.S.; Ferry, D.; Drakaki, A.; Bendell, J.; et al. Pegilodecakin as monotherapy or in combination with anti-PD-1 or tyrosine kinase inhibitor in heavily pretreated patients with advanced renal cell carcinoma: Final results of cohorts A, G, H and I of IVY Phase I study. Int. J. Cancer 2021, 149, 403–408. [Google Scholar] [CrossRef]
- Autio, K.; Naing, A.; Hung, A.; Oft, M.; Leveque, J.; Falchook, G. Durability of clinical benefit in metastatic epithelial ovarian cancer patients treated with pegilodecakin monotherapy or in combination with platinum plus taxane-based chemotherapy. Ann. Oncol. 2018, 29, viii408. [Google Scholar] [CrossRef]
- Spigel, D.; Jotte, R.; Nemunaitis, J.; Shum, M.; Schneider, J.; Goldschmidt, J.; Eisenstein, J.; Berz, D.; Seneviratne, L.; Socoteanu, M.; et al. Randomized Phase 2 Studies of Checkpoint Inhibitors Alone or in Combination with Pegilodecakin in Patients With Metastatic NSCLC (CYPRESS 1 and CYPRESS 2). J. Thorac. Oncol. 2021, 16, 327–333. [Google Scholar] [CrossRef]
- Hecht, J.R.; Lonardi, S.; Bendell, J.; Sim, H.W.; Macarulla, T.; Lopez, C.D.; Van Cutsem, E.; Munoz Martin, A.J.; Park, J.O.; Greil, R.; et al. Randomized Phase III Study of FOLFOX Alone or With Pegilodecakin as Second-Line Therapy in Patients With Metastatic Pancreatic Cancer That Progressed After Gemcitabine (SEQUOIA). J. Clin. Oncol. 2021, 39, 1108–1118. [Google Scholar] [CrossRef]
- Hecht, J.R.; Papadopoulos, K.P.; Falchook, G.S.; Patel, M.R.; Infante, J.R.; Aljumaily, R.; Wong, D.J.; Autio, K.A.; Wainberg, Z.A.; Bauer, T.M.; et al. Immunologic and tumor responses of pegilodecakin with 5-FU/LV and oxaliplatin (FOLFOX) in pancreatic ductal adenocarcinoma (PDAC). Invest. New Drugs 2021, 39, 182–192. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Hoos, A.; O’Day, S.; Weber, J.S.; Hamid, O.; Lebbe, C.; Maio, M.; Binder, M.; Bohnsack, O.; Nichol, G.; et al. Guidelines for the evaluation of immune therapy activity in solid tumors: Immune-related response criteria. Clin. Cancer Res. 2009, 15, 7412–7420. [Google Scholar] [CrossRef]
- Weber, J.S.; Hodi, F.S.; Wolchok, J.D.; Topalian, S.L.; Schadendorf, D.; Larkin, J.; Sznol, M.; Long, G.V.; Li, H.; Waxman, I.M.; et al. Safety Profile of Nivolumab Monotherapy: A Pooled Analysis of Patients with Advanced Melanoma. J. Clin. Oncol. 2017, 35, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.B.; Lotze, M.T.; Dutcher, J.P.; Fisher, R.I.; Weiss, G.; Margolin, K.; Abrams, J.; Sznol, M.; Parkinson, D.; Hawkins, M.; et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: Analysis of 270 patients treated between 1985 and 1993. J. Clin. Oncol. 1999, 17, 2105–2116. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Rini, B.I.; McDermott, D.F.; Redman, B.G.; Kuzel, T.M.; Harrison, M.R.; Vaishampayan, U.N.; Drabkin, H.A.; George, S.; Logan, T.F.; et al. Nivolumab for Metastatic Renal Cell Carcinoma: Results of a Randomized Phase II Trial. J. Clin. Oncol. 2015, 33, 1430–1437. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Shemi, A.G.; Alqurashi, A.; Abdulrahman, J.A.; Alzahrani, H.D.; Almwalad, K.S.; Felfilan, H.H.; Alomiri, W.S.; Aloufi, J.A.; Madkhali, G.H.; Maqliyah, S.A.; et al. IL-10-Directed Cancer Immunotherapy: Preclinical Advances, Clinical Insights, and Future Perspectives. Cancers 2025, 17, 1012. https://doi.org/10.3390/cancers17061012
El-Shemi AG, Alqurashi A, Abdulrahman JA, Alzahrani HD, Almwalad KS, Felfilan HH, Alomiri WS, Aloufi JA, Madkhali GH, Maqliyah SA, et al. IL-10-Directed Cancer Immunotherapy: Preclinical Advances, Clinical Insights, and Future Perspectives. Cancers. 2025; 17(6):1012. https://doi.org/10.3390/cancers17061012
Chicago/Turabian StyleEl-Shemi, Adel G., Afnan Alqurashi, Jihan Abdullah Abdulrahman, Hanin Dhaifallah Alzahrani, Khawlah Saad Almwalad, Hadeel Hisham Felfilan, Wahaj Saud Alomiri, Jana Ahmed Aloufi, Ghadeer Hassn Madkhali, Sarah Adel Maqliyah, and et al. 2025. "IL-10-Directed Cancer Immunotherapy: Preclinical Advances, Clinical Insights, and Future Perspectives" Cancers 17, no. 6: 1012. https://doi.org/10.3390/cancers17061012
APA StyleEl-Shemi, A. G., Alqurashi, A., Abdulrahman, J. A., Alzahrani, H. D., Almwalad, K. S., Felfilan, H. H., Alomiri, W. S., Aloufi, J. A., Madkhali, G. H., Maqliyah, S. A., Alshahrani, J. B., Kamal, H. T., Daghistani, S. H., Refaat, B., & Minshawi, F. (2025). IL-10-Directed Cancer Immunotherapy: Preclinical Advances, Clinical Insights, and Future Perspectives. Cancers, 17(6), 1012. https://doi.org/10.3390/cancers17061012