Robust Cluster Prediction Across Data Types Validates Association of Sex and Therapy Response in GBM

 , , , , , and
, , , , , and 
Simple Summary
Abstract
1. Introduction
2. Approach
2.1. Statistical Decision-Making Approach
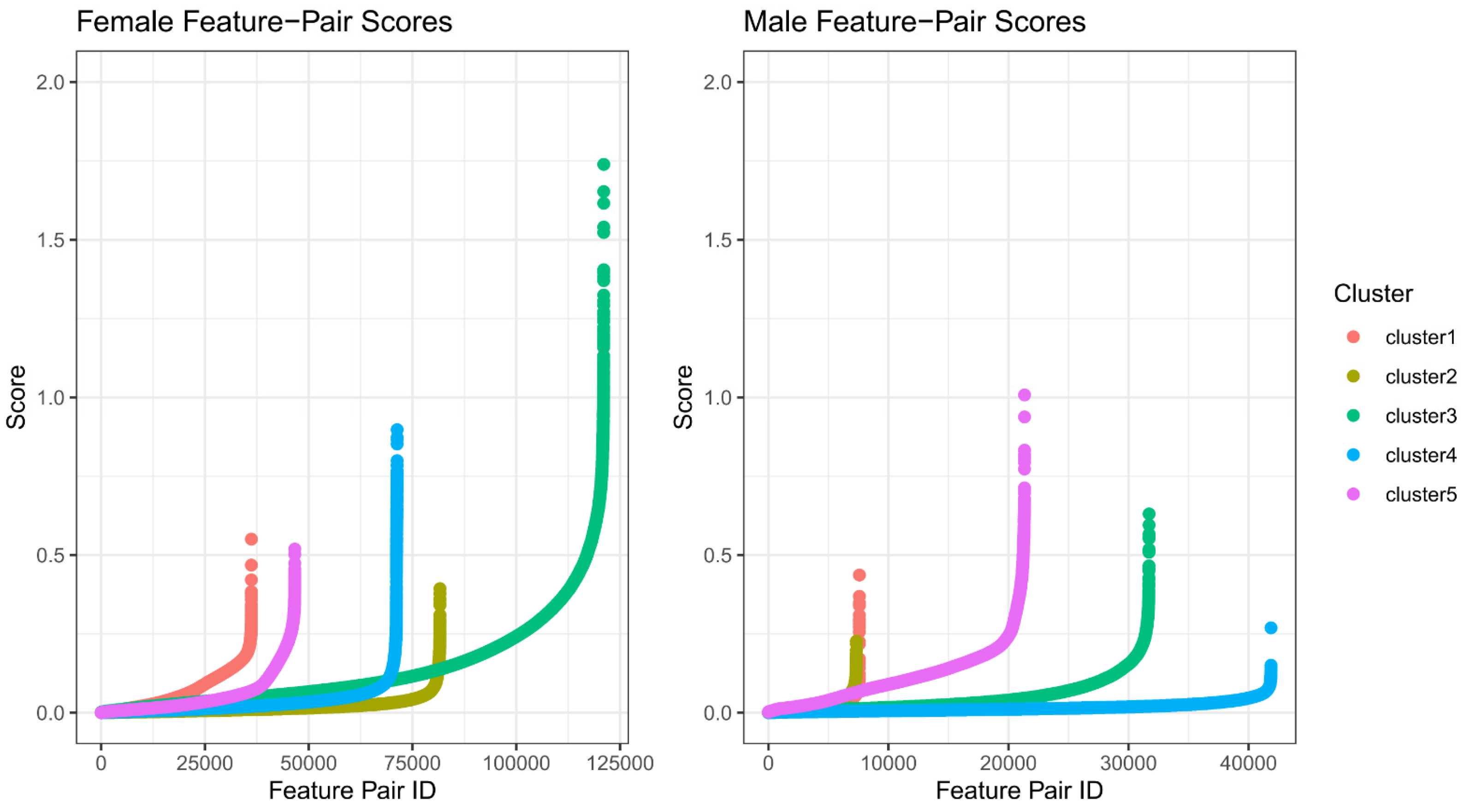
2.2. Feature Selection for Cluster Prediction
2.3. Robust Ensemble Model Training
3. Results
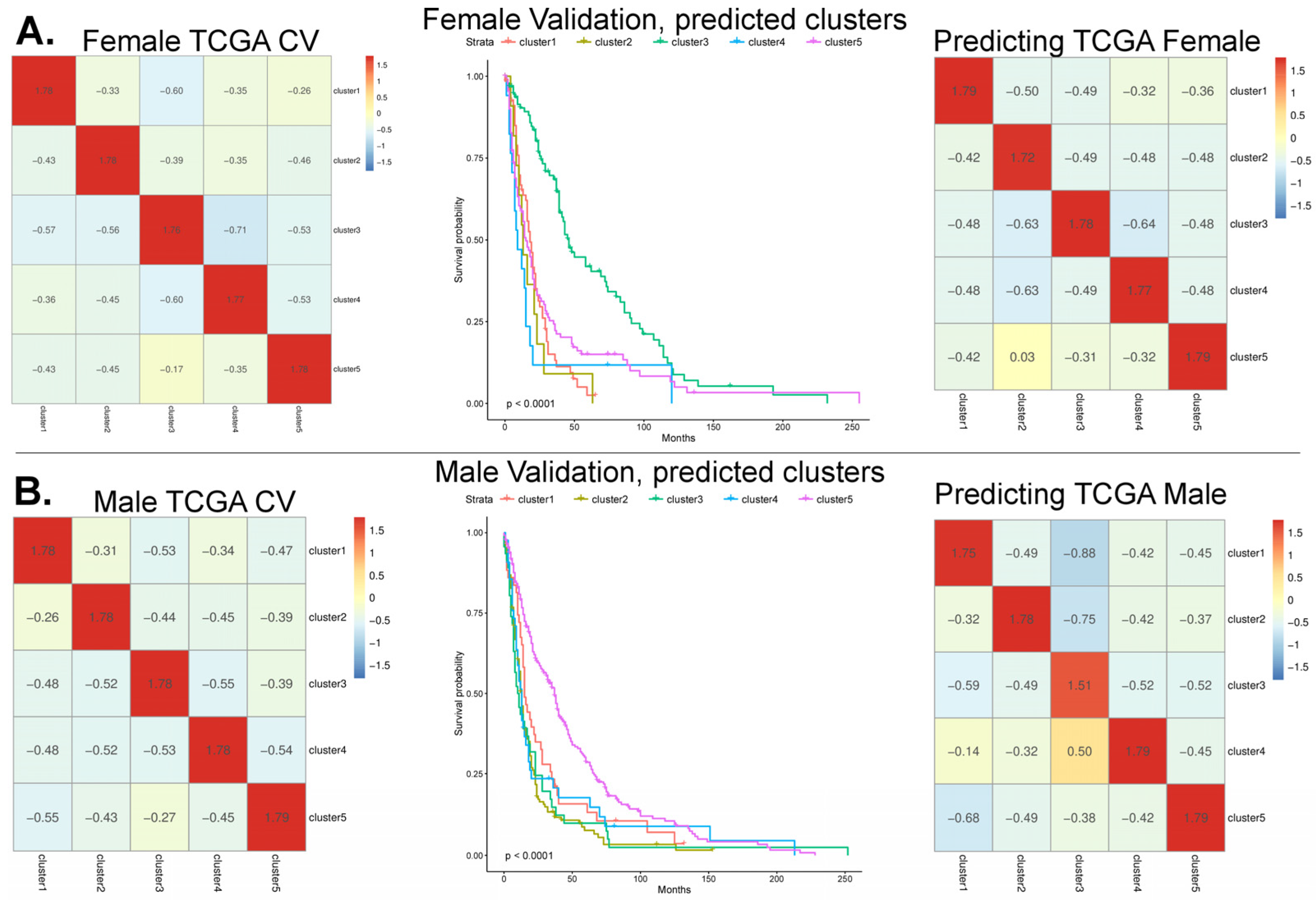
3.1. Model Cross-Validation Using TCGA-GBM Array-Based Data
3.2. Validation of the Array-Based Model with Three Additional Datasets
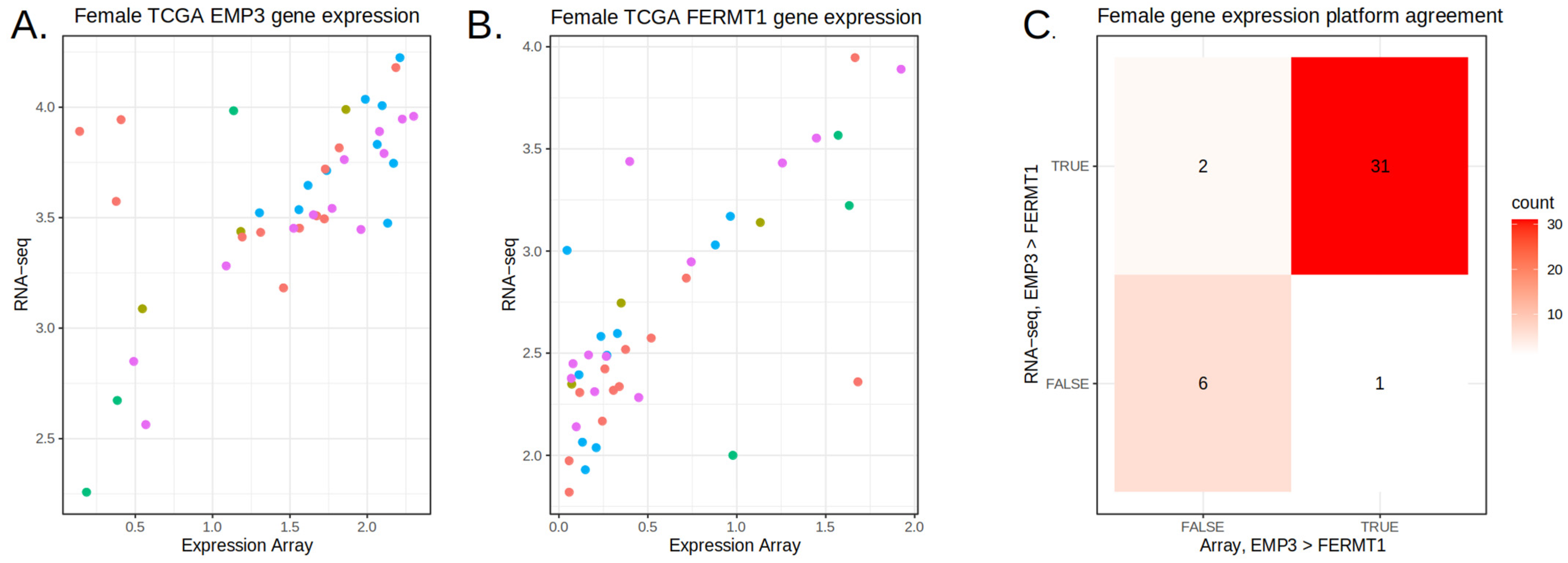
3.3. Feature Refinement Improves Generalizability of Classification
3.4. Prediction on the TEMPUS and CPTAC3 RNA-Seq Datasets
3.5. Aggregating Information Rankings Across Datasets
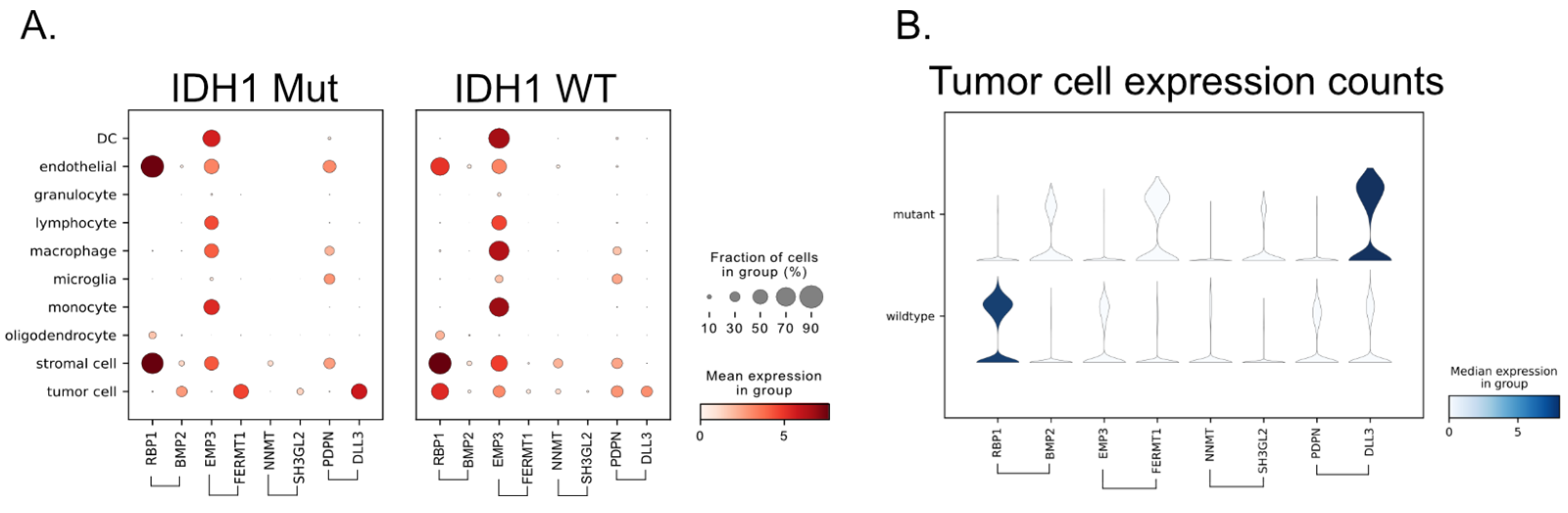
3.6. Single-Cell RNA-Seq Provides the Source of the Signal
4. Discussion
5. Conclusions
6. Methods
6.1. Data Sources
6.2. Gene Microarray Data Processing
6.3. RNA-Seq Processing and Acquisition
6.4. Feature Selection
6.5. Param Search
6.6. Survival Curves
6.7. Classification Task Order
6.8. Feature Alignment
6.9. Single-Cell RNA-Seq
6.10. Code Repository
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhao, L.; Lee, V.H.F.; Ng, M.K.; Yan, H.; Bijlsma, M.F. Molecular Subtyping of Cancer: Current Status and Moving toward Clinical Applications. Brief. Bioinform. 2019, 20, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.M.; Yau, C.; Wulfkuhle, J.; Brown-Swigart, L.; Gallagher, R.I.; Lee, P.R.E.; Zhu, Z.; Magbanua, M.J.; Sayaman, R.; O’Grady, N.; et al. Redefining Breast Cancer Subtypes to Guide Treatment Prioritization and Maximize Response: Predictive Biomarkers across 10 Cancer Therapies. Cancer Cell 2022, 40, 609–623.e6. [Google Scholar] [CrossRef] [PubMed]
- Benam, K.H.; Gilchrist, S.; Kleensang, A.; Satz, A.B.; Willett, C.; Zhang, Q. Exploring New Technologies in Biomedical Research. Drug Discov. Today 2019, 24, 1242–1247. [Google Scholar] [CrossRef] [PubMed]
- Hartl, D.; de Luca, V.; Kostikova, A.; Laramie, J.; Kennedy, S.; Ferrero, E.; Siegel, R.; Fink, M.; Ahmed, S.; Millholland, J.; et al. Translational Precision Medicine: An Industry Perspective. J. Transl. Med. 2021, 19, 245. [Google Scholar] [CrossRef] [PubMed]
- Clough, E.; Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; et al. NCBI GEO: Archive for Gene Expression and Epigenomics Data Sets: 23-Year Update. Nucleic Acids Res. 2023, 52, D138–D144. [Google Scholar] [CrossRef]
- Kim, Y.-M.; Poline, J.-B.; Dumas, G. Experimenting with Reproducibility: A Case Study of Robustness in Bioinformatics. GigaScience 2018, 7, giy077. [Google Scholar] [CrossRef]
- Kanderi, T.; Munakomi, S.; Gupta, V. Glioblastoma Multiforme. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.M.; Lu, C.; Ward, P.S.; et al. IDH1 Mutation Is Sufficient to Establish the Glioma Hypermethylator Phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef]
- Pirozzi, C.J.; Yan, H. The Implications of IDH Mutations for Cancer Development and Therapy. Nat. Rev. Clin. Oncol. 2021, 18, 645–661. [Google Scholar] [CrossRef]
- Yang, W.; Warrington, N.M.; Taylor, S.J.; Whitmire, P.; Carrasco, E.; Singleton, K.W.; Wu, N.; Lathia, J.D.; Berens, M.E.; Kim, A.H.; et al. Sex Differences in GBM Revealed by Analysis of Patient Imaging, Transcriptome, and Survival Data. Sci. Transl. Med. 2019, 11, eaao5253. [Google Scholar] [CrossRef]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG Island Methylator Phenotype That Defines a Distinct Subgroup of Glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef]
- Lee, Y.; Scheck, A.C.; Cloughesy, T.F.; Lai, A.; Dong, J.; Farooqi, H.K.; Liau, L.M.; Horvath, S.; Mischel, P.S.; Nelson, S.F. Gene Expression Analysis of Glioblastomas Identifies the Major Molecular Basis for the Prognostic Benefit of Younger Age. BMC Med. Genom. 2008, 1, 52. [Google Scholar] [CrossRef] [PubMed]
- Gravendeel, L.A.M.; Kouwenhoven, M.C.M.; Gevaert, O.; de Rooi, J.J.; Stubbs, A.P.; Duijm, J.E.; Daemen, A.; Bleeker, F.E.; Bralten, L.B.C.; Kloosterhof, N.K.; et al. Intrinsic Gene Expression Profiles of Gliomas Are a Better Predictor of Survival than Histology. Cancer Res. 2009, 69, 9065–9072. [Google Scholar] [CrossRef] [PubMed]
- Gusev, Y.; Bhuvaneshwar, K.; Song, L.; Zenklusen, J.-C.; Fine, H.; Madhavan, S. The REMBRANDT Study, a Large Collection of Genomic Data from Brain Cancer Patients. Sci. Data 2018, 5, 180158. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Whiteaker, J.R.; Halusa, G.N.; Hoofnagle, A.N.; Sharma, V.; MacLean, B.; Yan, P.; Wrobel, J.A.; Kennedy, J.; Mani, D.R.; Zimmerman, L.J.; et al. CPTAC Assay Portal: A Repository of Targeted Proteomic Assays. Nat. Methods 2014, 11, 703–704. [Google Scholar] [CrossRef]
- Shah, N.; Park, H.J.; Sonpatki, P.; Schroeder, B.; Han, K.Y.; Kim, H.J.; Chowdhury, T.; Hwang, J.H.; Nam, S.M.; Byun, Y.H.; et al. A Spatially Resolved Human Glioblastoma Atlas Reveals Distinct Cellular and Molecular Patterns of Anatomical Niche 2024. preprint 2024. [Google Scholar] [CrossRef]
- Geman, D.; d’Avignon, C.; Naiman, D.Q.; Winslow, R.L. Classifying Gene Expression Profiles from Pairwise MRNA Comparisons. Stat. Appl. Genet. Mol. Biol. 2004, 3, 19. [Google Scholar] [CrossRef]
- Tan, A.C.; Naiman, D.Q.; Xu, L.; Winslow, R.L.; Geman, D. Simple Decision Rules for Classifying Human Cancers from Gene Expression Profiles. Bioinformatics 2005, 21, 3896–3904. [Google Scholar] [CrossRef]
- Eddy, J.A.; Geman, D.; Price, N.D. Relative Expression Analysis for Identifying Perturbed Pathways. In Proceedings of the 2009 Annual International Conference of the IEEE Engineering in Medicine and Biology Society, Minneapolis, MN, USA, 3–6 September 2009; pp. 5456–5459. [Google Scholar] [CrossRef]
- Tong, M.; Liu, K.-H.; Xu, C.; Ju, W. An Ensemble of SVM Classifiers Based on Gene Pairs. Comput. Biol. Med. 2013, 43, 729–737. [Google Scholar] [CrossRef]
- Yoon, S.; Kim, S. K-Top Scoring Pair Algorithm for Feature Selection in SVM with Applications to Microarray Data Classification. Soft Comput. 2010, 14, 151–159. [Google Scholar] [CrossRef]
- Leek, J.T. The Tspair Package for Finding Top Scoring Pair Classifiers in R. Bioinformatics 2009, 25, 1203–1204. [Google Scholar] [CrossRef] [PubMed]
- Magis, A.T.; Price, N.D. The Top-Scoring “N” Algorithm: A Generalized Relative Expression Classification Method from Small Numbers of Biomolecules. BMC Bioinform. 2012, 13, 227. [Google Scholar] [CrossRef] [PubMed]
- Czajkowski, M.; Krȩtowski, M. Top Scoring Pair Decision Tree for Gene Expression Data Analysis. Adv. Exp. Med. Biol. 2011, 696, 27–35. [Google Scholar] [PubMed]
- Marzouka, N.-A.-D.; Eriksson, P. MulticlassPairs: An R Package to Train Multiclass Pair-Based Classifier. Bioinformatics 2021, 37, 3043–3044. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, D.L. Robust Classification of Immune Subtypes in Cancer. bioRxiv 2020. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef]
- Tu, Y.; Stolovitzky, G.; Klein, U. Quantitative Noise Analysis for Gene Expression Microarray Experiments. Proc. Natl. Acad. Sci. USA 2002, 99, 14031–14036. [Google Scholar] [CrossRef]
- Marioni, J.C.; Mason, C.E.; Mane, S.M.; Stephens, M.; Gilad, Y. RNA-Seq: An Assessment of Technical Reproducibility and Comparison with Gene Expression Arrays. Genome Res. 2008, 18, 1509–1517. [Google Scholar] [CrossRef]
- Agarwal, A.; Koppstein, D.; Rozowsky, J.; Sboner, A.; Habegger, L.; Hillier, L.W.; Sasidharan, R.; Reinke, V.; Waterston, R.H.; Gerstein, M. Comparison and Calibration of Transcriptome Data from RNA-Seq and Tiling Arrays. BMC Genom. 2010, 11, 383. [Google Scholar] [CrossRef]
- Seidl, F.; Hagen, L.; Wilson, J.; Aguilar, B.; Bleich, D.; Wolfe, L.; Gundluru, P.; Venkatesan, P.; Tian, M.; Paquette, S.; et al. The ISB Cancer Gateway in the Cloud (ISB-CGC): Access, Explore and Analyze Large-Scale Cancer Data through the Google Cloud. Cancer Res. 2024, 84, 3547. [Google Scholar] [CrossRef]
- Reynolds, S.M.; Miller, M.; Lee, P.; Leinonen, K.; Paquette, S.M.; Rodebaugh, Z.; Hahn, A.; Gibbs, D.L.; Slagel, J.; Longabaugh, W.J.; et al. The ISB Cancer Genomics Cloud: A Flexible Cloud-Based Platform for Cancer Genomics Research. Cancer Res. 2017, 77, e7–e10. [Google Scholar] [CrossRef] [PubMed]
- Andrieux, G.; Das, T.; Griffin, M.; Straehle, J.; Paine, S.M.L.; Beck, J.; Boerries, M.; Heiland, D.H.; Smith, S.J.; Rahman, R.; et al. Spatially Resolved Transcriptomic Profiles Reveal Unique Defining Molecular Features of Infiltrative 5ALA-Metabolizing Cells Associated with Glioblastoma Recurrence. Genome Med. 2023, 15, 48. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Davidsen, T.; Davis-Dusenbery, B.N.; Baumann, A.; Maggio, A.; Chen, Z.; Meerzaman, D.; Casas-Silva, E.; Pot, D.; Pihl, T.; et al. NCI Cancer Research Data Commons: Lessons Learned and Future State. Cancer Res. 2024, 84, 1404–1409. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Davidsen, T.M.; Kuffel, G.R.; Addepalli, K.; Bell, A.; Casas-Silva, E.; Dingerdissen, H.; Farahani, K.; Fedorov, A.; Gaheen, S.; et al. NCI Cancer Research Data Commons: Resources to Share Key Cancer Data. Cancer Res. 2024, 84, 1388–1395. [Google Scholar] [CrossRef]
- Fedorov, A.; Longabaugh, W.J.; Pot, D.; Clunie, D.A.; Pieper, S.; Aerts, H.J.; Homeyer, A.; Lewis, R.; Akbarzadeh, A.; Bontempi, D. NCI Imaging Data Commons. Cancer Res. 2021, 81, 4188–4193. [Google Scholar] [CrossRef]
- Thangudu, R.R.; Rudnick, P.A.; Holck, M.; Singhal, D.; MacCoss, M.J.; Edwards, N.J.; Ketchum, K.A.; Kinsinger, C.R.; Kim, E.; Basu, A. Abstract LB-242: Proteomic Data Commons: A Resource for Proteogenomic Analysis. Cancer Res. 2020, 80, LB-242. [Google Scholar] [CrossRef]
- Zhang, Z.; Hernandez, K.; Savage, J.; Li, S.; Miller, D.; Agrawal, S.; Ortuno, F.; Staudt, L.M.; Heath, A.; Grossman, R.L. Uniform Genomic Data Analysis in the NCI Genomic Data Commons. Nat. Commun. 2021, 12, 1226. [Google Scholar] [CrossRef]
- Wolf, S.; Melo, D.; Garske, K.M.; Pallares, L.F.; Lea, A.J.; Ayroles, J.F. Characterizing the Landscape of Gene Expression Variance in Humans. PLoS Genet. 2023, 19, e1010833. [Google Scholar] [CrossRef]
- Foltz, S.M.; Greene, C.S.; Taroni, J.N. Cross-Platform Normalization Enables Machine Learning Model Training on Microarray and RNA-Seq Data Simultaneously. Commun. Biol. 2023, 6, 222. [Google Scholar] [CrossRef]
- Yu, J.; Perri, M.; Jones, J.W.; Pierzchalski, K.; Ceaicovscaia, N.; Cione, E.; Kane, M.A. Altered RBP1 Gene Expression Impacts Epithelial Cell Retinoic Acid, Proliferation, and Microenvironment. Cells 2022, 11, 792. [Google Scholar] [CrossRef]
- Ji, P.; Shan, X.; Wang, J.; Zhang, P.; Cai, Z. Integrative Analysis of CBR1 as a Prognostic Factor Associated with IDH-Mutant Glioblastoma in the Chinese Population. Am. J. Transl. Res. 2022, 14, 5394–5408. [Google Scholar] [PubMed]
- Sintupisut, N.; Liu, P.-L.; Yeang, C.-H. An Integrative Characterization of Recurrent Molecular Aberrations in Glioblastoma Genomes. Nucleic Acids Res. 2013, 41, 8803–8821. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, D.; Cheng, S.; Fan, K.; Sheng, L.; Zhang, J.; Feng, B.; Xu, Z. The Correlation of Bone Morphogenetic Protein 2 with Poor Prognosis in Glioma Patients. Tumor Biol. 2014, 35, 11091–11095. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Zhao, Z.; Li, S.; Liu, Y.; Li, G.; Jiang, T. A New Glioma Grading Model Based on Histopathology and Bone Morphogenetic Protein 2 MRNA Expression. Sci. Rep. 2020, 10, 18420. [Google Scholar] [CrossRef]
- Modrek, A.S.; Eskilsson, E.; Ezhilarasan, R.; Wang, Q.; Goodman, L.D.; Ding, Y.; Zhang, Z.-Y.; Bhat, K.P.L.; Le, T.-T.T.; Barthel, F.P.; et al. PDPN Marks a Subset of Aggressive and Radiation-Resistant Glioblastoma Cells. Front. Oncol. 2022, 12, 941657. [Google Scholar] [CrossRef]
- He, J.; Zhang, G.; Yuan, Q.; Wang, S.; Liu, Z.; Wang, M.; Cai, H.; Wan, J.; Zhao, B. Overexpression of Podoplanin Predicts Poor Prognosis in Patients With Glioma. Appl. Immunohistochem. Mol. Morphol. 2023, 31, 295. [Google Scholar] [CrossRef]
- Wang, X.; Wang, X.; Li, J.; Liang, J.; Ren, X.; Yun, D.; Liu, J.; Fan, J.; Zhang, Y.; Zhang, J.; et al. PDPN Contributes to Constructing Immunosuppressive Microenvironment in IDH Wildtype Glioma. Cancer Gene Ther. 2023, 30, 345–357. [Google Scholar] [CrossRef]
- Noor, H.; Whittaker, S.; McDonald, K.L. DLL3 Expression and Methylation Are Associated with Lower-Grade Glioma Immune Microenvironment and Prognosis. Genomics 2022, 114, 110289. [Google Scholar] [CrossRef]
- Martija, A.A.; Pusch, S. The Multifunctional Role of EMP3 in the Regulation of Membrane Receptors Associated with IDH-Wild-Type Glioblastoma. Int. J. Mol. Sci. 2021, 22, 5261. [Google Scholar] [CrossRef]
- Chen, Q.; Jin, J.; Huang, X.; Wu, F.; Huang, H.; Zhan, R. EMP3 Mediates Glioblastoma-associated Macrophage Infiltration to Drive T Cell Exclusion. J. Exp. Clin. Cancer Res. 2021, 40, 160. [Google Scholar] [CrossRef]
- Ernst, A.; Hofmann, S.; Ahmadi, R.; Becker, N.; Korshunov, A.; Engel, F.; Hartmann, C.; Felsberg, J.; Sabel, M.; Peterziel, H.; et al. Genomic and Expression Profiling of Glioblastoma Stem Cell–Like Spheroid Cultures Identifies Novel Tumor-Relevant Genes Associated with Survival. Clin. Cancer Res. 2009, 15, 6541–6550. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.-H.; Wei, S.-T.; Liu, J.-J.; Chang, Y.-J.; Lin, Y.-F.; Yu, C.-S.; Chang, S.L.-Y. Recognition of a Novel Gene Signature for Human Glioblastoma. Int. J. Mol. Sci. 2022, 23, 4157. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Shim, J.-K.; Yoon, S.-J.; Kim, S.H.; Chang, J.H.; Kang, S.-G. Transcriptome Profiling-Based Identification of Prognostic Subtypes and Multi-Omics Signatures of Glioblastoma. Sci. Rep. 2019, 9, 10555. [Google Scholar] [CrossRef] [PubMed]
- Menyhárt, O.; Fekete, J.T.; Győrffy, B. Gene Expression-Based Biomarkers Designating Glioblastomas Resistant to Multiple Treatment Strategies. Carcinogenesis 2021, 42, 804–813. [Google Scholar] [CrossRef] [PubMed]
- Teng, L.; Nakada, M.; Furuyama, N.; Sabit, H.; Furuta, T.; Hayashi, Y.; Takino, T.; Dong, Y.; Sato, H.; Sai, Y.; et al. Ligand-Dependent EphB1 Signaling Suppresses Glioma Invasion and Correlates with Patient Survival. Neuro-Oncology 2013, 15, 1710–1720. [Google Scholar] [CrossRef]
- Yuan, Q.; Cai, H.-Q.; Zhong, Y.; Zhang, M.-J.; Cheng, Z.-J.; Hao, J.-J.; Wang, M.-R.; Wan, J.-H. Overexpression of IGFBP2 MRNA Predicts Poor Survival in Patients with Glioblastoma. Biosci. Rep. 2019, 39, BSR20190045. [Google Scholar] [CrossRef]
- Sun, W.; Zou, Y.; Cai, Z.; Huang, J.; Hong, X.; Liang, Q.; Jin, W. Overexpression of NNMT in Glioma Aggravates Tumor Cell Progression: An Emerging Therapeutic Target. Cancers 2022, 14, 3538. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, X.; Wang, L.; Ji, Z.; Xie, M.; Zhou, X.; Liu, Z.; Shi, H.; Yu, R. Loss of SH3GL2 Promotes the Migration and Invasion Behaviours of Glioblastoma Cells through Activating the STAT3/MMP2 Signalling. J. Cell. Mol. Med. 2017, 21, 2685–2694. [Google Scholar] [CrossRef]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinforma. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef]
- Piccolo, S.R.; Sun, Y.; Campbell, J.D.; Lenburg, M.E.; Bild, A.H.; Johnson, W.E. A Single-Sample Microarray Normalization Method to Facilitate Personalized-Medicine Workflows. Genomics 2012, 100, 337–344. [Google Scholar] [CrossRef]
- Cieslik, M.; Chugh, R.; Wu, Y.-M.; Wu, M.; Brennan, C.; Lonigro, R.; Su, F.; Wang, R.; Siddiqui, J.; Mehra, R.; et al. The Use of Exome Capture RNA-Seq for Highly Degraded RNA with Application to Clinical Cancer Sequencing. Genome Res. 2015, 25, 1372–1381. [Google Scholar] [CrossRef] [PubMed]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a Shared Vision for Cancer Genomic Data. N. Engl. J. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Wolf, F.A.; Angerer, P.; Theis, F.J. SCANPY: Large-Scale Single-Cell Gene Expression Data Analysis. Genome Biol. 2018, 19, 15. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ClusterLabel | GeneA | GeneB | Datasets | Array PD (A < B) | RNA-Seq PD (A < B) |
|---|---|---|---|---|---|
| cluster1 | POSTN | C1QL1 | 3 | −0.51 | −0.46 |
| cluster1 | CD46 | PDGFRA | 3 | −0.6 | −0.36 |
| cluster1 | ACSL3 | TF | 3 | −0.51 | −0.42 |
| cluster1 | DCX | LGR4 | 2 | 0.61 | 0.38 |
| cluster1 | EGFR | PMP2 | 2 | −0.52 | −0.27 |
| cluster1 | TMSB15A | LXN | 2 | 0.53 | 0.48 |
| cluster1 | TCEAL2 | MYO5C | 2 | 0.5 | 0.44 |
| cluster2 | HILPDA | BANF1 | 3 | −0.67 | −0.59 |
| cluster2 | NDRG1 | APLP2 | 3 | −0.7 | −0.61 |
| cluster2 | IFITM3 | BNIP3 | 2 | 0.57 | 0.53 |
| cluster2 | MRFAP1L1 | ZNF395 | 2 | 0.68 | 0.48 |
| cluster3 | PDPN | DLL3 | 3 | 0.9 | 0.56 |
| cluster3 | TMEM100 | DYNLT3 | 3 | −0.86 | −0.81 |
| cluster3 | EMP3 | FERMT1 | 3 | 0.7 | 0.53 |
| cluster3 | RBP1 | BMP2 | 2 | 0.88 | 0.64 |
| cluster3 | EPHB1 | IGFBP2 | 2 | −0.75 | −0.64 |
| cluster3 | NNMT | SH3GL2 | 2 | 0.71 | 0.5 |
| cluster4 | P4HA1 | LMO3 | 3 | 0.61 | 0.57 |
| cluster4 | CDKN2A | MYC | 3 | −0.51 | −0.3 |
| cluster4 | MAOB | VEGFA | 3 | −0.61 | −0.6 |
| cluster4 | C21orf62 | SEMA5A | 2 | −0.79 | −0.77 |
| cluster4 | APLNR | PHLDA1 | 2 | −0.64 | −0.7 |
| cluster5 | GULP1 | CA10 | 3 | 0.69 | 0.69 |
| cluster5 | SLC7A11 | NKX2-2 | 3 | 0.57 | 0.41 |
| cluster5 | MYO5C | UGT8 | 3 | 0.58 | 0.48 |
| cluster5 | C1QL1 | LTBP1 | 3 | −0.56 | −0.49 |
| cluster5 | ECM2 | GPR17 | 2 | 0.51 | 0.32 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gibbs, D.L.; Cioffi, G.; Aguilar, B.; Waite, K.A.; Pan, E.; Mandel, J.; Umemura, Y.; Luo, J.; Rubin, J.B.; Pot, D.; et al. Robust Cluster Prediction Across Data Types Validates Association of Sex and Therapy Response in GBM. Cancers 2025, 17, 445. https://doi.org/10.3390/cancers17030445
Gibbs DL, Cioffi G, Aguilar B, Waite KA, Pan E, Mandel J, Umemura Y, Luo J, Rubin JB, Pot D, et al. Robust Cluster Prediction Across Data Types Validates Association of Sex and Therapy Response in GBM. Cancers. 2025; 17(3):445. https://doi.org/10.3390/cancers17030445
Chicago/Turabian StyleGibbs, David L., Gino Cioffi, Boris Aguilar, Kristin A. Waite, Edward Pan, Jacob Mandel, Yoshie Umemura, Jingqin Luo, Joshua B. Rubin, David Pot, and et al. 2025. "Robust Cluster Prediction Across Data Types Validates Association of Sex and Therapy Response in GBM" Cancers 17, no. 3: 445. https://doi.org/10.3390/cancers17030445
APA StyleGibbs, D. L., Cioffi, G., Aguilar, B., Waite, K. A., Pan, E., Mandel, J., Umemura, Y., Luo, J., Rubin, J. B., Pot, D., & Barnholtz-Sloan, J. (2025). Robust Cluster Prediction Across Data Types Validates Association of Sex and Therapy Response in GBM. Cancers, 17(3), 445. https://doi.org/10.3390/cancers17030445