

Role of Rac1 in p53-Related Proliferation and Drug Sensitivity in Multiple Myeloma †

 , , , , , ,
, , , , , , 
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Patients
2.3. Reagents
2.4. Expression of Wild-Type p53 Using Tet-On System
2.5. Gene Knockdown
2.6. Isolation of Nucleic Acids and RNA Expression Analysis Using PCR
2.7. Western Blotting Analysis
2.8. Flow Cytometry Analysis
2.9. RNA Sequencing
2.10. Statistical Analysis
3. Results
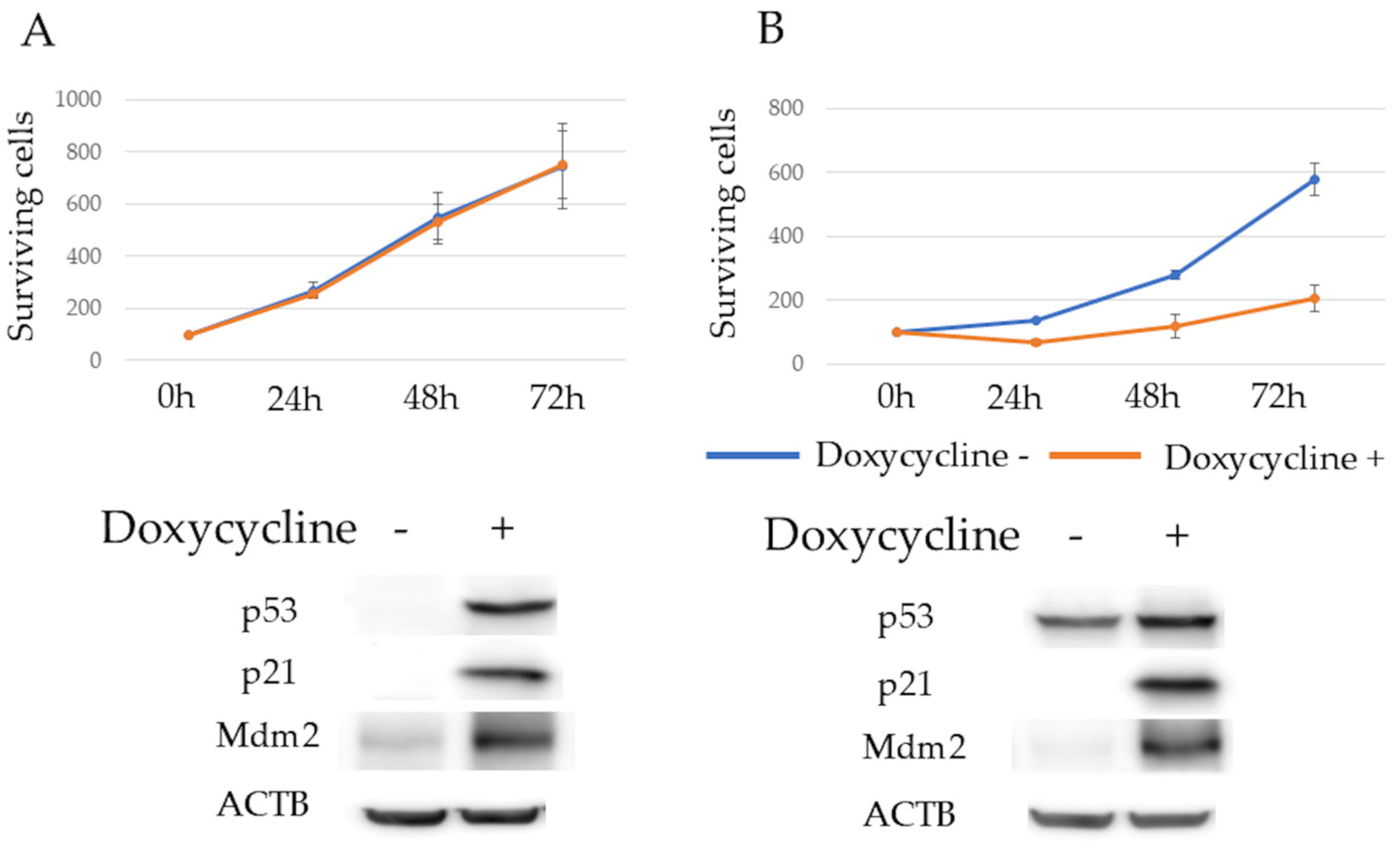
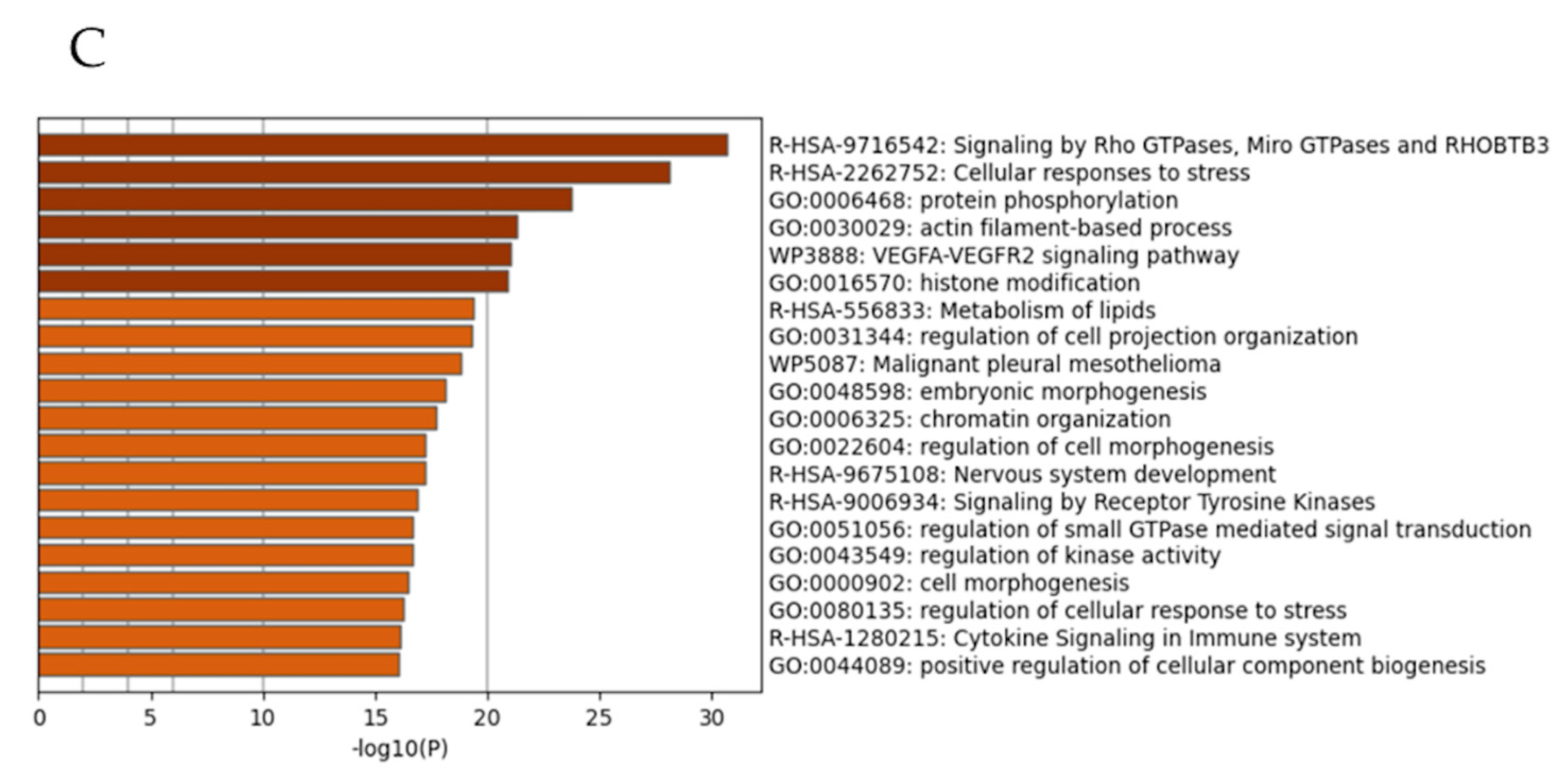
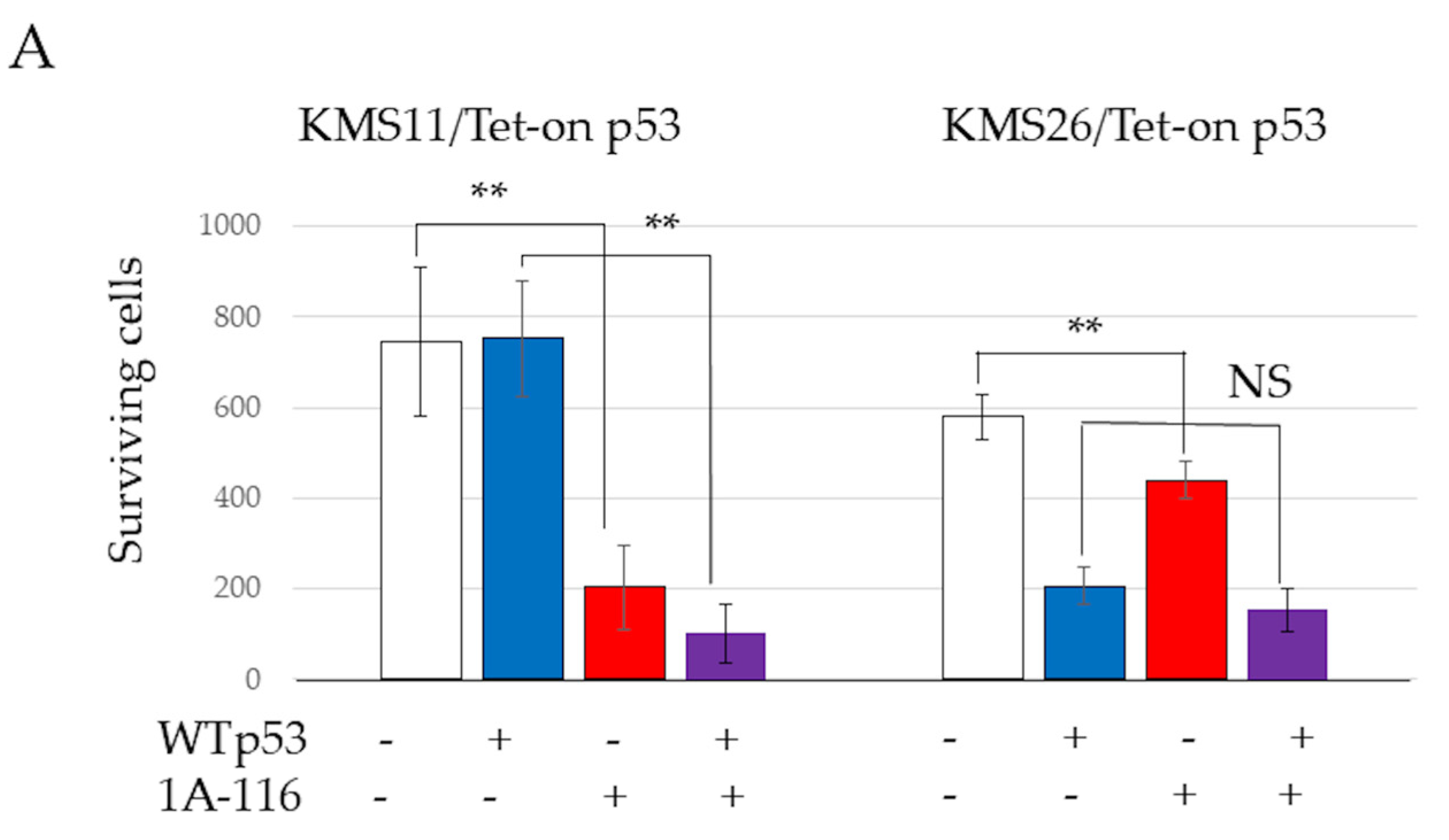
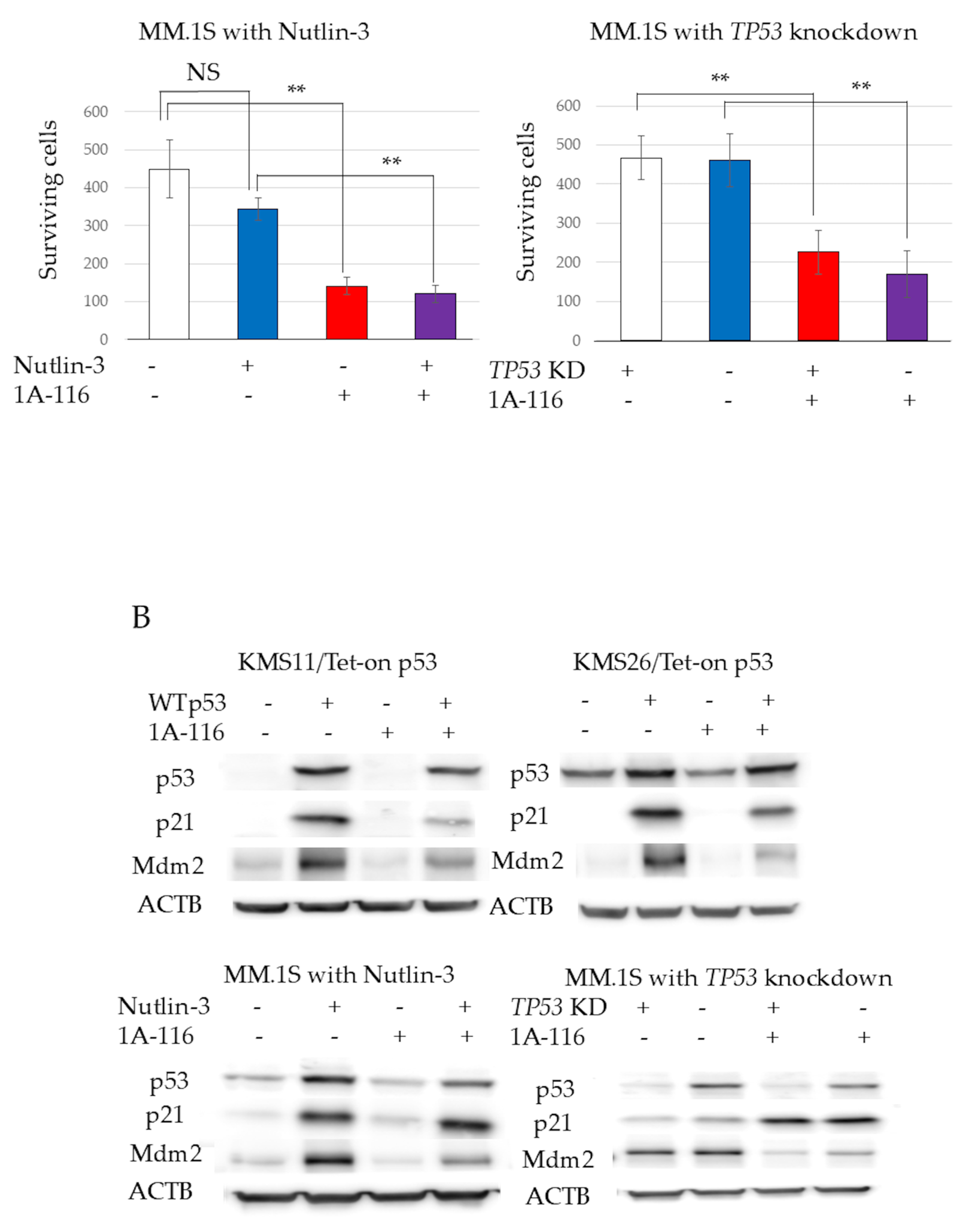
3.1. Gene Ontology Analysis of p53, p21, and Mdm2 Protein Expression and Proliferation in Tet-On p53 HMCLs
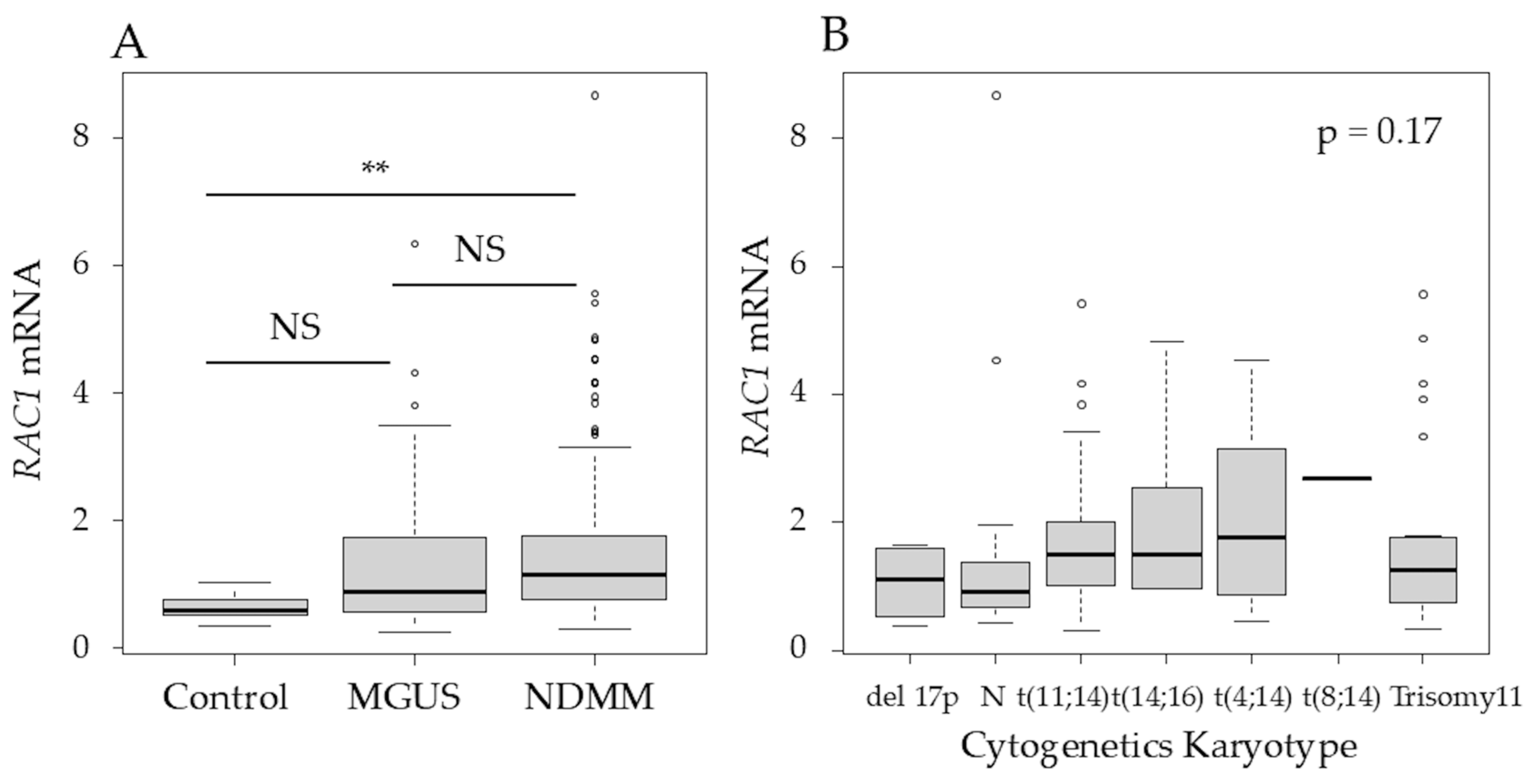
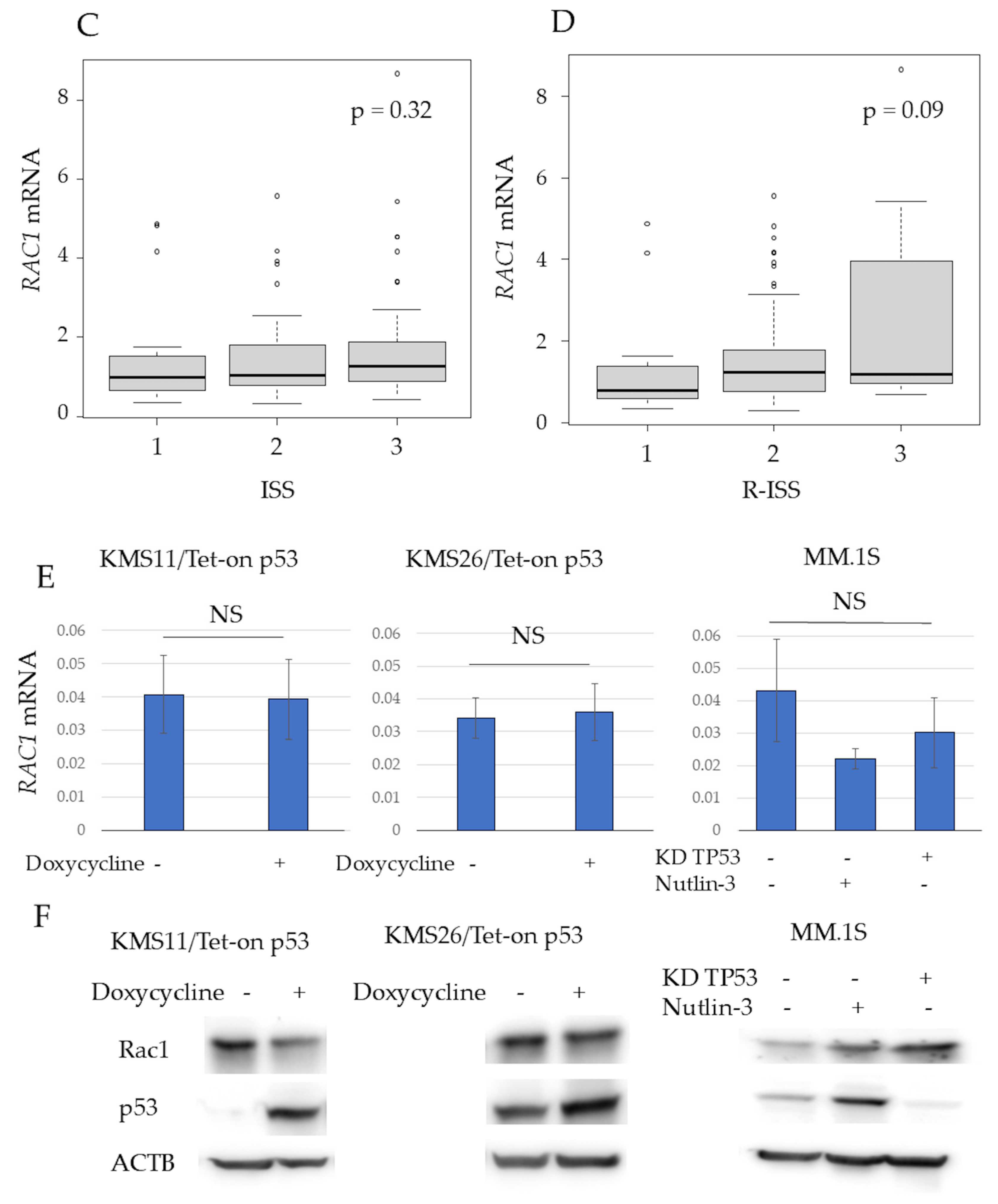
3.2. Expression of RAC1 in Multiple Myeloma Patient Samples and HMCLs
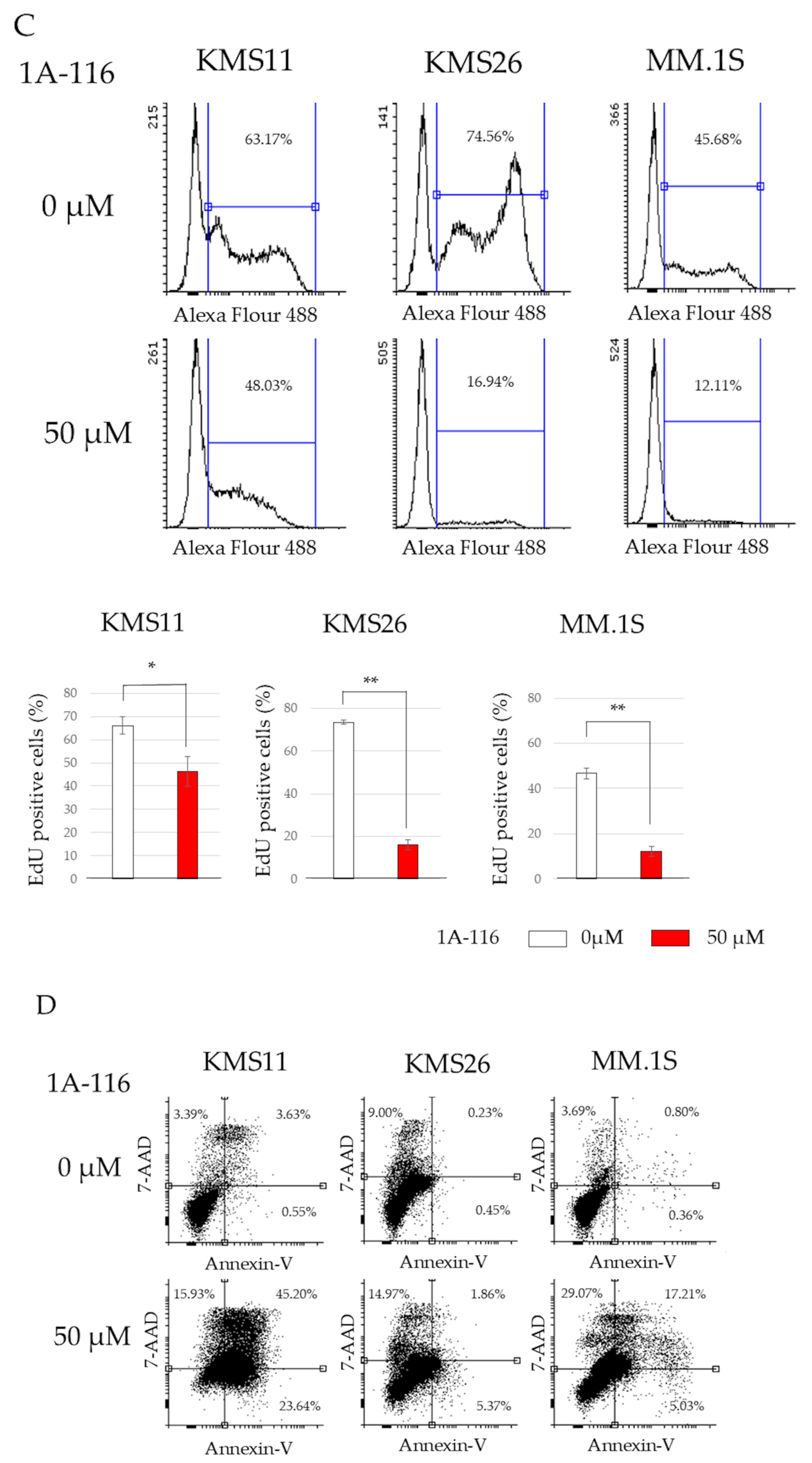
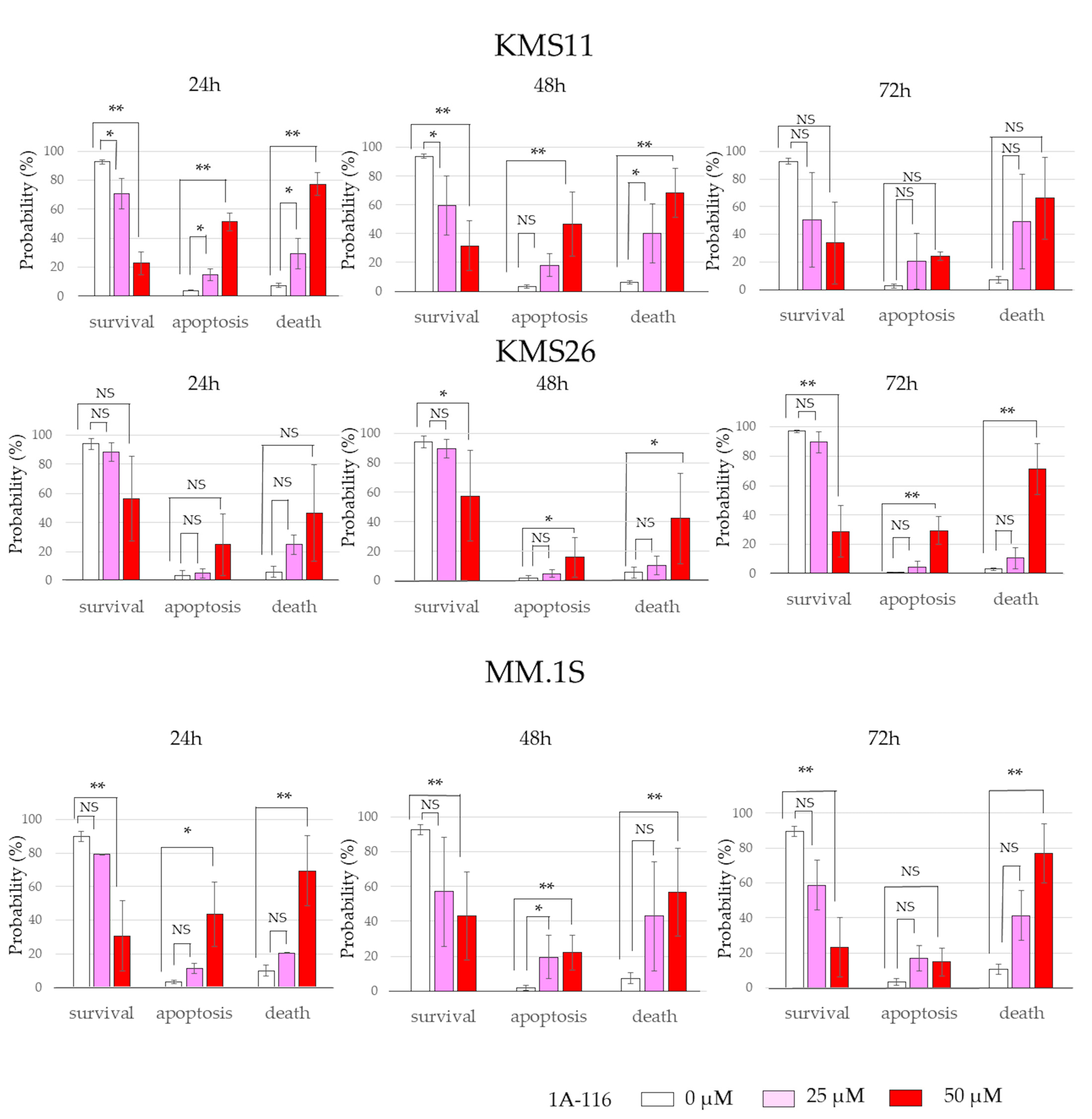
3.3. MM Cell Proliferation and Apoptosis After Treatment with 1A-116
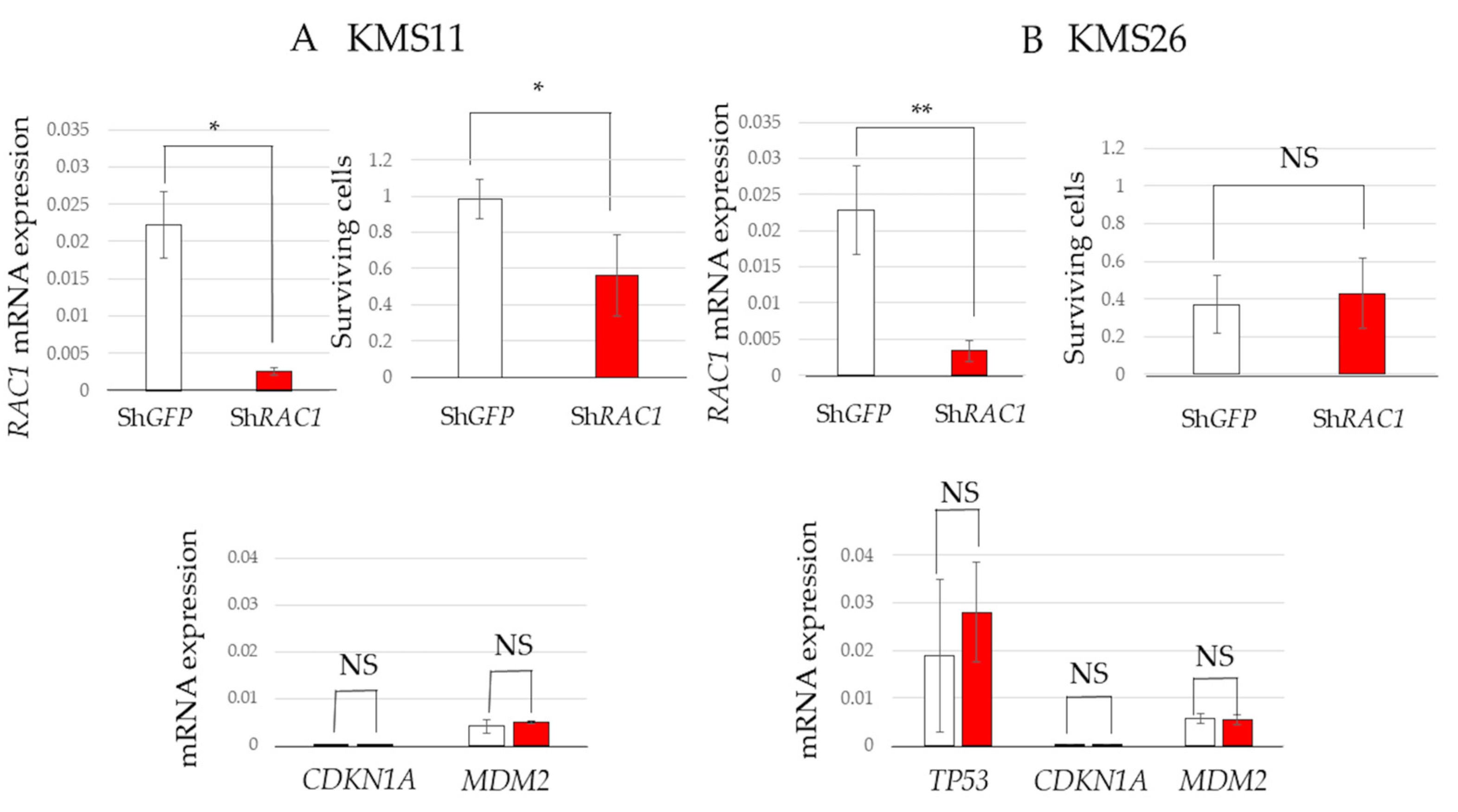
3.4. Cell Survival in RAC1 Knockdown MM Cells and Transcriptome Analysis
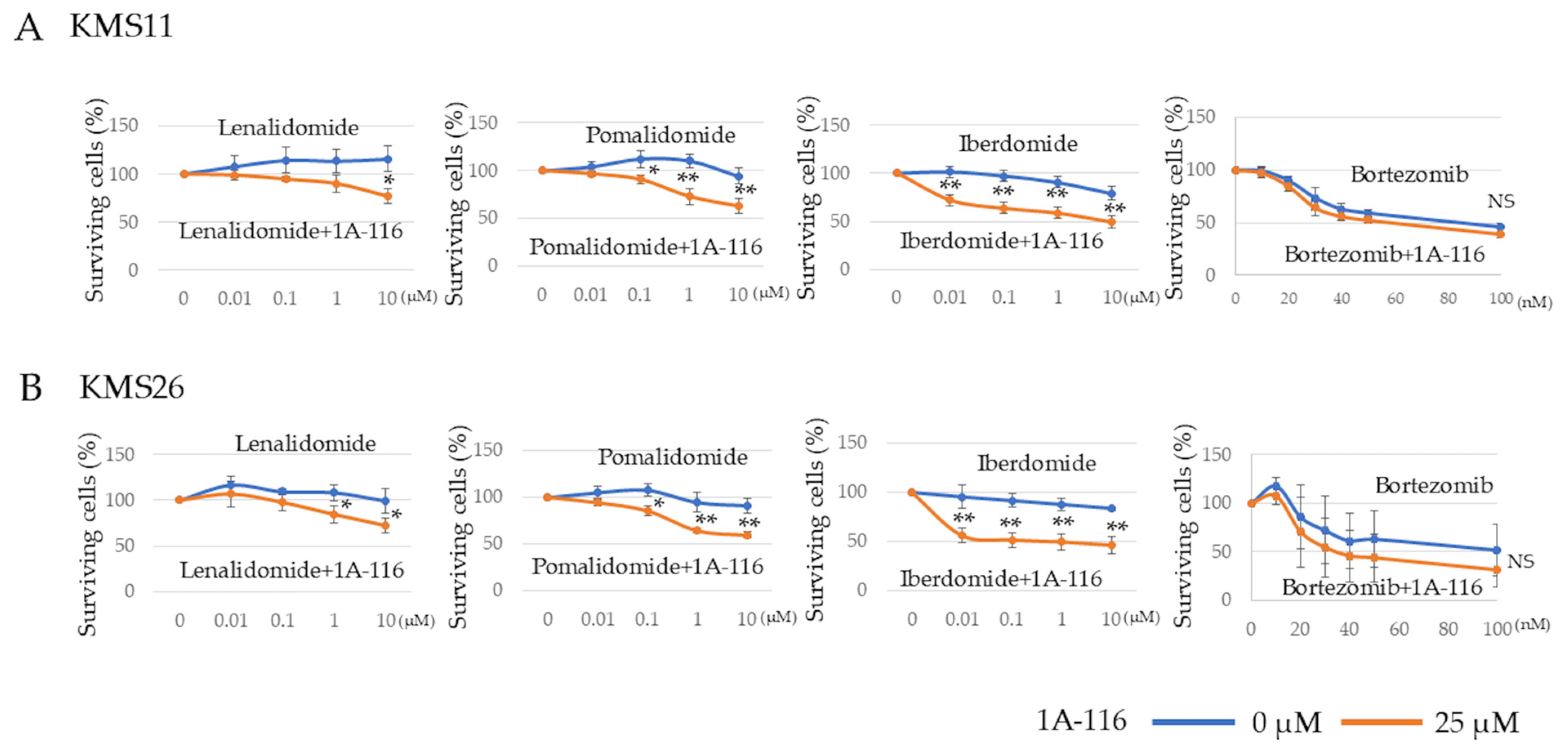
3.5. Effect of Rac1 on Sensitivity Against Cereblon Modulators and Transcriptome Analysis
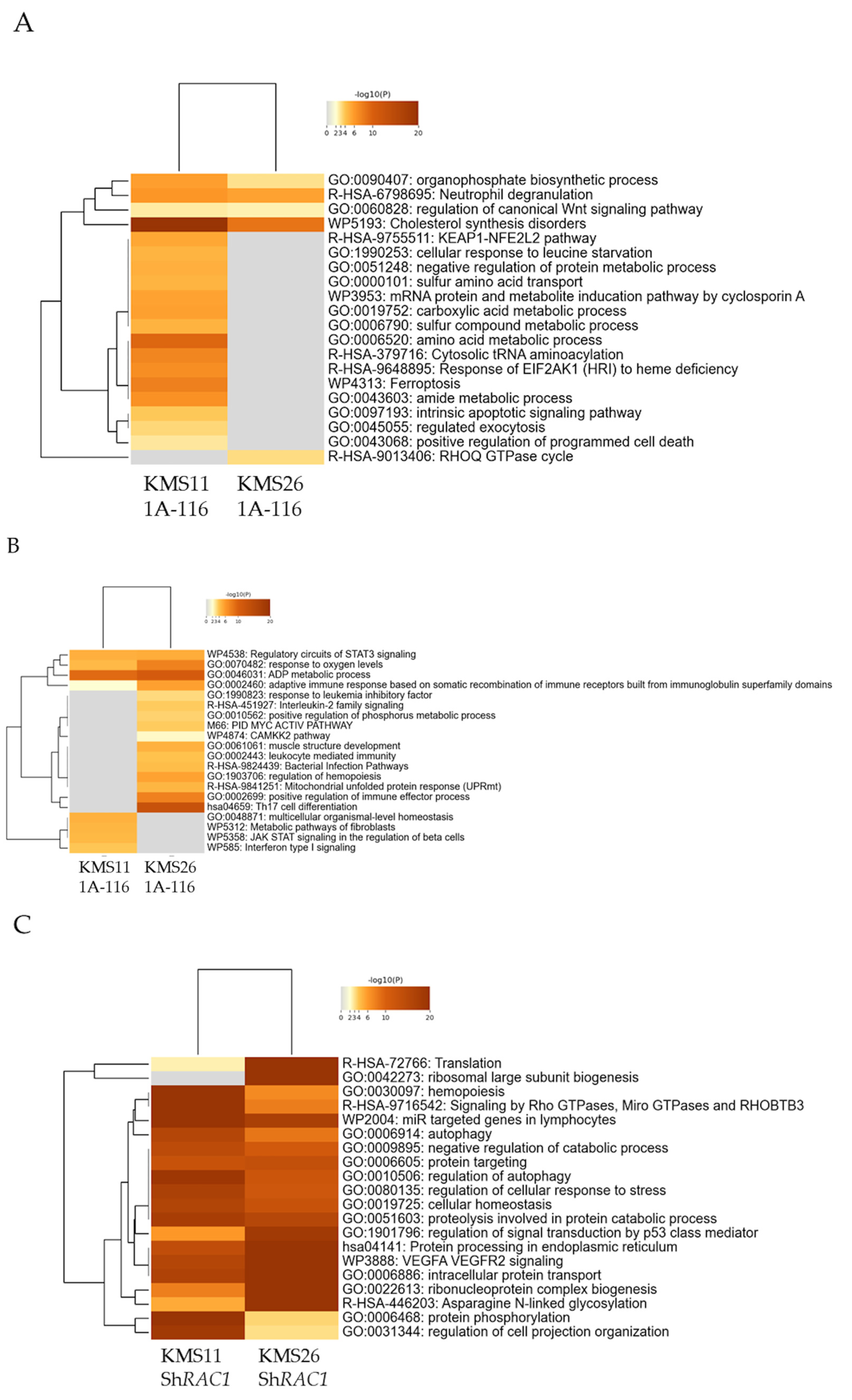
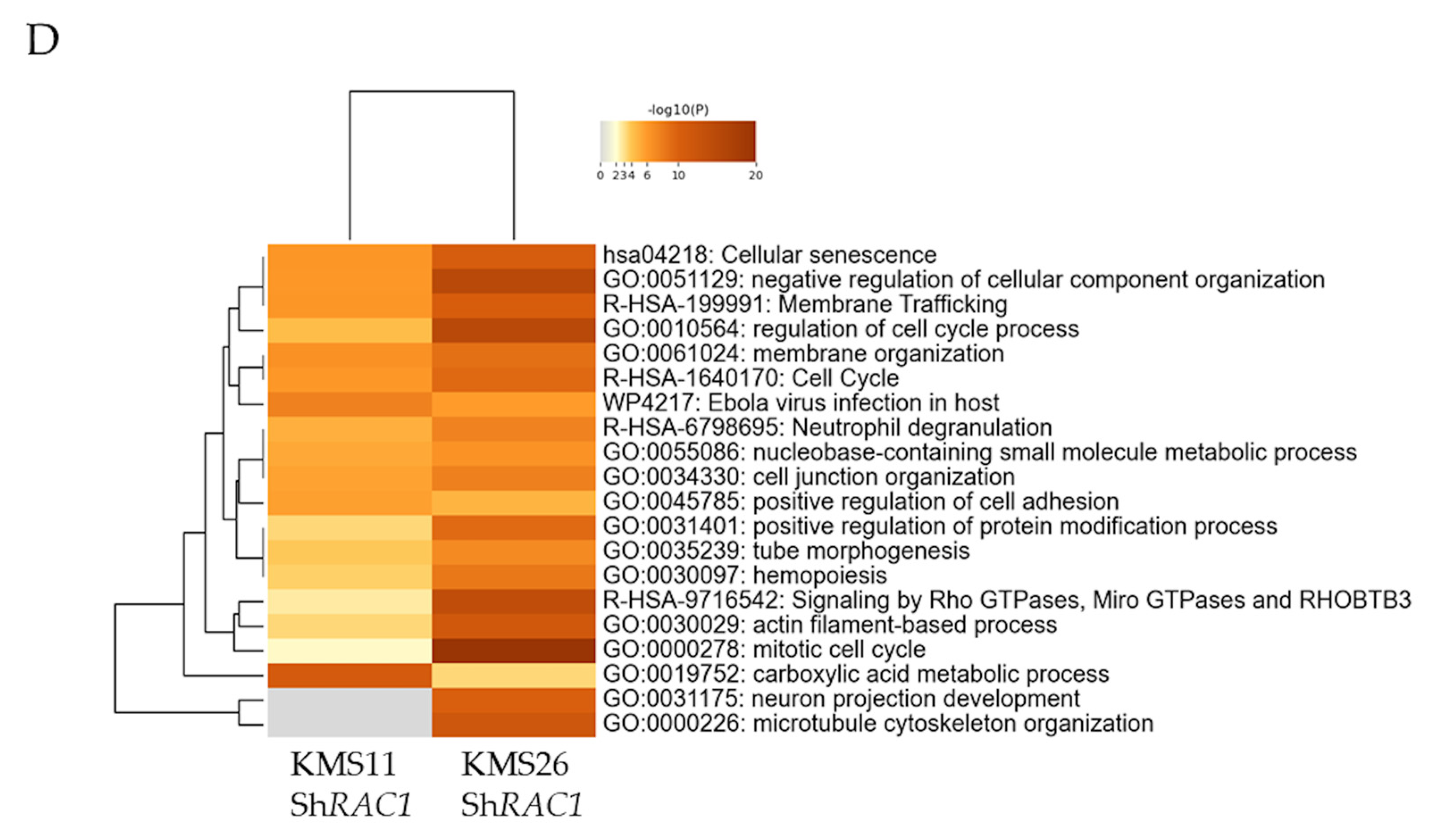
3.6. Alteration of Gene Expression Profile by Rac1 Inhibition
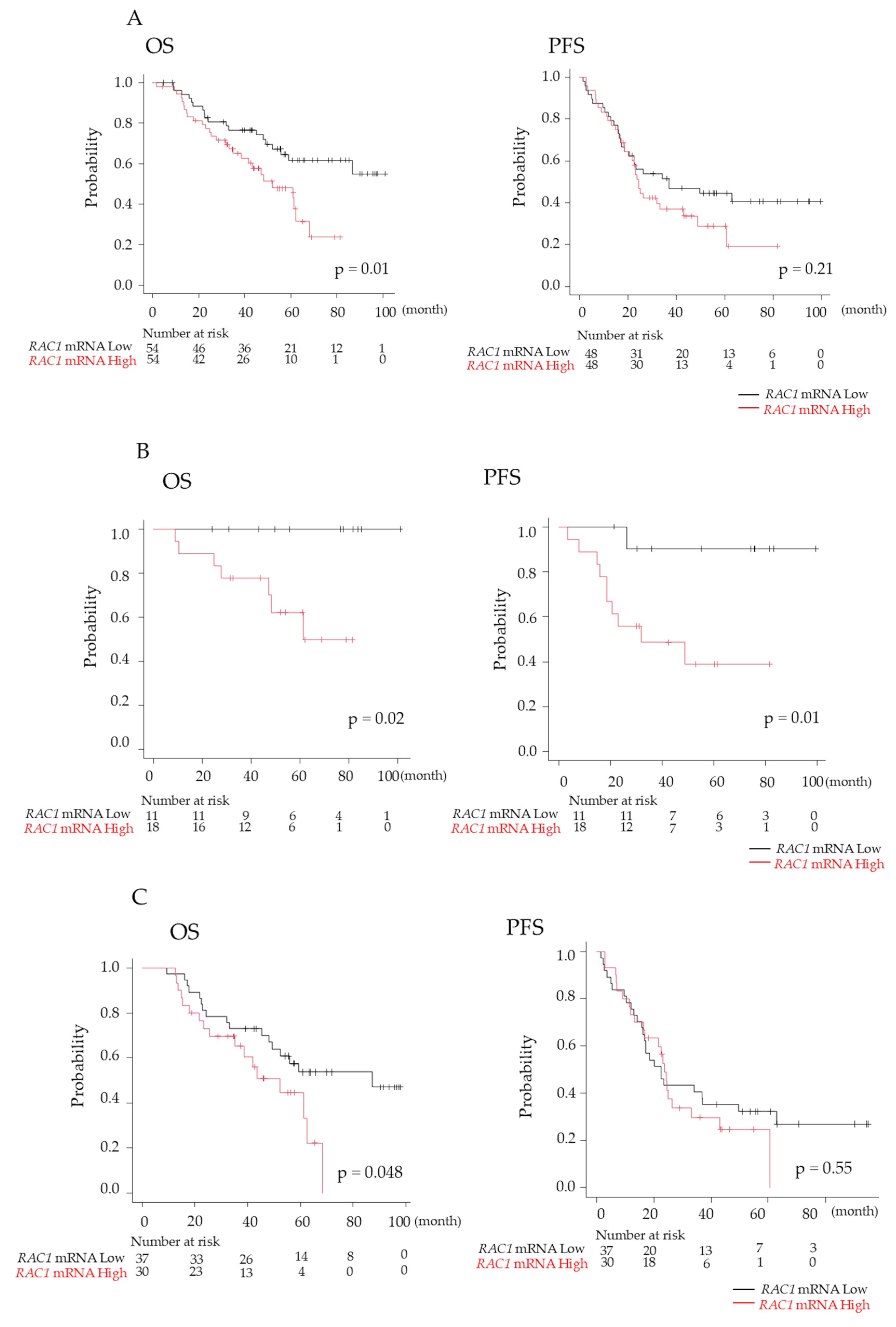
3.7. Prognosis of Patients with MM
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Matsumura, I.; Oda, T.; Kasamatsu, T.; Murakami, Y.; Ishihara, R.; Ohmori, A.; Gotoh, N.; Matsumoto, A.; Kobayashi, N.; Miyazawa, Y.; et al. Role of Ras-Related C3 Botulinus Toxin Substrate 1 in p53-Related Proliferation and Drug Sensitivity in Multiple Myeloma. In Proceedings of the 66th American Society of Hematology Annual Meeting and Exposition, San Diego, CA, USA, 8 December 2024. [Google Scholar]
- Kyle, R.A.; Rajkumar, S.V. Multiple myeloma. Blood 2008, 111, 2962–2972. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, A.; Anderson, K. Multiple myeloma. N. Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar] [CrossRef]
- Chng, W.J.; Dispenzieri, A.; Chim, C.S.; Fonseca, R.; Goldschmidt, H.; Lentzsch, S.; Munshi, N.; Palumbo, A.; Miguel, J.S.; Sonneveld, P.; et al. IMWG consensus on risk stratification in multiple myeloma. Leukemia 2014, 28, 269–277. [Google Scholar] [CrossRef]
- Kanamori, T.; Sanada, M.; Ri, M.; Ueno, H.; Nishijima, D.; Yasuda, T.; Tachita, T.; Narita, T.; Kusumoto, S.; Inagaki, A.; et al. Genomic analysis of multiple myeloma using targeted capture sequencing in the Japanese cohort. Br. J. Haematol. 2020, 191, 755–763. [Google Scholar] [CrossRef]
- Corre, J.; Perrot, A.; Hulin, C.; Caillot, D.; Stoppa, A.M.; Facon, T.; Leleu, X.; Dib, M.; Karlin, L.; Moreau, P.; et al. Improved survival in multiple myeloma during the 2005–2009 and 2010–2014 periods. Leukemia 2021, 35, 3600–3603. [Google Scholar] [CrossRef] [PubMed]
- Boureux, A.; Vignal, E.; Faure, S.; Fort, P. Evolution of the Rho family of ras-like GTPases in eukaryotes. Mol. Biol. Evol. 2007, 24, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Crosas-Molist, E.; Samain, R.; Kohlhammer, L.; Orgaz, J.L.; George, S.L.; Maiques, O.; Barcelo, J.; Sanz-Moreno, V. Rho GTPase signaling in cancer progression and dissemination. Physiol. Rev. 2022, 102, 455–510. [Google Scholar] [CrossRef]
- Feng, X.; Zhang, L.; Nie, S.; Zhuang, L.; Wang, W.; Huang, J.; Yan, X.; Meng, F. The Effect of Ras Homolog C/Rho-Associated Coiled-Protein Kinase (Rho/ROCK) Signaling Pathways on Proliferation and Apoptosis of Human Myeloma Cells. Med. Sci. Monit. 2019, 25, 7605–7616. [Google Scholar] [CrossRef]
- Bosco, E.E.; Ni, W.; Wang, L.; Guo, F.; Johnson, J.F.; Zheng, Y. Rac1 targeting suppresses p53 deficiency-mediated lymphomagenesis. Blood 2010, 115, 3320–3328. [Google Scholar] [CrossRef]
- Murakami, Y.; Kimura-Masuda, K.; Oda, T.; Matsumura, I.; Masuda, Y.; Ishihara, R.; Watanabe, S.; Kuroda, Y.; Kasamatsu, T.; Gotoh, N.; et al. MYC Causes Multiple Myeloma Progression via Attenuating TP53-Induced MicroRNA-34 Expression. Genes 2022, 14, 100. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, R.; Oda, T.; Murakami, Y.; Matsumura, I.; Watanabe, S.; Asao, Y.; Masuda, Y.; Gotoh, N.; Kasamatsu, T.; Takei, H.; et al. Myeloma Microenvironmental TIMP1 Induces the Invasive Phenotype in Fibroblasts to Modulate Disease Progression. Int. J. Mol. Sci. 2023, 24, 2216. [Google Scholar] [CrossRef] [PubMed]
- Kanda, Y. Investigation of the freely available easy-to-use software ’EZR’ for medical statistics. Bone Marrow Transplant. 2013, 48, 452–458. [Google Scholar] [CrossRef]
- Attal, M.; Lauwers-Cances, V.; Hulin, C.; Leleu, X.; Caillot, D.; Escoffre, M.; Arnulf, B.; Macro, M.; Belhadj, K.; Garderet, L.; et al. Lenalidomide, Bortezomib, and Dexamethasone with Transplantation for Myeloma. N. Engl. J. Med. 2017, 376, 1311–1320. [Google Scholar] [CrossRef]
- Klein, E.A.; Campbell, L.E.; Kothapalli, D.; Fournier, A.K.; Assoian, R.K. Joint requirement for Rac and ERK activities underlies the mid-G1 phase induction of cyclin D1 and S phase entry in both epithelial and mesenchymal cells. J. Biol. Chem. 2008, 283, 30911–30918. [Google Scholar] [CrossRef]
- Yan, Y.; Greer, P.M.; Cao, P.T.; Kolb, R.H.; Cowan, K.H. RAC1 GTPase plays an important role in γ-irradiation induced G2/M checkpoint activation. Breast Cancer Res. 2012, 14, R60. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhang, Y.; Shacter, E. Rac1 inhibits apoptosis in human lymphoma cells by stimulating Bad phosphorylation on Ser-75. Mol. Cell. Biol. 2004, 24, 6205–6214. [Google Scholar] [CrossRef]
- Cardama, G.A.; Alonso, D.F.; Gonzalez, N.; Maggio, J.; Gomez, D.E.; Rolfo, C.; Menna, P.L. Relevance of small GTPase Rac1 pathway in drug and radio-resistance mechanisms: Opportunities in cancer therapeutics. Crit. Rev. Oncol. Hematol. 2018, 124, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Hofbauer, S.W.; Krenn, P.W.; Ganghammer, S.; Asslaber, D.; Pichler, U.; Oberascher, K.; Henschler, R.; Wallner, M.; Kerschbaum, H.; Greil, R.; et al. Tiam1/Rac1 signals contribute to the proliferation and chemoresistance, but not motility, of chronic lymphocytic leukemia cells. Blood 2014, 123, 2181–2188. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Yu, P.; Chen, S.; Xing, H.; Chen, Y.; Wang, M.; Tang, K.; Tian, Z.; Rao, Q.; Wang, J. Activation of Rac1 GTPase promotes leukemia cell chemotherapy resistance, quiescence and niche interaction. Mol. Oncol. 2013, 7, 907–916. [Google Scholar] [CrossRef]
- Velaithan, R.; Kang, J.; Hirpara, J.L.; Loh, T.; Goh, B.C.; Le Bras, M.; Brenner, C.; Clement, M.V.; Pervaiz, S. The small GTPase Rac1 is a novel binding partner of Bcl-2 and stabilizes its antiapoptotic activity. Blood 2011, 117, 6214–6226. [Google Scholar] [CrossRef]
- Yamamoto, T.; Nakayama, J.; Yamamoto, Y.; Kuroda, M.; Hattori, Y.; Ochiya, T. SORT1/LAMP2-mediated extracellular vesicle secretion and cell adhesion are linked to lenalidomide resistance in multiple myeloma. Blood Adv. 2022, 6, 2480–2495. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.X.; Braggio, E.; Shi, C.X.; Bruins, L.A.; Schmidt, J.E.; Van Wier, S.; Chang, X.B.; Bjorklund, C.C.; Fonseca, R.; Bergsagel, P.L.; et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood 2011, 118, 4771–4779. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Girona, A.; Mendy, D.; Ito, T.; Miller, K.; Gandhi, A.K.; Kang, J.; Karasawa, S.; Carmel, G.; Jackson, P.; Abbasian, M.; et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia 2012, 26, 2326–2335. [Google Scholar] [CrossRef]
- Bjorklund, C.C.; Kang, J.; Amatangelo, M.; Polonskaia, A.; Katz, M.; Chiu, H.; Couto, S.; Wang, M.; Ren, Y.; Ortiz, M.; et al. Iberdomide (CC-220) is a potent cereblon E3 ligase modulator with antitumor immunostimulatory activities in, l.e.n.a.l.i.d.o.m.i.d.e.-; pomalidomide-resistant multiple myeloma cells with dysregulated CRBN. Leukemia 2020, 34, 1197–1201. [Google Scholar] [CrossRef]
- Keifer, J.A.; Guttridge, D.C.; Ashburner, B.P.; Baldwin, A.S. Inhibition of NF-kappa B activity by thalidomide through suppression of IkappaB kinase activity. J. Biol. Chem. 2001, 276, 22382–22387. [Google Scholar] [CrossRef] [PubMed]
- Mitsiades, N.; Mitsiades, C.S.; Poulaki, V.; Chauhan, D.; Richardson, P.G.; Hideshima, T.; Munshi, N.C.; Treon, S.P.; Anderson, K.C. Apoptotic signaling induced by immunomodulatory thalidomide analogs in human multiple myeloma cells: Therapeutic implications. Blood 2002, 99, 4525–4530. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, G.; Oliva, L.; Cascio, P.; Pengo, N.; Fontana, F.; Cerruti, F.; Orsi, A.; Pasqualetto, E.; Mezghrani, A.; Calbi, V.; et al. The proteasome load versus capacity balance determines apoptotic sensitivity of multiple myeloma cells to proteasome inhibition. Blood 2009, 113, 3040–3049. [Google Scholar] [CrossRef]
- Menu, E.; Vanderkerken, K. Exosomes in multiple myeloma: From bench to bedside. Blood 2022, 140, 2429–2442. [Google Scholar] [CrossRef]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, S.; Tagawa, H. Impact of hypoxia on the pathogenesis and therapy resistance in multiple myeloma. Cancer Sci. 2021, 112, 3995–4004. [Google Scholar] [CrossRef]
- Zhang, J.G.; Zhou, H.M.; Zhang, X.; Mu, W.; Hu, J.N.; Liu, G.L.; Li, Q. Hypoxic induction of vasculogenic mimicry in hepatocellular carcinoma: Role of HIF-1 α, RhoA/ROCK and Rac1/PAK signaling. BMC Cancer 2020, 20, 32. [Google Scholar] [CrossRef] [PubMed]
- Azab, A.K.; Azab, F.; Blotta, S.; Pitsillides, C.M.; Thompson, B.; Runnels, J.M.; Roccaro, A.M.; Ngo, H.T.; Melhem, M.R.; Sacco, A.; et al. RhoA and Rac1 GTPases play major and differential roles in stromal cell-derived factor-1-induced cell adhesion and chemotaxis in multiple myeloma. Blood 2009, 114, 619–629. [Google Scholar] [CrossRef]
- Floerchinger, A.; Murphy, K.J.; Latham, S.L.; Warren, S.C.; McCulloch, A.T.; Lee, Y.K.; Stoehr, J.; Mélénec, P.; Guaman, C.S.; Metcalf, X.L.; et al. Optimizing metastatic-cascade-dependent Rac1 targeting in breast cancer: Guidance using optical window intravital FRET imaging. Cell Rep. 2021, 36, 109689. [Google Scholar] [CrossRef]
- Cardama, G.A.; Maggio, J.; Valdez Capuccino, L.; Gonzalez, N.; Matiller, V.; Ortega, H.H.; Perez, G.R.; Demarco, I.A.; Spitzer, E.; Gomez, D.E.; et al. Preclinical Efficacy and Toxicology Evaluation of RAC1 Inhibitor 1A-116 in Human Glioblastoma Models. Cancers 2022, 14, 4810. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RAC1 High (n = 57) | RAC1 Low (n = 57) | p-Value | |
|---|---|---|---|
| Age | 71 (41–87) | 71 (42–86) | 1.00 |
| Sex | 0.70 | ||
| Male | 24 (42.1%) | 21 (36.8%) | |
| Female | 33 (57.9%) | 36 (63.2%) | |
| ALB | 3.45 (2.0–4.8) | 3.60 (2.2–4.6) | 0.73 |
| Hb | 10.65 (5.9–15.7) | 10.10 (6.3–14.6) | 0.17 |
| LDH | 177 (92–353) | 169 (99–497) | 0.78 |
| β2MG | 3.8 (1.4–27.0) | 4.4 (1.8–29.1) | 0.21 |
| IgH | 0.43 | ||
| IgG | 37 (66.1%) | 32 (56.1%) | |
| IgA | 12 (21.4%) | 16 (28.1%) | |
| IgD | 0 (0.0%) | 2 (3.5%) | |
| IgE | 0 (0.0%) | 1 (1.8%) | |
| BJ | 7 (12.5%) | 5 (8.8%) | |
| Non | 0 (0.0%) | 1 (1.8%) | |
| IgL | 0.16 | ||
| κ | 35 (62.5%) | 27 (47.4%) | |
| λ | 21 (37.5%) | 29 (50.9%) | |
| Non | 0 (0.0%) | 1 (1.8%) | |
| Cytogenetic Risk | 0.28 | ||
| Standard | 43 (79.6%) | 40 (70.2%) | |
| High | 11 (20.4%) | 17 (29.8%) | |
| ISS | 0.59 | ||
| 1 | 15 (27.3%) | 11 (21.2%) | |
| 2 | 24 (43.6%) | 21 (40.4%) | |
| 3 | 16 (29.1%) | 20 (38.5%) | |
| R-ISS | 0.49 | ||
| 1 | 12 (22.6%) | 7 (13.2%) | |
| 2 | 36 (67.9%) | 40 (75.5%) | |
| 3 | 5 (9.4%) | 6 (11.3%) | |
| ASCT | 0.18 | ||
| Yes | 11 (22.9%) | 18 (37.5%) | |
| No | 37 (77.1%) | 30 (62.5%) |
| OS Hazard Ratio (95% CI) | p-Value | PFS Hazard Ratio (95% CI) | p-Value | ||
|---|---|---|---|---|---|
| R-ISS | ≥2 | 1.751 | 0.30 | 1.628 | 0.25 |
| (0.608–5.039) | (0.710–3.730) | ||||
| ASCT | Yes | 0.409 | 0.04 | 0.405 | 0.01 |
| (0.175–0.954) | (0.201–0.814) | ||||
| RAC1 mRNA | High | 2.211 | 0.02 | 1.287 | 0.37 |
| (1.123–4.350) | (0.738–2.243) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsumura, I.; Oda, T.; Kasamatsu, T.; Murakami, Y.; Ishihara, R.; Ohmori, A.; Matsumoto, A.; Gotoh, N.; Kobayashi, N.; Miyazawa, Y.; et al. Role of Rac1 in p53-Related Proliferation and Drug Sensitivity in Multiple Myeloma. Cancers 2025, 17, 461. https://doi.org/10.3390/cancers17030461
Matsumura I, Oda T, Kasamatsu T, Murakami Y, Ishihara R, Ohmori A, Matsumoto A, Gotoh N, Kobayashi N, Miyazawa Y, et al. Role of Rac1 in p53-Related Proliferation and Drug Sensitivity in Multiple Myeloma. Cancers. 2025; 17(3):461. https://doi.org/10.3390/cancers17030461
Chicago/Turabian StyleMatsumura, Ikuko, Tsukasa Oda, Tetsuhiro Kasamatsu, Yuki Murakami, Rei Ishihara, Ayane Ohmori, Akira Matsumoto, Nanami Gotoh, Nobuhiko Kobayashi, Yuri Miyazawa, and et al. 2025. "Role of Rac1 in p53-Related Proliferation and Drug Sensitivity in Multiple Myeloma" Cancers 17, no. 3: 461. https://doi.org/10.3390/cancers17030461
APA StyleMatsumura, I., Oda, T., Kasamatsu, T., Murakami, Y., Ishihara, R., Ohmori, A., Matsumoto, A., Gotoh, N., Kobayashi, N., Miyazawa, Y., Ogawa, Y., Yokohama, A., Sasaki, N., Saitoh, T., & Handa, H. (2025). Role of Rac1 in p53-Related Proliferation and Drug Sensitivity in Multiple Myeloma. Cancers, 17(3), 461. https://doi.org/10.3390/cancers17030461