Simple Summary
Pancreatic ductal adenocarcinoma is very hard to treat, and most patients do not benefit from current immune-based drugs. One reason may be that we are looking for the wrong targets. Proteins in cancer cells often undergo small chemical changes after they are made. These changes (PTMs) can create new “flags” that the immune system can see, but usual tools do not catch them. We describe a clear plan to find these PTM targets with modern lab methods, check them in patient-derived models, and connect them to tests and treatments like vaccines or engineered T cells. This could make immunotherapy work for more people with PDAC.
Abstract
Background: Pancreatic ductal adenocarcinoma (PDAC) exhibits marked resistance to immunotherapy. Beyond its characteristically low tumor mutational burden, post-translational modifications (PTMs) remodel the immunopeptidome and promote immune escape through reversible, enzyme-driven programs. Subject Matter: We synthesize evidence that aberrant glycosylation, O-GlcNAcylation, phosphorylation, and citrullination constitute core determinants of antigen visibility operating within spatially discrete tumor niches and a desmoplastic stroma. In hypoxic regions, HIF-linked hexosamine metabolism and OGT activity stabilize immune checkpoints and attenuate antigen processing; at tumor margins, sialylated mucins engage inhibitory Siglec receptors on innate and adaptive lymphocytes; within the stroma, PAD4-dependent NET formation enforces T cell exclusion. We also delineate technical barriers to discovering PTM antigens labile chemistry, low stoichiometry, and method-embedded biases and outline practical solutions: ETD/EThcD/AI-ETD fragmentation, PTM-aware database searching and machine-learning models, and autologous validation in patient-derived organoid–T cell co-cultures. Finally, we highlight therapeutic strategies that either immunize against PTM neoepitopes or inhibit PTM machinery (e.g., PAD4, OGT, ST6GAL1), with stromal remodeling as an enabling adjunct. Conclusions: PTM biology, spatial omics, and patient sample models can uncover targetable niches and speed up PDAC vaccination, TCR, and enzyme-directed treatment development.
1. Introduction
1.1. Pancreatic Cancer: Immune Evasion Beyond Mutational Load
Pancreatic ductal adenocarcinoma (PDAC) remains exceptionally challenging to treat; even with contemporary multimodal regimens, the five-year survival rate is below 12% [1]. This grim outlook largely reflects presentation at advanced stages and robust tumor-intrinsic immune evasion [2]. Although recurrent oncogenic lesions, most notably KRAS substitutions at G12 (G12D/V/R) in nearly 90% of cases [3] and TP53 (identified in about 77%) [4], represent prototypical tumor-specific antigens (TSAs), efforts to therapeutically target them have achieved limited success. Clinical studies of KRAS-directed mRNA vaccines (e.g., mRNA-5671; NCT03948763) and TP53-focused synthetic long-peptide vaccines have failed to produce objective responses in more than 90% of recipients, underscoring the constraints of mutation-centric immunotherapies in PDAC [5,6].
The pronounced immunoresistance of PDAC is largely driven by its dense, desmoplastic, and immunosuppressive tumor microenvironment (TME) [7,8]. Stromal components, particularly cancer-associated fibroblasts (CAFs), can constitute up to 80% of the tumor mass and secrete immunomodulators such as TGF-β, CXCL12, and IL-10 [9]. Collectively, these mediators impede the infiltration and effector function of cytotoxic CD8+ T cells while promoting the expansion of immunosuppressive subsets, including FOXP3+ regulatory T cells and CD11b+Gr1+ myeloid-derived suppressor cells [10]. These immune evasion mechanisms, beyond just low TMB, are depicted in Figure 1 Compounding this, PDAC typically exhibits one of the lowest tumor mutational burdens among solid tumors, often fewer than one nonsynonymous mutation per megabase which correlates with poor responsiveness to immune checkpoint inhibitors [11,12]. Accordingly, anti-PD-1/PD-L1 monotherapy yields consistent response rates of under 5%, and phase III trials have not demonstrated durable therapeutic benefit in PDAC patients [13,14,15,16,17]. Recent studies have highlighted the essential function of PTMs in the immunopeptidome of PDAC. Ely et al. demonstrated that PDAC tumors possess concealed antigens resulting from PTMs that are undetectable by conventional genomic assays. PTM-derived antigens may serve as targets for immunotherapy, a promising approach for treating PDAC patients with limited mutations [18].
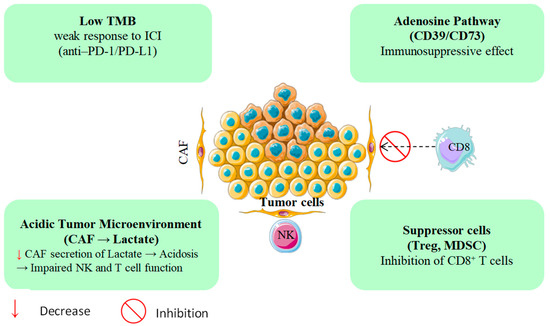
Figure 1.
PDAC’s immune evasion mechanisms beyond low TMB.
Beyond its structural barriers, PDAC exhibits profound metabolic dysregulation that further entrenches immune escape [19]. Tumor hypoxia drives extracellular adenosine accumulation via the CD39/CD73 ectonucleotidase axis, suppressing dendritic cell maturation and compromising T cell priming [20,21,22]. In parallel, the heightened glycolytic flux of CAFs generates excess lactate, acidifying the tumor milieu, and blunting natural killer (NK) cell cytotoxicity [23]. In keeping with these constraints, recent trials of PDAC immunotherapy show limited activity: no complete responses to TSA-directed vaccines or checkpoint inhibitors and a median progression-free survival under 2.5 months, a pattern largely attributable to an immunosuppressive microenvironment enriched for myeloid-derived suppressor cells and CAFs (Table 1). Taken together, these data underscore the need for innovative therapeutic strategies [24,25].

Table 1.
Clinical failures of mutation-targeted immunotherapies in PDAC.
Collectively, these observations argue for a reorientation of antigen discovery away from an exclusive focus on mutational neoantigens and toward non-mutational targets, particularly PTMs such as glycosylation, phosphorylation, and citrullination. These alterations are frequent in PDAC and may be recognized through non-canonical, MHC-independent pathways, potentially circumventing both stromal barriers and the immunologic impediments that limit conventional immunotherapies.
1.2. Post-Translational Modifications: The Subsequent Frontier in Antigen Discovery
PTMs including glycosylation, phosphorylation, citrullination, and ubiquitination govern protein function and reconfigure the antigenic landscape of malignant cells [34]. Unlike DNA mutations, PTMs generate neoantigenic determinants by chemically altering self-proteins, thereby providing a non-mutational route to immune recognition [35]. PDAC; such modifications both facilitate immune escape and present actionable targets for therapy [36].
Aberrant glycosylation, particularly of mucins such as MUC1, drives the expression of tumor-associated carbohydrate antigens (e.g., Tn and sialyl-Tn [STn]) [37]. These glycans can sterically shield immunodominant peptide epitopes, diminishing detection by cytotoxic CD8+ T cells [38]. Immunopeptidomic analyses using mass spectrometry on patient-derived xenografts substantiate this masking effect in PDAC [39,40]. Conversely, citrullination catalyzed by peptidylarginine deiminases (PAD2 and PAD4) can create immunogenic neoepitopes exemplified by citrullinated vimentin that are efficiently presented on MHCI and recognized by autoreactive T cells, even in tumors with very low mutational burdens [41,42].
Spatial proteomics further indicates that PTMs are patterned rather than randomly dispersed within the tumor microenvironment: hypoxic niches exhibit elevated PAD4 and O-GlcNAc transferase (OGT) activity, forming localized PTM “hotspots” that coincide with immune-exclusion zones in PDAC (Figure 2) [43,44,45]. These distributions have functional consequences. Phosphorylation of tumor antigens, for instance, ENO1 at serine 419, can enhance proteasomal processing and promote efficient MHC-I loading, whereas O-GlcNAcylation of components of the antigen-processing machinery impairs peptide presentation [46,47]. Reflecting this context dependence, sialylated MUC16 dampens immunity through the engagement of inhibitory Siglec receptors on natural killer (NK) cells, while phosphorylated HER2-derived peptides elicit robust T cell responses in breast and pancreatic cancers [48,49].
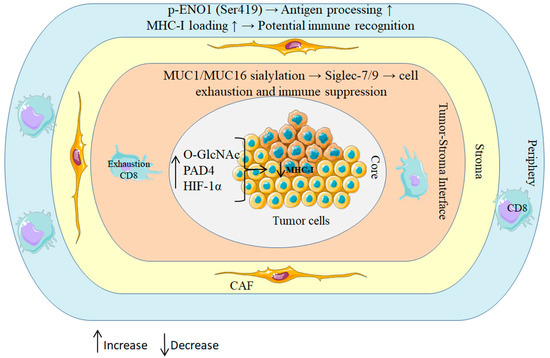
Figure 2.
Spatial regulation of PTMs in PDAC shapes immune outcomes.
Clinical validation of PTM-derived targets is gaining momentum. In preclinical models, a chemically defined synthetic MUC1 glycopeptide vaccine directed at tumor-associated epitopes such as sTn elicited strong Th1-biased responses and activated cytotoxic T cells in murine PDAC [50]. Nevertheless, substantial hurdles remain: many PTM-modified peptides are scarce and short-lived, rendering them difficult to detect with conventional mass spectrometry [51,52,53]. Moreover, distinguishing biologically consequential (“driver”) PTM events such as PAD4-mediated histone citrullination linked to neutrophil extracellular trap formation from incidental changes requires single-cell PTM-omics with high spatial and temporal resolution [54,55].
Overcoming these barriers could open a new class of immunotherapies for antigenically “cold” tumors like PDAC. PTM-focused interventions may enable next-generation vaccines, T cell receptor-engineered cellular therapies, and selective small-molecule agents, for example, PAD4 inhibitors, to disrupt citrullination-driven immunosuppression. As the PTM-centered immunobiology of PDAC is mapped with greater precision, these alterations are poised to become integral design parameters for tailored immunotherapeutic strategies.
This review focusses on four PTM axes (glycosylation, O-GlcNAcylation, phosphorylation, citrullination) to provide depth in accordance with PDAC evidence; additional PTM programs (e.g., SUMOylation, ubiquitination, acetylation, β-hydroxybutyrylation, methylation, ferroptosis) are summarized in Table 2.
Table 2.
PTM-mediated immune regulation in PDAC: mechanisms, biomarkers, and therapeutic opportunities.
Schematic of concentric tumor zones illustrating how patterned PTMs rewire antigen processing and immune surveillance. Hypoxic core: HIF-1α–driven OGT activity elevates O-GlcNAcylation and PAD4-mediated citrullination, jointly reducing MHC-I peptide display. Tumor stroma interface: aberrant MUC1/MUC16 sialylation ligates inhibitory Siglec-7/9 on T and NK cells, promoting functional exhaustion; a CAF-rich stroma further enforces exclusion. Peripheral rim: context-dependent ENO1 phosphorylation (Ser419) enhances antigen processing and MHC-I loading, enabling CD8+ T cell recognition. Collectively, these spatial PTM programs create immunosuppressive niches centrally while permitting immune detection at the margins.
2. The Hypothesis of Antigenic Dark Matter
Antigenic black matter refers to the PTM-rich part of the PDAC immunopeptidome that is undoubtedly quite large but is often missed since acquisition settings, database searches, and immune-tolerance filters all down-weight fragile, low-abundance PTMs. Data from PDAC and other malignancies indicate that glycosylated, O-GlcNAcylated, phosphorylated, and citrullinated ligands facilitate immune evasion, whereas PTM-aware mass spectrometry, incorporating ETD/EThcD, DIA, multi-engine rescoring, and machine-learning inference is progressively enhancing detection capabilities. This “hidden” layer thus presents a promising reservoir of targets for vaccines, T cell receptors, and enzyme- or pathway-directed treatments in PDAC.
The “antigenic dark matter” hypothesis holds that PTMs constitute a vast, underexplored reservoir of tumor antigens that occupy a substantial share of the immunopeptidome [69] but largely escape detection because of methodological blind spots and layers of immunological tolerance (Figure 3a). Unlike mutation-derived neoantigens, PTM-bearing peptides arise through dynamic, enzyme-mediated processes and can markedly expand the antigenic diversity of cancers [35]. In PDAC, mass spectrometry-based immunopeptidomics indicates that a considerable proportion of MHC class I-presented peptides carry PTMs, broadening the antigenic repertoire and yielding putatively tumor-specific targets with clear relevance for precision immunotherapy [36,70]. These observations emphasize that PTMs can modulate tumor immunogenicity independently of TMB.
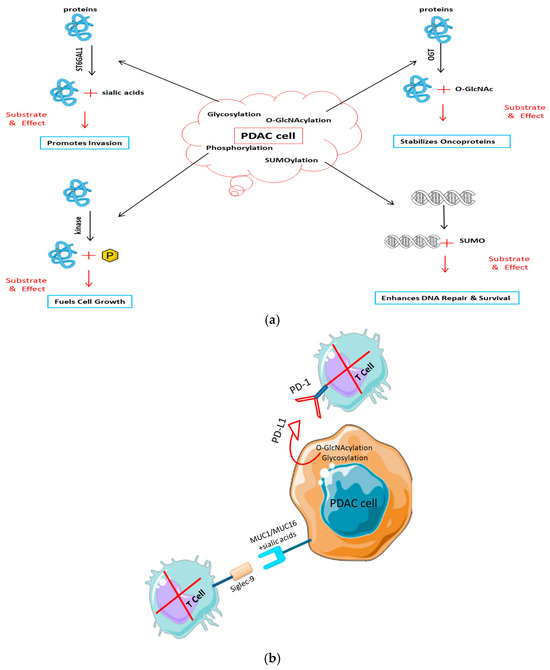
Figure 3.
Post-translational modifications drive pancreatic cancer progression and immune evasion. (a) PTMs in tumor development and signaling. Activated key PTM pathways in PDAC cells lead to cancer growth: glycosylation (e.g., by ST6GAL1) of receptors like EGFR promotes invasion; O-GlcNAcylation by OGT stabilizes oncoproteins such as β-catenin to drive proliferation; and kinase-mediated phosphorylation (e.g., MAPK/ERK) drives uncontrolled growth. (b) PTMs in avoiding the immune system. PTMs change the surface of tumor cells to make them less immune: Sialylated mucins (MUC1/MUC16) bind to inhibitory Siglec receptors on T and NK cells. O-GlcNAcylation and glycosylation stabilize the PD-L1 checkpoint protein, which stops T cells from entering.
2.1. Mechanistic Bases of PTM-Mediated Immune Evasion
PTMs promote immune escape in PDAC through multiple antigen-focused mechanisms:
- Glycosylation: Aberrant hypersialylation of mucins (e.g., MUC1, MUC16) generates a dense glycan shield that interferes with T cell receptor engagement, while simultaneously triggering inhibitory Siglec receptors on NK cells to dampen cytotoxicity [36,71,72].
- Citrullination: PAD4-driven conversion of arginine to citrulline in proteins such as vimentin and histones can create neoepitopes; however, central tolerance particularly thymic deletion of autoreactive clones limits effective recognition [73,74].
- Phosphorylation: Hypoxia-associated phosphorylation of metabolic enzymes (e.g., ENO1 S419) enhances peptide processing and MHC-I display [46]. Paradoxically, increased antigen presentation may recruit CD73+ regulatory T cells via adenosine signaling [75], establishing localized immune tolerance.
2.2. Neoantigen Diversity Beyond the Genome
In contrast to fixed genetic alterations, PTMs provide a reversible and time-sensitive layer of antigenic plasticity [76]. This metabolic agility enables tumors to rapidly recalibrate their antigenic surface under immune selection [77,78].
- Dynamic Glycosylation: Real-time modulation of sialylation by enzymes such as ST6GAL1 can reconfigure immune visibility within hours [79,80].
- Contextual Immunogenicity: Although many PTMs favor immune evasion, some enhance recognition. Notably, citrullinated peptides derived from oncogenic KRAS (G12D) elicit CD8+ T cell responses in PDAC patients who do not respond to the corresponding unmodified peptides, revealing fresh avenues for neoantigen targeting [33,81,82].
2.3. Technical Barriers and Emerging Solutions
Despite their immunological importance, PTM-derived antigens have been difficult to access because of several practical hurdles:
- Detection Limitations: Conventional mass spectrometry often fails to capture PTM-modified peptides owing to low abundance and chemical lability [83,84]. Advances in MS-based glycoproteomics including size-exclusion-based enrichment are improving the detection of sialylated epitopes that are particularly relevant in PDAC, including within the stromal compartment [85,86].
- Functional Validation: Establishing bona fide immunogenicity requires stringent in vitro and in vivo testing. CRISPR-engineered PDAC organoids, including constructs that recapitulate specific PTMs (e.g., p53 O-GlcNAcylation), are being leveraged to probe T cell reactivity and antigen processing [87,88].
- Clinical Proof-of-Concept: Early clinical experience with vaccines targeting citrullinated vimentin in PDAC, together with preclinical studies of PTM-focused vaccines, points to therapeutic promise. PTMs such as citrullination can yield tumor-associated antigens capable of driving immune responses [34,77,89]. A cohesive strategy to decode this “dark matter” of the immunopeptidome from PTM-compatible discovery platforms to the prioritization and translation of targetable epitopes is therefore essential (Figure 4).Collectively, these data position PTMs as a critical yet underutilized dimension of cancer immunotherapy. Systematically harnessing this antigenic “dark matter” may provide new strategies to overcome immune resistance in antigenically cold tumors such as PDAC.
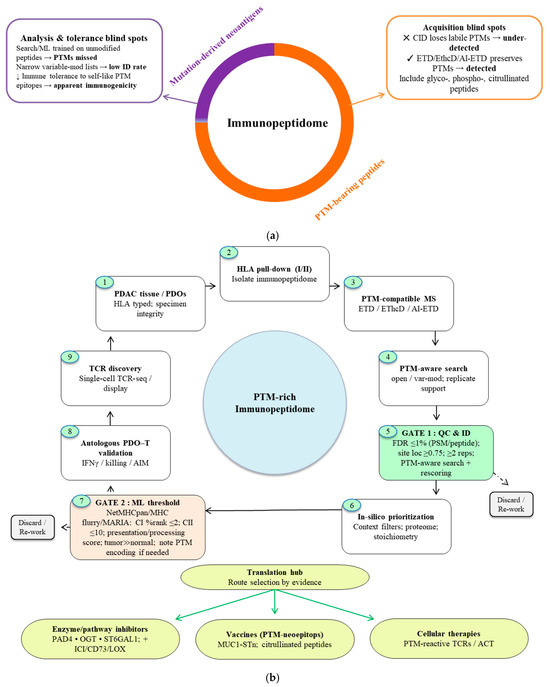 Figure 4. Decoding antigenic dark matter in PDAC. (a) Composition and detectability of the PDAC immunopeptidome. PTM-bearing peptides (orange) constitute the predominant fraction, whereas mutation-derived neoantigens represent a smaller subset (purple). This PTM-rich space is systematically under-recovered by (i) acquisition methods that strip labile groups (CID), (ii) analysis biases (search/ML models trained on unmodified peptides; restricted variable-mod settings), and (iii) immunological tolerance to self-like PTM epitopes together yielding the illusion of low immunogenicity. Electron-based fragmentation (ETD, EThcD, AI-ETD) preserves fragile modifications and improves detection and site localization. (b) Gated decoding pipeline from discovery to translation. PDAC tissue/PDOs → HLA pulldown → PTM-compatible MS → PTM-aware search → Gate 1 applies standard QC (≤ 1% FDR, site localization ≥ 0.75, replicate support, PTM-aware search/rescoring) → Gate 2 retains peptides meeting HLA-binding rank thresholds (Class I ≤ 2%, Class II ≤ 10%), favorable presentation/processing scores, and tumor-restricted expression, with surrogate PTM encodings documented where predictors lack native support. Candidates passing both gates are routed via a Translation Hub to (i) enzyme/pathway inhibitors (PAD4, OGT, ST6GAL1; often combined with ICI/CD73/LOX), (ii) PTM-neoepitope vaccines (e.g., MUC1-STn, citrullinated peptides), or (iii) cellular therapies (PTM-reactive TCRs/ACT).
Figure 4. Decoding antigenic dark matter in PDAC. (a) Composition and detectability of the PDAC immunopeptidome. PTM-bearing peptides (orange) constitute the predominant fraction, whereas mutation-derived neoantigens represent a smaller subset (purple). This PTM-rich space is systematically under-recovered by (i) acquisition methods that strip labile groups (CID), (ii) analysis biases (search/ML models trained on unmodified peptides; restricted variable-mod settings), and (iii) immunological tolerance to self-like PTM epitopes together yielding the illusion of low immunogenicity. Electron-based fragmentation (ETD, EThcD, AI-ETD) preserves fragile modifications and improves detection and site localization. (b) Gated decoding pipeline from discovery to translation. PDAC tissue/PDOs → HLA pulldown → PTM-compatible MS → PTM-aware search → Gate 1 applies standard QC (≤ 1% FDR, site localization ≥ 0.75, replicate support, PTM-aware search/rescoring) → Gate 2 retains peptides meeting HLA-binding rank thresholds (Class I ≤ 2%, Class II ≤ 10%), favorable presentation/processing scores, and tumor-restricted expression, with surrogate PTM encodings documented where predictors lack native support. Candidates passing both gates are routed via a Translation Hub to (i) enzyme/pathway inhibitors (PAD4, OGT, ST6GAL1; often combined with ICI/CD73/LOX), (ii) PTM-neoepitope vaccines (e.g., MUC1-STn, citrullinated peptides), or (iii) cellular therapies (PTM-reactive TCRs/ACT).
Numerous studies indicate that PTM-modified HLA peptides are a reliable and detectable element of the immunopeptidome, with their prevalence significantly influenced by sample type, PTM class, and search approach [90,91]. Extensive re-analyses and targeted methodological publications illustrate a significant quantity of phosphorylated and glycosylated HLA ligands, highlighting that open/PTM-aware searches significantly enhance modified peptide identification, but PDAC-specific proportions remain indeterminate. These data together validate PTM-aware discovery in PDAC without excessive extrapolation of numerical fractions.
3. PTMs in Cancer Immunology: Mechanisms and Consequences
3.1. Modifying the Antigenic Code: The Role of PTMs in Epitope Diversification
PTMs operate as molecular switches that remodel the tumor immunopeptidome in real time, generating antigenic heterogeneity that exceeds the limits of the static genome [34,92]. In PDAC, three PTM classes glycosylation, phosphorylation, and citrullination emerge as dominant regulators of immune visibility, shaping antigen processing, presentation, and recognition in both immunostimulatory and immunosuppressive directions [34]. PDAC cells exhibit increased terminal sialylation (e.g., on MUC1/MUC16), which activates inhibitory Siglec-7/-9 on myeloid cells, resulting in antigen-presentation deficiencies and T cell suppression. Additionally, CAFs provide sialylated ligands, broadening this checkpoint beyond tumor cells [56]. Stabilizing N-glycans on PD-L1 extends surface residency and inhibits cytotoxic T cell activity, establishing a direct correlation between glycosylation and checkpoint efficacy [93]. These glyco-circuits, which come from tumors and stroma, are now known in PDAC as immune-evasion nodes that can be worked with [94].
3.2. Regulation of PTM Landscapes by Microenvironmental Factors
The spatial heterogeneity of the PDAC microenvironment imposes strong region-specific pressures on PTM patterning and its immunologic consequences [95]. Spatially resolved proteomics delineate discrete PTM gradients within tumors:
- Hypoxic cores show the enrichment of O-GlcNAcylated proteins including HIF-1α which perturbs peptide loading and complex stability on MHC-I, thereby diminishing antigen presentation [87,96].
- Stromal interfaces harbor abundant citrullinated extracellular matrix proteins that promote neutrophil extracellular trap (NET) formation; these structures foster immune evasion and facilitate metastatic dissemination through NET-associated immunomodulation [97,98].
3.3. PTM Interference and Immune Regulation
PTMs integrate metabolic, epigenetic, and stromal cues into coordinated immune-evasion programs. In PDAC, these interconnected circuits create context-dependent vulnerabilities that enhance tumor adaptability [36]. Three principal axes link extrinsic stress signals to immune regulation [99].
3.4. Metabolic Hypoxic Axis: Interplay Between O-GlcNAcylation and Phosphorylation
Under hypoxia, HIF-1α upregulates the hexosamine biosynthetic pathway, elevating UDP-GlcNAc and driving O-GlcNAcylation of key signaling proteins [87,100]. In PDAC, O-GlcNAcylation of β-catenin at serine 552 blocks GSK3β-mediated phosphorylation, preventing proteasomal degradation [101]. The resulting stabilization enhances β-catenin nuclear accumulation and transcriptional output, promoting metastatic behavior [87,101]. Concurrently, O-GlcNAcylation of pyruvate kinase M2 (PKM2) at threonine 405 suppresses its kinase activity, redirecting glucose flux toward glycosylation precursors and further reinforcing O-GlcNAcylation [102]. O-GlcNAcylation of β-catenin may block GSK3β-mediated phosphorylation, leading to β-catenin stabilization. This, in turn, could increase its presentation as an MHC-I epitope, potentially influencing immune recognition.
Hypoxia and metabolic rewiring (GFPT1/2 flux) enhance O-GlcNAc cycling in PDAC, altering signaling and antigen-processing proteins, which, therefore, diminishes effective MHC-I visibility in immune-excluded niches [103]. Recent PDAC results reveal an HBP–O-GlcNAc–YBX1 axis that alters cytokine production (e.g., IL-18) and the tumor immunological milieu, highlighting O-GlcNAcylation as a context-dependent immune modulator [59]. O-GlcNAc integrates hypoxia metabolism with immune editing in PDAC [104].
3.5. Epigenetic Immunogenic Axis: Circuits of Acetylation and Ubiquitination
Epigenetic control intersects with PTM-dependent antigen presentation. Histone deacetylase inhibitors (e.g., panobinostat) induce hyperacetylation of HSP90 at lysine 294 [105,106], which flags the chaperone for CHIP-mediated ubiquitination, proteasomal turnover, and exposure of otherwise cryptic client oncoproteins such as HER2/neu as immunogenic targets [107]. PDAC cells counter that by overexpressing the glycosyltransferase ST6GAL1, increasing PD-L1 sialylation, enhancing engagement of Siglec-9 on T cells, and dampening activation [57]. This layered resistance where epigenetic inputs drive PTM-based modulation of immune checkpoints illustrates a broader paradigm of PTM-enabled immune escape in PDAC [36].
3.6. Stromal Tumor Axis: Networks of SUMOylation and Citrullination
Within PDAC’s desmoplastic stroma, CAFs orchestrate immunosuppression through coordinated PTM signaling [108]. The activation of STAT3 shaped by microenvironmental cues including hypoxia can downregulate components of the MHC-I antigen-processing machinery (e.g., TAP1/TAP2), thereby impairing antigen display; activated SUMOylation can limit MHC-I antigen presentation and reduce tumor immunogenicity; preliminary preclinical studies on PDAC with SUMO-pathway blockage indicate the activation of antitumor immunity [109,110,111]. At the matrix interface, PAD4-driven citrullination of proteins such as fibronectin (e.g., arginine 38) promotes NET formation, fueling metastasis and establishing immunosuppressive niches [34,97]. The genetic inhibition of PAD4 in the KPC mouse model markedly reduces NETs and constrains metastatic progression, underscoring the functional importance of stromal tumor PTM crosstalk [112,113]. The creation of PAD4-dependent NETs leads to stromal entrapment, vascular problems, and T cell exclusion in PDAC. It also makes citrullinated self-peptides that can act as neoepitopes [98]. Targeting PAD4/NETs serves a dual purpose: it breaks down physical and chemical barriers to infiltration and reveals citrullinated antigens for vaccination or T cell treatments [98,114].
3.7. Evolutionary Consequences of PTM Interactions
Single-cell analyses of PTM states indicate that immunologic pressure can shape clonal selection according to specific modification patterns, evidencing adaptive evolution in malignant populations [115].
- Hybrid glyco-phospho-signaling on mucins: In PDAC, atypical glycosylation and phosphorylation of MUC1 are recurrent and contribute to immune escape. Hybrid glyco-phospho-PTMs on mucins remodel molecular conformation and binding interfaces, creating steric barriers to immune recognition and altering antigen processing and presentation [116,117].
- Enzymatic crosstalk among PAD4 and OGT: Tumors with altered PAD4 activity including putative loss-of-function variants frequently display concomitant O-GlcNAc transferase (OGT) overexpression, revealing coordinated dysregulation across PTM enzymes. The resulting imbalance between citrullination (PAD4) and O-GlcNAcylation (OGT) fosters a protumor immunosuppressive niche across multiple cancer types [58,118].
3.8. Immune Editing Pressures and Post-Translational Modification-Driven Antigenic Landscapes
PTMs provide a rapid, reversible layer of antigenic plasticity that operates independently of DNA sequence change, enabling tumors to recalibrate their immune visibility under the selective pressures of immune editing [76]. Unlike mutation-fixed neoantigens, PTM programs are enzymatically tunable, conferring swift immuno-adaptation and a survival advantage in hostile microenvironments [35].
3.9. Immune Evasion Induced by Hypoxia
In PDAC, hypoxia reprograms cellular metabolism and elevates O-GlcNAcylation. O-GlcNAcylation of β-catenin stabilizes the protein often by antagonizing phosphorylation, leading to its accumulation, activation of Wnt pathway transcriptional outputs, and promotion of proliferation and metastatic competence [101]. These post-translational changes also reshape the antigenic display of β-catenin, with downstream effects on antigen presentation and T cell recognition [119]. Moreover, changes in β-catenin after translation, such as O-GlcNAcylation, can affect its expression as a tumor antigen. Deviant β-catenin signaling is recognized for its role in immune evasion mechanisms in cancer, encompassing effects on antigen presentation and T cell identification [87,120]. Comprehensive investigations of PTMs and glycoproteomics in pancreatic cancer have elucidated their significant roles in tumor advancement and immunological regulation. Integrative PTM and glycoproteomic studies in pancreatic cancer link such β-catenin modifications to weakened antitumor immunity, including reduced CD8+ T cell recognition; these effects may be accentuated in individuals whose HLA alleles present β-catenin-derived epitopes [121,122].
3.10. Thymic Tolerance and Context-Dependent Immunogenicity
Citrullination mediated by PAD4 can generate novel self-peptides, some of which become MHC I-restricted neoepitopes. Central tolerance in the thymus typically purges high-affinity T cell clones against self, including many PTM-modified self-antigens, thereby constraining baseline immunogenicity [34,123]. Under inflammatory stress such as that induced by cytotoxic therapy, these tolerance barriers may be relaxed [124,125]. In PDAC, regimens like FOLFIRINOX can modulate immunity and increase tumor cell death, potentially unveiling previously concealed or newly modified epitopes [126]. Therapy- or disease-associated tissue damage further remodels the proteome via PTMs (e.g., citrullination), enabling the presentation of PTM-derived peptides by MHC molecules and, in defined contexts, reshaping T cell responses [34,127,128]. Functional data confirms the existence of PTM-reactive T cells in PDAC, including T cells targeting citrullinated enolase and vimentin in patients, alongside glycopeptide vaccines that elicit MUC1-specific T cell responses in models [50,129].
4. The PTM Landscape in Pancreatic Cancer
4.1. The PDAC Paradox: Minimal Mutational Load, Elevated Antigenic Complexity
PDAC presents a striking immunological paradox: it exhibits an exceptionally low TMB (often <1 nonsynonymous mutation per megabase) that is, typically associated with poor responsiveness to checkpoint blockade [130]; yet demonstrates pronounced antigenic complexity largely sculpted by diverse PTMs [36,131]. This discrepancy challenges neoantigen-centric models of tumor immunogenicity and underscores PTMs as key determinants of the PDAC immune milieu [132,133].
4.2. Universal Hyperactivation of PTM-Regulating Enzymes
A non-mutational hallmark of PDAC is the widespread hyperactivation of PTM-controlling enzymes, driven by convergent oncogenic and microenvironmental inputs [134]. Mutant KRAS, via MAPK MYC signaling, upregulates multiple PTM enzymes including the glycosyltransferase ST6GAL1, thereby reprogramming metabolism and signaling independently of genomic change [57,135].
Severe hypoxia engages the hexosamine biosynthetic pathway (HBP), increasing O-GlcNAcylation and fueling OGT activity; this in turn stabilizes HIF-1α and sustains hypoxic signaling [100,136]. Although direct evidence for PAD4 and ST6GAL1 as explicit downstream effectors within this HIF-1α axis is limited, they remain as plausible targets. A self-reinforcing circuit marked by OGT-mediated O-GlcNAcylation and tumor hypersialylation through ST3GALs drives proteome-wide remodeling and immune evasion, enhancing PD-L1 stability and promoting Siglec-mediated polarization of tumor-associated macrophages (TAMs) [58,94]. While PTM-driven, non-mutational neoantigen generation highlights these enzymes as attractive immunotherapeutic targets, direct evidence specifically implicating PAD4 in this loop is still lacking [137].
4.3. Spatial Heterogeneity of Post-Translational Modifications in the Tumor Microenvironment
Spatial profiling reveals a regionally organized PDAC microenvironment with discrete immune neighborhoods and tertiary lymphoid-like structures, suggesting localized immunologic niches. Although PTMs are likely regulated within this architecture, patterned distributions remain to be directly demonstrated [138,139].
Spatial proteomics identifies hypoxic zones in PDAC, and hypoxic cores can downregulate MHC-I presentation through HIF-1α–dependent and autophagy-mediated mechanisms [96,140]. However, direct accumulation of O-GlcNAc-modified HIF-1α or phospho-metabolic enzymes in these regions has yet to be shown [141]. PAD4 activity promotes histone citrullination and NET formation phenomena linked to immune exclusion and progression—yet stromal ECM-specific citrullination and its creation of physical barriers to T cell entry remain to be established [142,143,144]. CAFs in PDAC display enriched sialylation; sialic acids on CAFs can engage Siglec-7/-9/-10 on immune cells to enforce immunosuppression. While spatial omics has mapped immune niches and stromal architecture, direct visualization of sialylated mucin gradients at tumor–stroma borders still needs confirmation [141,145,146,147]. Collectively, these observations indicate that PDAC immune escape is spatially programmed by compartmentalized PTM networks, now accessible only through advanced spatial omics.
4.4. Stromal Contributions and Tumor Microenvironmental Influences
The desmoplastic stroma, particularly CAFs, actively reshapes the PTM landscape through metabolic and signaling crosstalk, including CAF-derived exosomes [148,149]. Although ST6GAL1-mediated α-2,6 sialylation is increased in PDAC and generates immunosuppressive glyco-ligands that can engage inhibitory Siglecs, a direct link from CAF exosomal miR-155 to ST6GAL1 upregulation remains to be shown [57,150]. In parallel, CAF-derived TGF-β suppresses antigen-processing genes (e.g., TAP2, ERAP1, β2-microglobulin) via Smad3, reducing MHC-I surface expression and dampening tumor immunogenicity [151]. Activation of SUMOylation pathways has likewise been implicated in restricting MHC-I presentation, raising the possibility of TGF-β–SUMOylation crosstalk in PDAC immune evasion, though direct SUMOylation of TAP/LMP components has not been demonstrated [111,152]. PTMs directly influence diagnosis. The CA19-9 biomarker is a sialylated glycoform, with its levels and detectability regulated by tumor-specific glycosylation mechanisms. Certain glycoforms exhibit superior diagnostic capabilities compared to others [39,85].
4.5. Case Studies and Notable PTM-Modified Antigens in PDAC
Glycosylated MUC1: Truncated O-glycans (Tn, STn) create a bulky glycocalyx that sterically shields peptide epitopes from CD8+ T cell surveillance [153]. The sialylated STn variant also binds inhibitory Siglec-7/-9 on NK and CD8+ T cells, promoting functional exhaustion and helping explain the limited efficacy of MUC1-targeted vaccines [154,155,156].
Phosphorylated p53: Beyond mutation-derived neoantigens, phospho-p53 (e.g., Ser392) yields MHC-I-presented phosphopeptides that broaden targetable epitopes [157,158]. Although phosphopeptide presentation is documented in several cancers, context-specific effects such as Treg recruitment by these epitopes remain hypothetical and unproven in PDAC [159,160].
Annexin A2 / CXCL1: Annexin A2 supports EMT, invasion, and metastasis, but roles for its S-nitrosylation under hypoxia- or iNOS-driven stress are not yet characterized. Separately, CXCL1 promotes MDSC expansion and recruitment, fostering immune-excluded niches [161].
Together, these exemplars illustrate how discrete PTMs can rewire antigen visibility and immune outcomes, reinforcing the need for PTM-aware antigen discovery and therapeutic design in PDAC.
5. Challenges: Why PTMs Remain Dark Matter in Oncology
5.1. Challenges in Technical Detection
Deciphering the PTM-bearing immunopeptidome remains technically difficult because standard mass spectrometry workflows are poorly suited to capture these species [162,163,164]. Modified peptides typically occur at low stoichiometry, ionize inefficiently, and frequently undergo neutral loss during fragmentation [165,166]. As a result, labile modifications such as O-GlcNAcylation and citrullination are often cleaved under collision-induced dissociation (CID), yielding underrepresentation or misassignment [167,168]. The wide dynamic range of the proteome further obscures rare PTM peptides beneath abundant unmodified counterparts [169]. Although enrichment strategies (e.g., lectin affinity for glycopeptides; antibody-based capture such as PTMScan) improve recovery, they introduce method-specific biases and remain limited for uncommon or emergent PTMs [170,171]. Compounding this, conventional immunopeptidomics pipelines are optimized for unmodified ligands, systematically under-detecting PTM-derived antigens [172,173,174]. Together, these challenges create a persistent analytical blind spot that conceals much of the antigenic dark matter in PDAC.
Table 3 contrasts traditional CID/HCD with PTM-compatible electron-based processes (ETD/EThcD) and the newer AI-ETD to show how real trade-offs can be measured.
Table 3.
PTM peptide fragmentation procedures.
5.2. Complexities of HLA Presentation and Immune Recognition
A central barrier to harnessing PTM neoepitopes is that current HLA-binding predictors trained largely on unmodified peptides poorly capture how modifications alter peptide–MHC stability: a given PTM may disrupt anchor contacts, be neutral, or enhance binding to generate strong neoepitopes [181,182,183]. While central tolerance typically deletes T cells with high affinity for self, accumulating evidence indicates that T cells specific for PTM-derived epitopes (e.g., phosphorylation-induced targets) may escape thymic deletion; however, such clones often exhibit low functional avidity, limiting cytotoxic efficacy [173]. PTMs can also reconfigure TCR recognition geometry phosphate or glycan moieties may become dominant contact features in the peptide–MHC–TCR interface, shifting signaling outcomes from activation to anergy or tolerance [157,184,185,186]. These constraints underscore the need to prioritize PTMs that are tumor-restricted or abundantly presented to ensure sufficient T cell engagement despite tolerance.
5.3. Telling the Difference Between Driver and Passenger PTMs
Not every PTM in PDAC has a functional meaning. We propose an evidence-weighted checklist to focus on probable drivers rather than incidental passengers: (i) Specificity and perturbability of the enzyme as a writer or eraser of PTMs (e.g., OGT, ST6GAL1, PAD4, kinases), where genetic or pharmacologic modulation can alter the PTM and influence the downstream immune phenotype (e.g., OGT, ST6GAL1, PAD4, kinases) whose genetic or pharmacologic modulation shifts the PTM and the downstream immune phenotype (Table 2 entries and enzyme sections); (ii) tumor-specific enrichment versus matched normal across independent cohorts, ideally with measurable stoichiometry or dynamic changes along hypoxic or metabolic gradients [94,135,136,147]; (iii) spatial co-localization with immune components (e.g., Siglec+ myeloid cells, Tregs, PD-L1high niches) shown by spatial PTMs that fulfill many criteria, including enzyme-linked causality [145], tumor-restricted enrichment, spatial immune coupling, and CRISPR/inhibitor validation, should be prioritized as key drivers and progressed towards vaccines, TCR treatments, or enzyme-targeted therapeutics.
6. Novel Solutions and Innovations
New analytical and functional platforms are beginning to illuminate PTM-derived epitopes. Hybrid fragmentation strategies that combine electron transfer with higher-energy collision dissociation (EThcD) better preserve fragile modifications (e.g., O-glycans) and enable confident site localization [168,187]. Electron-based methods such as ETD and AI-ETD similarly minimize PTM loss, improving the identification of phosphosites and citrullinated residues [188,189,190]. On the computational side, machine-learning models trained on PTM-enriched immunopeptidome datasets are improving HLA-binding and presentation predictions for modified peptides, outperforming models built solely on unmodified sequences [191,192,193,194]. Incorporating these additional layers of information may enhance models’ predictive accuracy for PTM-driven immune targets. This indicates that they are not solely dependent on peptide sequences, hence enhancing the specificity of predictions for therapeutic applications. Recent advancements in deep learning have shown that including local sequence context and post-translational modification-specific characteristics significantly enhances forecast accuracy [195]. For functional validation, patient-derived organoid T cell co-cultures provide autologous, HLA-matched systems to test PTM neoantigen immunogenicity under physiologic constraints [196,197]. In parallel, high-throughput TCR discovery pipelines are uncovering rare PTM-reactive receptors from tumor-infiltrating lymphocytes or donor repertoires, supporting the development of engineered TCR therapies [196,197,198,199,200]. Together, these advances outline a practical path to render PTM biology actionable in oncology.
7. Therapeutic Horizons: Targeting PTM Antigens
To prevent mixing up maturity levels, we categorized PTM-anchored therapies into three groups: tested in humans, promising preclinical, and conceptual/early discovery (Table 4).
Table 4.
PTM-anchored immunotherapy approaches by translational stage.
7.1. Vaccines Aimed at PTM Neoepitopes
Vaccines targeting PTM-derived epitopes (notably aberrant glycosylation) offer a rational path to overcome tolerance and enhance antitumour immunity in PDAC [208,209]. The principal obstacle is that many PTM-modified epitopes originate from self-proteins, invoking B and T cell tolerance mechanisms [210]. Nonetheless, PTM-targeted vaccines have the potential to overcome these barriers and strengthen tumor-specific immunity in PDAC [211,212]. Second-generation synthetic glycopeptide platforms such as MUC1-STn constructs linked to T-helper epitopes or formulated on nanoparticle carriers have elicited class-switched IgG and cytolytic T cell responses that recognize glycosylated MUC1 on tumor cells [50,211,212,213,214]. In parallel, vaccines directed at citrullinated antigens (e.g., vimentin, α-enolase) have advanced into early-phase clinical or preclinical testing; by engaging HLA class II, these constructs can circumvent central tolerance and induce robust CD4+ and, in some contexts, CD8+ responses in poorly immunogenic settings [74,129,215,216,217]. Although the dominant signal is often Th1-biased CD4+ immunity, efficacy in low-immunogenicity tumors indicates potential for breaking tolerance and supporting CD8+ activation [129]. Importantly, PTM-focused vaccines appear most effective when combined with checkpoint blockade: preclinical studies show that responses to PTM-derived epitopes depend on relief from PD-1/CTLA-4-mediated suppression to maintain durable antitumor activity [218,219,220,221]. Collectively, these data chart a transition from proof-of-concept glycopeptide vaccines toward a new generation of PTM-informed immunization strategies that broaden PDAC’s targetable antigenic space. If PTM-based vaccines disrupt immunological tolerance, the resulting T cells may rapidly become fatigued within the tumor microenvironment (TME), which inhibits the immune system. This highlights the importance of combining these vaccinations with immune checkpoint inhibitors (ICIs) to prevent T cell exhaustion and enhance sustained antitumor responses [222].
7.2. Enzyme Inhibitors to Block PTM Machinery
An alternative to antigen-directed vaccination is to pharmacologically disrupt the enzymatic programs that generate immunosuppressive PTM signatures. In PDAC models, PAD4 inhibition (e.g., GSK484) reduces NET formation and curtails tumor growth [113,223,224], while PAD4 deletion or treatment with JBI-589 impairs metastasis and markedly improves responses to immune checkpoint blockade [113,225]. Key transcriptional regulators, such as β-catenin, are affected by OGT inhibition, which prevents hyper-O-GlcNAcylation, while OGT inhibitors restore cGAS–STING-dependent antigen presentation and elicit potent CD8+ T cell-mediated antitumor immunity [58,226]. Inhibiting tumor-associated sialyltransferases such as ST6GAL1 reduces hypersialylation, disrupts Siglec–sialoglycan interactions that dampen effector function, and enhances NK and T cell cytotoxicity [155,227,228,229,230,231]. Finally, stromal-targeted approaches such as lysyl oxidase (LOX) inhibition can soften the ECM, improve immune infiltration, and complement PTM-directed strategies [232,233,234]. Together, inhibitors of PTM enzymes constitute a class of metabolic immunotherapeutic agents capable of reprogramming PDAC from an immune-excluded state to an immune-responsive state.
7.3. Risks of Clinical Translation
Alongside these opportunities, PTM-targeted approaches come with clear safety trade-offs. Hitting broadly expressed epitopes like citrullinated peptides can break tolerance and cause autoimmunity [235]. This risk may go up when ICIs are added [236]. Enzyme-directed medicines (e.g., OGT or PAD4 inhibitors) can also have effects on-target and off-tumor because these enzymes help normal tissue work [100,237]. To mitigate these risks, candidates must demonstrate significant tumor restriction, validated HLA presentation, and T cell specificity in autologous systems. They should also participate in stepwise, closely monitored trials (with careful dose escalation, predefined stopping rules, and serial assessments of autoantibodies, cytokines, and organ function). To protect important physiology, context-dependent or localized techniques including targeted administration, transient/inducible regimens, and selective enzyme inhibition (isoform/complex-specific) should be used whenever possible.
8. Outlook and Strategic Recommendations
8.1. Thorough Mapping of PTM–Immune Interfaces
Targeting the stroma offers a complementary route to unlock antitumor immunity in PDAC. Inhibiting lysyl oxidase (LOX) can lessen collagen crosslinking and extracellular matrix (ECM) stiffening hallmarks of PDAC, thereby improving immune cell ingress [233,238,239,240]. Although direct PDAC evidence remains limited, studies on other solid tumors show that LOX blockade reduces matrix rigidity, facilitates CD8+ T cell trafficking, and augments responses to anti–PD-1 therapy [241,242]. In parallel, advances in deep immunopeptidomics such as applying EThcD or AI-ETD fragmentation to HLA pulldown samples increase recovery and confident localization of labile PTM-modified peptides that commonly escape conventional workflows [180,243,244]. Critically, machine-learning models trained on PTM-enriched immunopeptidome datasets are beginning to improve the prediction of PTM–HLA interactions. Together, these developments motivate the creation of a PTM antigen atlas analogous to TCGA or CPTAC in proteogenomics to convert disparate PTM observations into a standardized resource for therapeutic target triage.
8.2. Immunogenicity Assessment in Patient-Derived Organoids and Models
A definitive evaluation of PTM neoantigen function requires models that preserve human tumor context. Patient-derived organoids (PDOs) co-cultured with autologous peripheral or tumor-infiltrating immune cells provide an HLA-matched platform to measure antigen-specific T cell priming, effector function, and cytotoxicity under physiologically relevant conditions [197,245,246]. These immuno-organoid systems can be precisely edited with CRISPR/Cas9 to delete key PTM enzymes (e.g., PAD4, OGT) or to introduce defined PTM neoantigen constructs, enabling causal tests of how specific modifications alter immune recognition [247,248]. Because PDOs retain each patient’s native HLA background and, thus, their history of thymic tolerance, they also help prioritize modifications with genuine translational potential [196,248]. When combined with high-throughput TCR discovery pipelines, this framework links antigen identification to epitope prioritization, ensuring that only actionable PTM targets advance.
8.3. Collaborative Frameworks: International PTM Antigen Discovery Consortia
Systematically charting the PTM “dark matter” in cancer immunology will require coordinated, multi-institutional efforts modeled on initiatives such as the Human Immuno-Peptidome Project (HIPP). An international PTM Antigen Discovery Consortium could harmonize PTM-focused immunopeptidomics protocols, build centralized repositories of PTM–HLA ligands, and enable the sharing of scarce patient samples and specialized reagent libraries [249,250]. Such a structure would reduce duplication, enable cross-cohort validation, and generate a robust reference atlas of PTM epitopes. By integrating immunopeptidomics with spatial proteomics and clinical HLA typing, these networks could stratify patients by HLA allotype and PTM phenotype, informing the rational design of off-the-shelf vaccines and adoptive cell therapies [251,252]. Establishing this collaborative infrastructure is essential to move PTM antigens from isolated findings to clinically actionable immunotherapy targets.
9. Additional Regulatory Mechanisms in Pancreatic Cancer
In PDAC, the SUMO conjugation machinery is often overexpressed, which leads to better transcriptional regulation, faster DNA repair, continued cell-cycle progression, and more resistance to therapy, especially in aggressive tumor phenotypes [253]. Pharmacologic inhibition of this system using TAK-981 impairs normal mitotic processes, resulting in G2/M arrest in PDAC cells and significantly inhibiting tumor growth in animal models by simultaneously enhancing antitumor immunity [254]. Additionally, oncogenic KRAS has been demonstrated to facilitate SUMOylation-dependent release of extracellular vesicles and promote lymphangiogenesis, hence enhancing the interplay between intracellular oncogenic signaling and the tumor microenvironment [255]. More recent findings reveal that SUMOylation also promotes immune evasion in PDAC by modulating pathways such as TIGIT/CD155, underlining potential intersections with immunotherapeutic methods [256].
β-Hydroxybutyrylation: The ketone body β-hydroxybutyrate (β-HB) can trigger histone β-hydroxybutyrylation, a change that boosts the transcription of antioxidant genes and protects pancreatic cells from oxidative stress linked to ferroptosis [257]. In malignant contexts, β-HB has been demonstrated to facilitate ferroptotic cell death by inhibiting caveolin-1 (CAV1) production, hence illustrating its dual regulatory role in pancreatic disease [258].
Methylation: In PDAC, many protein methyltransferases, such as CARM1 and PRMT1, are crucial in regulating cellular metabolism and gene expression [259]. Dysregulation of histone methylation, especially via hyperactive EZH2 and G9a enzymes, facilitates chromatin remodeling that enhances tumor aggressiveness and is associated with poor patient outcomes [260].
Ferroptosis: Lipid peroxidation-induced ferroptosis represents a unique susceptibility in pancreatic cancer cells [261]. The glutathione peroxidase GPX4, the exchange of cystine and glutamate, and the balance of iron inside the cell all tightly control this process. Inducing ferroptosis impairs redox homeostasis, inhibits tumor proliferation, and increases the sensitivity of resistant pancreatic ductal adenocarcinoma (PDAC) to current therapy strategies [262].
10. Conclusions
PTMs represent a dominant yet incompletely charted dimension of antigenicity in PDAC. Spatially organized modification programs O-GlcNAcylation and citrullination enriched in hypoxic cores, heightened sialylation at tumor–stroma interfaces, and context-dependent phosphorylation at the periphery converge to enforce immune exclusion by suppressing MHC-I display, stabilizing PD-L1, and fostering NET-mediated physical barriers. Overcoming this immune refractoriness calls for a PTM-aware, end-to-end pipeline: (i) preserve labile PTMs during HLA-peptidomics using ETD/EThcD/AI-ETD; (ii) analyze spectra with machine-learning models trained on PTM-enriched datasets; (iii) functionally triage candidates in autologous PD-T-cell co-cultures; and (iv) translate prioritized targets into PTM-directed vaccines and enzyme inhibitors (e.g., OGT, PAD4, ST6GAL1), coupled with stromal remodeling where indicated. By prioritizing PDAC-relevant PTM immune axes and standardizing PTM-centric workflows, the field can enable robust target discovery and rational combinations with checkpoint blockade moving PTM antigens from “dark matter” to a tractable therapeutic class.
Author Contributions
Conceptualization, A.S.; data curation, A.S., I.V., and M.G.; writing original draft preparation, A.S.; writing review and editing, A.S., I.V., and M.G.; supervision, G.T.; project administration, A.S. and G.T. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Italian Ministry of Health (Ricerca Corrente).
Data Availability Statement
There is no new information in this review article. All evidence that backs up the discussion and conclusions come from studies that have already been published and are cited in the manuscript.
Acknowledgments
The author acknowledges Bioicons.com for the pictures that were used in the illustrations. The author utilized ChatGPT v4.5 (OpenAI) to check the grammar. They also went over and corrected all of the content; therefore, they are fully responsible for the final document.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ACT | Adoptive Cell Therapy |
| APM | Antigen-Processing Machinery |
| β-HB | β-hydroxybutyrate |
| β2M | β2-microglobulin |
| CA19-9 | Carbohydrate Antigen 19-9 |
| CAF | Cancer-Associated Fibroblast |
| CARM1 | Coactivator-Associated Arginine Methyltransferase 1 |
| CAV1 | Caveolin-1 |
| CD | Cluster of Differentiation |
| CID | Collision-Induced Dissociation |
| CTLA-4 | Cytotoxic T-Lymphocyte-Associated Protein 4 |
| CXCL | C-X-C Motif Chemokine Ligand |
| DIA | Data-Independent Acquisition |
| DNA | Deoxyribonucleic Acid |
| DUB | Deubiquitinating Enzyme |
| ECM | Extracellular Matrix |
| EGFR | Epidermal Growth Factor Receptor |
| EMT | Epithelial–Mesenchymal Transition |
| ENO1 | Enolase 1 |
| ERAP1 | Endoplasmic Reticulum Aminopeptidase 1 |
| ERK | Extracellular Signal-Regulated Kinase |
| ETD | Electron Transfer Dissociation |
| EThcD | Electron Transfer/Higher-Energy Collision Dissociation |
| EZH2 | Enhancer of Zeste Homolog 2 |
| FDR | False Discovery Rate |
| FOXP3 | Forkhead Box P3 |
| FOLFIRINOX | Folinic Acid, Fluorouracil, Irinotecan, Oxaliplatin |
| GALNTs | Polypeptide N-Acetylgalactosaminyltransferases |
| GFAT | Glutamine:Fructose-6-Phosphate Aminotransferase |
| GPX4 | Glutathione Peroxidase 4 |
| GR | Glucocorticoid Receptor |
| GSK3β | Glycogen Synthase Kinase 3 Beta |
| HBP | Hexosamine Biosynthetic Pathway |
| HCD | Higher-Energy Collisional Dissociation |
| HDAC | Histone Deacetylase |
| HER2 | Human Epidermal Growth Factor Receptor 2 |
| HIF | Hypoxia-Inducible Factor |
| HIPP | Human Immuno-Peptidome Project |
| HLA | Human Leukocyte Antigen |
| HSP | Heat Shock Protein |
| ICI | Immune Checkpoint Inhibitor |
| IL | Interleukin |
| iNOS | Inducible Nitric Oxide Synthase |
| JAK | Janus Kinase |
| KRAS | Kirsten Rat Sarcoma Viral Oncogene Homolog |
| LOX | Lysyl Oxidase |
| MAPK | Mitogen-Activated Protein Kinase |
| MDSC | Myeloid-Derived Suppressor Cell |
| MHC | Major Histocompatibility Complex |
| MS | Mass Spectrometry |
| MSS | Microsatellite Stable |
| MSI-H | Microsatellite Instability-High |
| MUC | Mucin |
| NET | Neutrophil Extracellular Trap |
| NF-κB | Nuclear Factor Kappa B |
| NK | Natural Killer |
| NO | Nitric Oxide |
| NOS | Nitric Oxide Synthase |
| OGT | O-GlcNAc Transferase |
| ORR | Objective Response Rate |
| PAD | Peptidylarginine Deiminase |
| PD-1 | Programmed Cell Death Protein 1 |
| PD-L1 | Programmed Death-Ligand 1 |
| PDAC | Pancreatic Ductal Adenocarcinoma |
| PDO | Patient-Derived Organoid |
| PIAS | Protein Inhibitor of Activated STAT |
| PKM2 | Pyruvate Kinase M2 |
| PRMT | Protein Arginine Methyltransferase |
| PTM | Post-Translational Modification |
| QC | Quality Control |
| RAF1 | Raf-1 Proto-Oncogene, Serine/Threonine Kinase |
| SER | Serine |
| STAT | Signal Transducer and Activator of Transcription |
| ST6GAL1 | β-Galactoside α-2,6-Sialyltransferase 1 |
| ST3GAL | β-Galactoside α-2,3-Sialyltransferase |
| STING | Stimulator of Interferon Genes |
| SUMO | Small Ubiquitin-like Modifier |
| TAM | Tumor-Associated Macrophage |
| TAP | Transporter Associated with Antigen Processing |
| TCR | T cell Receptor |
| TGF-β | Transforming Growth Factor Beta |
| Th | T Helper Cell |
| TIGIT | T Cell Immunoreceptor with Ig and ITIM Domains |
| TMB | Tumor Mutational Burden |
| TME | Tumor Microenvironment |
| Tn | Thomsen-nouvelle Antigen |
| TP53 | Tumor Protein P53 |
| Treg | Regulatory T Cell |
| TSA | Tumor-Specific Antigen |
| UBA2 | Ubiquitin-Like Modifier Activating Enzyme 2 |
| UBC9 | Ubiquitin Conjugating Enzyme 9 |
| UDP | Uridine Diphosphate |
| USP | Ubiquitin Specific Peptidase |
References
- Muaddi, H.; Kearse, L.; Warner, S. Multimodal Approaches to Patient Selection for Pancreas Cancer Surgery. Curr. Oncol. 2024, 31, 2260–2273. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.; Xu, D.; Liao, M.-M.; Sun, Y.; Bao, W.-D.; Yao, F.; Ma, L. Barriers and opportunities in pancreatic cancer immunotherapy. Npj Precis. Oncol. 2024, 8, 199. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Zhang, T.; Feng, L.; de Silva, N.; Greenspun, B.; Wang, X.; Moyer, J.; Martin, M.L.; Chandwani, R.; Elemento, O.; et al. A pancreatic cancer organoid platform identifies an inhibitor specific to mutant KRAS. Cell Stem Cell 2023, 31, 71–88.e8. [Google Scholar] [CrossRef]
- Stefanoudakis, D.; Frountzas, M.; Schizas, D.; Michalopoulos, N.V.; Drakaki, A.; Toutouzas, K.G. Significance of TP53, CDKN2A, SMAD4 and KRAS in Pancreatic Cancer. Curr. Issues Mol. Biol. 2024, 46, 2827–2844. [Google Scholar] [CrossRef]
- Lorentzen, C.L.; Haanen, J.B.; Met, Ö.; Svane, I.M. Clinical advances and ongoing trials of mRNA vaccines for cancer treatment. Lancet Oncol. 2022, 23, e450–e458. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Fan, C.; Zeng, Z.; Young, K.H.; Li, Y. Clinical and Immunological Effects of p53-Targeting Vaccines. Front. Cell Dev. Biol. 2021, 9, 762796. [Google Scholar] [CrossRef]
- Zhang, W.; Li, S.; Liu, Y.; Xing, R.; Jin, Z.; Yan, X.; Xue, H. Immunosuppressive microenvironment improvement and treatment of aggressive malignancy pancreatic ductal adenocarcinoma based on local administration of injectable hydrogel. Nano Today 2023, 50, 101832. [Google Scholar] [CrossRef]
- Vruzhaj, I.; Gambirasi, M.; Busato, D.; Giacomin, A.; Toffoli, G.; Safa, A. Gut Microbiota-Based Immunotherapy: Engineered Escherichia coli Nissle 1917 for Oral Delivery of Glypican-1 in Pancreatic Cancer. Medicina 2025, 61, 633. [Google Scholar] [CrossRef]
- Glapiński, F.; Zając, W.; Fudalej, M.; Deptała, A.; Czerw, A.; Sygit, K.; Kozłowski, R.; Badowska-Kozakiewicz, A. The Role of the Tumor Microenvironment in Pancreatic Ductal Adenocarcinoma: Recent Advancements and Emerging Therapeutic Strategies. Cancers 2025, 17, 1599. [Google Scholar] [CrossRef]
- Jia, H.; Chen, X.; Zhang, L.; Chen, M. Cancer associated fibroblasts in cancer development and therapy. J. Hematol. Oncol. 2025, 18, 36. [Google Scholar] [CrossRef]
- Turpin, A.; Neuzillet, C.; Colle, E.; Dusetti, N.; Nicolle, R.; Cros, J.; de Mestier, L.; Bachet, J.-B.; Hammel, P. Therapeutic advances in metastatic pancreatic cancer: A focus on targeted therapies. Ther. Adv. Med. Oncol. 2022, 14, 17588359221118019. [Google Scholar] [CrossRef] [PubMed]
- Bear, A.S.; Vonderheide, R.H.; O’Hara, M.H. Challenges and Opportunities for Pancreatic Cancer Immunotherapy. Cancer Cell 2020, 38, 788–802. [Google Scholar] [CrossRef]
- Qi, Y.; Zhang, L.; Wang, Z.; Kong, X.; Zhai, J.; Fang, Y.; Wang, J. Efficacy and Safety of Anti-PD-1/ PD-L1 Monotherapy for Metastatic Breast Cancer: Clinical Evidence. Front. Pharmacol. 2021, 12, 653521. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Huang, X.; Shi, F.; Song, J.; Guo, C.; Yang, J.; Liang, T.; Bai, X. Combination therapy for pancreatic cancer: Anti-PD-(L)1-based strategy. J. Exp. Clin. Cancer Res. 2022, 41, 56. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhou, Y.; Wu, Y.; Shi, M.; Sun, W.; Wang, R. Evaluation of the efficacy and predictive indicators of PD- 1 inhibitors combined with chemotherapy in advanced pancreatic cancer. Sci. Rep. 2025, 15, 12175. [Google Scholar] [CrossRef]
- Ye, X.; Yu, Y.; Zheng, X.; Ma, H. Clinical immunotherapy in pancreatic cancer. Cancer Immunol. Immunother. 2024, 73, 64. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Oh, D.-Y.; Dhani, N.; Renouf, D.J.; Lee, M.A.; Sun, W.; Fisher, G.; Hezel, A.; Chang, S.-C.; Vlahovic, G.; et al. Durvalumab With or Without Tremelimumab for Patients With Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 1431–1438. [Google Scholar] [CrossRef]
- Ely, Z.A.; Kulstad, Z.J.; Gunaydin, G.; Addepalli, S.; Verzani, E.K.; Casarrubios, M.; Clauser, K.R.; Wang, X.; Lippincott, I.E.; Louvet, C.; et al. Pancreatic cancer–restricted cryptic antigens are targets for T cell recognition. Science 2025, 388, eadk3487. [Google Scholar] [CrossRef]
- Vigano, S.; Alatzoglou, D.; Irving, M.; Ménétrier-Caux, C.; Caux, C.; Romero, P.; Coukos, G. Targeting Adenosine in Cancer Immunotherapy to Enhance T-Cell Function. Front. Immunol. 2019, 10, 925. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, Q.; Zhu, S.; Gong, X. Hypoxia promotes immune escape of pancreatic cancer cells by lncRNA NNT-AS1/METTL3-HuR-mediated ITGB1 m6A modification. Exp. Cell Res. 2023, 432, 113764. [Google Scholar] [CrossRef]
- Jacoberger-Foissac, C.; Cousineau, I.; Bareche, Y.; Allard, D.; Chrobak, P.; Allard, B.; Pommey, S.; Messaoudi, N.; McNicoll, Y.; Soucy, G.; et al. CD73 Inhibits cGAS–STING and Cooperates with CD39 to Promote Pancreatic Cancer. Cancer Immunol. Res. 2022, 11, 56–71. [Google Scholar] [CrossRef]
- Xia, C.; Yin, S.; To, K.K.W.; Fu, L. CD39/CD73/A2AR pathway and cancer immunotherapy. Mol. Cancer 2023, 22, 44. [Google Scholar] [CrossRef]
- Zhang, F.; Ma, Y.; Li, D.; Wei, J.; Chen, K.; Zhang, E.; Liu, G.; Chu, X.; Liu, X.; Liu, W.; et al. Cancer associated fibroblasts and metabolic reprogramming: Unraveling the intricate crosstalk in tumor evolution. J. Hematol. Oncol. 2024, 17, 80. [Google Scholar] [CrossRef]
- Zheng, R.; Liu, X.; Zhang, Y.; Liu, Y.; Wang, Y.; Guo, S.; Jin, X.; Zhang, J.; Guan, Y.; Liu, Y. Frontiers and future of immunotherapy for pancreatic cancer: From molecular mechanisms to clinical application. Front. Immunol. 2024, 15, 1383978. [Google Scholar] [CrossRef] [PubMed]
- Di Federico, A.; Mosca, M.; Pagani, R.; Carloni, R.; Frega, G.; De Giglio, A.; Rizzo, A.; Ricci, D.; Tavolari, S.; Di Marco, M.; et al. Immunotherapy in Pancreatic Cancer: Why Do We Keep Failing? A Focus on Tumor Immune Microenvironment, Predictive Biomarkers and Treatment Outcomes. Cancers 2022, 14, 2429. [Google Scholar] [CrossRef] [PubMed]
- Muscarella, P.; Bekaii-Saab, T.; McIntyre, K.; Rosemurgy, A.; Ross, S.B.; Richards, D.A.; Fisher, W.E.; Flynn, P.J.; Mattson, A.; Coeshott, C.; et al. A Phase 2 Randomized Placebo-Controlled Adjuvant Trial of GI-4000, a Recombinant Yeast Expressing Mutated RAS Proteins in Patients with Resected Pancreas Cancer. J. Pancreat. Cancer 2021, 7, 8–19. [Google Scholar] [CrossRef]
- Wang, B.; Pei, J.; Xu, S.; Liu, J.; Yu, J. Recent advances in mRNA cancer vaccines: Meeting challenges and embracing opportunities. Front. Immunol. 2023, 14, 1246682. [Google Scholar] [CrossRef] [PubMed]
- Wainberg, Z.A.; Weekes, C.D.; Furqan, M.; Kasi, P.M.; Devoe, C.E.; Leal, A.D.; Chung, V.; Perry, J.R.; Kheoh, T.; McNeil, L.K.; et al. Lymph node-targeted, mKRAS-specific amphiphile vaccine in pancreatic and colorectal cancer: Phase 1 AMPLIFY-201 trial final results. Nat. Med. 2025, 1–6. [Google Scholar] [CrossRef]
- Zhang, F.; Zhong, W.; Li, H.; Huang, K.; Yu, M.; Liu, Y. TP53 Mutational Status-Based Genomic Signature for Prognosis and Predicting Therapeutic Response in Pancreatic Cancer. Front. Cell Dev. Biol. 2021, 9, 665265. [Google Scholar] [CrossRef]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.-P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair–Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Rojas, L.A.; Sethna, Z.; Soares, K.C.; Olcese, C.; Pang, N.; Patterson, E.; Lihm, J.; Ceglia, N.; Guasp, P.; Chu, A.; et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature 2023, 618, 144–150. [Google Scholar] [CrossRef]
- Strickler, J.H.; Satake, H.; George, T.J.; Yaeger, R.; Hollebecque, A.; Garrido-Laguna, I.; Schuler, M.; Burns, T.F.; Coveler, A.L.; Falchook, G.S.; et al. Sotorasib in KRAS p.G12C–Mutated Advanced Pancreatic Cancer. N. Engl. J. Med. 2023, 388, 33–43. [Google Scholar] [CrossRef]
- Leidner, R.; Silva, N.S.; Huang, H.; Sprott, D.; Zheng, C.; Shih, Y.-P.; Leung, A.; Payne, R.; Sutcliffe, K.; Cramer, J.; et al. Neoantigen T-Cell Receptor Gene Therapy in Pancreatic Cancer. N. Engl. J. Med. 2022, 386, 2112–2119. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Guadagnin, G.; Cappello, P.; Novelli, F. Post-Translational Modifications in Tumor-Associated Antigens as a Platform for Novel Immuno-Oncology Therapies. Cancers 2022, 15, 138. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Shen, G.; Gao, W.; Huang, Z.; Huang, C.; Fu, L. Neoantigens: Promising targets for cancer therapy. Signal Transduct. Target. Ther. 2023, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Zhang, K.; Zhu, J.; Chen, Y.; Wang, Z.; Wang, P.; Xu, P.; Yao, J. Targeting protein modification: A new direction for immunotherapy of pancreatic cancer. Int. J. Biol. Sci. 2025, 21, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Radziejewska, I. Tumor-associated carbohydrate antigens of MUC1—Implication in cancer development. Biomed. Pharmacother. 2024, 174, 116619. [Google Scholar] [CrossRef]
- Rajesh, C.; Radhakrishnan, P. The (Sialyl) Tn antigen: Contributions to immunosuppression in gastrointestinal cancers. Front. Oncol. 2023, 12, 1093496. [Google Scholar] [CrossRef]
- Pienkowski, T.; Wawrzak-Pienkowska, K.; Tankiewicz-Kwedlo, A.; Ciborowski, M.; Kurek, K.; Pawlak, D. Leveraging glycosylation for early detection and therapeutic target discovery in pancreatic cancer. Cell Death Dis. 2025, 16, 227. [Google Scholar] [CrossRef]
- Sreejith, T.; Kamalasanan, K.; Sneha, S.; Keechilat, P.; Harb, H. Advancing Cancer Treatment Through Nanotechnology Driven Immunotherapy for Pancreatic Cancer. ACS Appl. Nano Mater. 2023, 6, 18670–18697. [Google Scholar] [CrossRef]
- Alghamdi, M.; Alasmari, D.; Assiri, A.; Mattar, E.; Aljaddawi, A.A.; Alattas, S.G.; Redwan, E.M. An Overview of the Intrinsic Role of Citrullination in Autoimmune Disorders. J. Immunol. Res. 2019, 2019, 7592851. [Google Scholar] [CrossRef]
- Chen, Y.; Teng, Y.; Xu, P.; Wang, S. The Role of Citrullination Modification in CD4+ T Cells in the Pathogenesis of Immune-Related Diseases. Biomolecules 2024, 14, 400. [Google Scholar] [CrossRef]
- Lee, J.B.; Pyo, K.-H.; Kim, H.R. Role and Function of O-GlcNAcylation in Cancer. Cancers 2021, 13, 5365. [Google Scholar] [CrossRef]
- Zhu, Q.; Zhou, H.; Wu, L.; Lai, Z.; Geng, D.; Yang, W.; Zhang, J.; Fan, Z.; Qin, W.; Wang, Y.; et al. O-GlcNAcylation promotes pancreatic tumor growth by regulating malate dehydrogenase 1. Nat. Chem. Biol. 2022, 18, 1087–1095. [Google Scholar] [CrossRef]
- Li, Y.; Patel, S.P.; Roszik, J.; Qin, Y. Hypoxia-Driven Immunosuppressive Metabolites in the Tumor Microenvironment: New Approaches for Combinational Immunotherapy. Front. Immunol. 2018, 9, 1591. [Google Scholar] [CrossRef] [PubMed]
- Marshall, M.L.; Fung, K.Y.; Jans, D.A.; Wagstaff, K.M. Tumour-specific phosphorylation of serine 419 drives alpha-enolase (ENO1) nuclear export in triple negative breast cancer progression. Cell Biosci. 2024, 14, 1591. [Google Scholar] [CrossRef]
- Tulamaiti, A.; Xiao, S.-Y.; Yang, Y.; Mutailifu, M.; Li, X.-Q.; Yin, S.-Q.; Ma, H.-T.; Yao, H.-F.; Yao, L.-L.; Hu, L.-P.; et al. ENO1 promotes PDAC progression by inhibiting CD8+ T cell infiltration through upregulating PD-L1 expression via HIF-1α signaling. Transl. Oncol. 2025, 52, 102261. [Google Scholar] [CrossRef]
- Belisle, J.A.; Horibata, S.; Jennifer, G.A.; Petrie, S.; Kapur, A.; André, S.; Gabius, H.-J.; Rancourt, C.; Connor, J.; Paulson, J.C.; et al. Identification of Siglec-9 as the receptor for MUC16 on human NK cells, B cells, and monocytes. Mol. Cancer 2010, 9, 118. [Google Scholar] [CrossRef]
- Rosenstock, P.; Kaufmann, T. Sialic Acids and Their Influence on Human NK Cell Function. Cells 2021, 10, 263. [Google Scholar] [CrossRef]
- Guerreiro, A.; Compañón, I.; Lazaris, F.S.; Labão-Almeida, C.; Oroz, P.; Ghirardello, M.; Marques, M.C.; Corzana, F.; Bernardes, G.J.L. Non-Natural MUC1 Glycopeptide Homogeneous Cancer Vaccine with Enhanced Immunogenicity and Therapeutic Activity. Angew. Chem. Int. Ed. Engl. 2024, 63, e202411009. [Google Scholar] [CrossRef]
- Mann, M.; Jensen, O.N. Proteomic analysis of post-translational modifications. Nat. Biotechnol. 2003, 21, 255–261. [Google Scholar] [CrossRef]
- Karpievitch, Y.V.; Polpitiya, A.D.; Anderson, G.A.; Smith, R.D.; Dabney, A.R. Liquid chromatography mass spectrometry-based proteomics: Biological and technological aspects. Ann. Appl. Stat. 2010, 4, 1797–1823. [Google Scholar] [CrossRef]
- Aebersold, R.; Mann, M. Mass-spectrometric exploration of proteome structure and function. Nature 2016, 537, 347–355. [Google Scholar] [CrossRef]
- Slavov, N. Single-cell protein analysis by mass spectrometry. Curr. Opin. Chem. Biol. 2021, 60, 1–9. [Google Scholar] [CrossRef]
- Liu, X.; Arfman, T.; Wichapong, K.; Reutelingsperger, C.P.; Voorberg, J.; Nicolaes, G.A. PAD4 takes charge during neutrophil activation: Impact of PAD4 mediated NET formation on immune-mediated disease. J. Thromb. Haemost. 2021, 19, 1607–1617. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, E.; Boelaars, K.; Brown, K.; Li, R.J.E.; Kruijssen, L.; Bruijns, S.C.M.; van Ee, T.; Schetters, S.T.T.; Crommentuijn, M.H.W.; van der Horst, J.C.; et al. Sialic acids in pancreatic cancer cells drive tumour-associated macrophage differentiation via the Siglec receptors Siglec-7 and Siglec-9. Nat. Commun. 2021, 12, 1270. [Google Scholar] [CrossRef]
- Bhalerao, N.; Chakraborty, A.; Marciel, M.P.; Hwang, J.; Britain, C.M.; Silva, A.D.; Eltoum, I.E.; Jones, R.B.; Alexander, K.L.; Smythies, L.E.; et al. ST6GAL1 sialyltransferase promotes acinar to ductal metaplasia and pancreatic cancer progression. J. Clin. Investig. 2023, 8, e161563. [Google Scholar] [CrossRef]
- Zhu, Q.; Wang, H.; Chai, S.; Xu, L.; Lin, B.; Yi, W.; Wu, L. O-GlcNAcylation promotes tumor immune evasion by inhibiting PD-L1 lysosomal degradation. Proc. Natl. Acad. Sci. USA 2023, 120, e2216796120. [Google Scholar] [CrossRef]
- Zhang, H.-R.; Li, T.-J.; Yu, X.-J.; Liu, C.; Wu, W.-D.; Ye, L.-Y.; Jin, K.-Z. The GFPT2-O-GlcNAcylation-YBX1 axis promotes IL-18 secretion to regulate the tumor immune microenvironment in pancreatic cancer. Cell Death Dis. 2024, 15, 224. [Google Scholar] [CrossRef]
- Uysal-Onganer, P.; D’alessio, S.; Mortoglou, M.; Kraev, I.; Lange, S. Peptidylarginine Deiminase Inhibitor Application, Using Cl-Amidine, PAD2, PAD3 and PAD4 Isozyme-Specific Inhibitors in Pancreatic Cancer Cells, Reveals Roles for PAD2 and PAD3 in Cancer Invasion and Modulation of Extracellular Vesicle Signatures. Int. J. Mol. Sci. 2021, 22, 1396. [Google Scholar] [CrossRef] [PubMed]
- Mahat, D.B.; Kumra, H.; Castro, S.A.; Metcalf, E.; Nguyen, K.; Morisue, R.; Ho, W.W.; Chen, I.; Sullivan, B.; Yim, L.H.; et al. Mutant p53 exploits enhancers to elevate immunosuppressive chemokine expression and impair immune checkpoint inhibitors in pancreatic cancer. Immunity 2025, 58, 1688–1705.e9. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Yang, L.V.; Abrams, S.L.; Steelman, L.S.; Follo, M.Y.; Cocco, L.; Ratti, S.; Martelli, A.M.; Augello, G.; Cervello, M. Effects of TP53 Mutations and miRs on Immune Responses in the Tumor Microenvironment Important in Pancreatic Cancer Progression. Cells 2022, 11, 2155. [Google Scholar] [CrossRef]
- Biederstädt, A.; Hassan, Z.; Schneeweis, C.; Schick, M.; Schneider, L.; Muckenhuber, A.; Hong, Y.; Siegers, G.; Nilsson, L.; Wirth, M.; et al. SUMO pathway inhibition targets an aggressive pancreatic cancer subtype. Gut 2020, 69, 1472–1482. [Google Scholar] [CrossRef]
- Ma, Y.; Xia, P.; Wang, Z.; Xu, J.; Zhang, L.; Jiang, Y. PDIA6 promotes pancreatic cancer progression and immune escape through CSN5-mediated deubiquitination of β-catenin and PD-L1. Neoplasia 2021, 23, 912–928. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, X.; Lao, M.; Sun, K.; He, L.; Xu, J.; Duan, Y.; Chen, Y.; Ying, H.; Li, M.; et al. Targeting ubiquitin-specific protease 8 sensitizes anti-programmed death-ligand 1 immunotherapy of pancreatic cancer. Cell Death Differ. 2022, 30, 560–575. [Google Scholar] [CrossRef] [PubMed]
- Looi, C.-K.; Gan, L.-L.; Sim, W.; Hii, L.-W.; Chung, F.F.-L.; Leong, C.-O.; Lim, W.-M.; Mai, C.-W. Histone Deacetylase Inhibitors Restore Cancer Cell Sensitivity towards T Lymphocytes Mediated Cytotoxicity in Pancreatic Cancer. Cancers 2022, 14, 3709. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Li, Y.; Huang, X.; Wei, M.; Huang, Y.; Tang, Z.; Huang, H.; Zhou, W.; Wang, Y.; Hu, J. Extensive protein S-nitrosylation associated with human pancreatic ductal adenocarcinoma pathogenesis. Cell Death Dis. 2019, 10, 914. [Google Scholar] [CrossRef]
- Pereira, P.M.; Edwards, K.J.; Mandleywala, K.; Carter, L.M.; Escorcia, F.E.; Campesato, L.F.; Cornejo, M.; Abma, L.; Mohsen, A.-A.; Iacobuzio-Donahue, C.A.; et al. iNOS Regulates the Therapeutic Response of Pancreatic Cancer Cells to Radiotherapy. Cancer Res. 2020, 80, 1681–1692. [Google Scholar] [CrossRef]
- Suskiewicz, M.J. The logic of protein post-translational modifications (PTMs): Chemistry, mechanisms and evolution of protein regulation through covalent attachments. BioEssays 2024, 46, e2300178. [Google Scholar] [CrossRef]
- Huang, X.; Gan, Z.; Cui, H.; Lan, T.; Liu, Y.; Caron, E.; Shao, W. The SysteMHC Atlas v2.0, an updated resource for mass spectrometry-based immunopeptidomics. Nucleic Acids Res. 2023, 52, D1062–D1071. [Google Scholar] [CrossRef]
- Beatson, R.; Tajadura-Ortega, V.; Achkova, D.; Picco, G.; Tsourouktsoglou, T.-D.; Klausing, S.; Hillier, M.; Maher, D.A.J.; Noll, S.K.T.; Crocker, P.R.; et al. The mucin MUC1 modulates the tumor immunological microenvironment through engagement of the lectin Siglec-9. Nat. Immunol. 2016, 17, 1273–1281. [Google Scholar] [CrossRef]
- Chen, X.; Sandrine, I.K.; Yang, M.; Tu, J.; Yuan, X. MUC1 and MUC16: Critical for immune modulation in cancer therapeutics. Front. Immunol. 2024, 15, 1356913. [Google Scholar] [CrossRef]
- Luo, X.; Chang, S.; Xiao, S.; Peng, Y.; Gao, Y.; Hu, F.; Liang, J.; Xu, Y.; Du, K.; Chen, Y.; et al. PAD4-dependent citrullination of nuclear translocation of GSK3β promotes colorectal cancer progression via the degradation of nuclear CDKN1A. Neoplasia 2022, 33, 100835. [Google Scholar] [CrossRef]
- Brentville, V.; Vankemmelbeke, M.; Metheringham, R.; Durrant, L. Post-translational modifications such as citrullination are excellent targets for cancer therapy. Semin. Immunol. 2020, 47, 101393. [Google Scholar] [CrossRef]
- Ehrentraut, H.; Clambey, E.T.; McNamee, E.N.; Brodsky, K.S.; Ehrentraut, S.F.; Poth, J.M.; Riegel, A.K.; Westrich, J.A.; Colgan, S.P.; Eltzschig, H.K. CD73 + regulatory T cells contribute to adenosine-mediated resolution of acute lung injury. FASEB J. 2013, 27, 2207–2219. [Google Scholar] [CrossRef]
- Zhong, Q.; Xiao, X.; Qiu, Y.; Xu, Z.; Chen, C.; Chong, B.; Zhao, X.; Hai, S.; Li, S.; An, Z.; et al. Protein posttranslational modifications in health and diseases: Functions, regulatory mechanisms, and therapeutic implications. MedComm 2023, 4, e261. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Zhang, J.; North, B.J.; Guo, J. Editorial: Post-translational modifications of proteins in cancer immunity and immunotherapy. Front. Immunol. 2022, 13, 1006145. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, N.; Galligan, J.J. A global view of the human post-translational modification landscape. Biochem. J. 2023, 480, 1241–1265. [Google Scholar] [CrossRef]
- Scott, E.; Goode, E.A.; Garnham, R.; Hodgson, K.; Orozco-Moreno, M.; Turner, H.; Livermore, K.; Nangkana, K.P.; Frame, F.M.; Bermudez, A.; et al. ST6GAL1-mediated aberrant sialylation promotes prostate cancer progression. J. Pathol. 2023, 261, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Garnham, R.; Scott, E.; Livermore, K.E.; Munkley, J. ST6GAL1: A key player in cancer (Review). Oncol. Lett. 2019, 18, 983–989. [Google Scholar] [CrossRef]
- Bannoura, S.F.; Khan, H.Y.; Azmi, A.S. KRAS G12D targeted therapies for pancreatic cancer: Has the fortress been conquered? Front. Oncol. 2022, 12, 1013902. [Google Scholar] [CrossRef]
- Doubleday, P.F.; Fornelli, L.; Ntai, I.; Kelleher, N.L. Oncogenic KRAS creates an aspartate metabolism signature in colorectal cancer cells. FEBS J. 2021, 288, 6683–6699. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Burlingame, A.L. Mass Spectrometry-Based Detection and Assignment of Protein Posttranslational Modifications. ACS Chem. Biol. 2014, 10, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.M.; Vitorino, R.; Domingues, M.M.; Spickett, C.M.; Domingues, P. Post-translational Modifications and Mass Spectrometry Detection. Free Radic. Biol. Med. 2013, 65, 925–941. [Google Scholar] [CrossRef]
- Tabang, D.N.; Ford, M.; Li, L. Recent Advances in Mass Spectrometry-Based Glycomic and Glycoproteomic Studies of Pancreatic Diseases. Front. Chem. 2021, 9, 707387. [Google Scholar] [CrossRef]
- Alvarez-Manilla, G.; Atwood, J.; Guo, Y.; Warren, N.L.; Orlando, R.; Pierce, M. Tools for Glycoproteomic Analysis: Size Exclusion Chromatography Facilitates Identification of Tryptic Glycopeptides with N-linked Glycosylation Sites. J. Proteome Res. 2006, 5, 701–708. [Google Scholar] [CrossRef]
- Ouyang, M.; Yu, C.; Deng, X.; Zhang, Y.; Zhang, X.; Duan, F. O-GlcNAcylation and Its Role in Cancer-Associated Inflammation. Front. Immunol. 2022, 13, 861559. [Google Scholar] [CrossRef]
- Yang, H.; Bailey, P.; Pilarsky, C. CRISPR Cas9 in Pancreatic Cancer Research. Front. Cell Dev. Biol. 2019, 7, 239. [Google Scholar] [CrossRef]
- Jardim-Perassi, B.V.; Irrera, P.; Oluwatola, O.E.; Abrahams, D.; Estrella, V.C.; Ordway, B.; Byrne, S.R.; Ojeda, A.A.; Whelan, C.J.; Kim, J.; et al. L-DOS47 Elevates Pancreatic Cancer Tumor pH and Enhances Response to Immunotherapy. Biomedicines 2024, 12, 461. [Google Scholar] [CrossRef]
- Molvi, Z.; Klatt, M.G.; Dao, T.; Urraca, J.; Scheinberg, D.A.; O’Reilly, R.J. The landscape of MHC-presented phosphopeptides yields actionable shared tumor antigens for cancer immunotherapy across multiple HLA alleles. J. Immunother. Cancer 2023, 11, e006889. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, K.E.; Reser, L.; Cuevas, M.V.R.; Abelin, J.G.; Shabanowitz, J.; Hunt, D.F.; Malaker, S.A. Identification of Post-translationally Modified MHC Class I–Associated Peptides as Potential Cancer Immunotherapeutic Targets. Mol. Cell. Proteom. 2025, 24, 100971. [Google Scholar] [CrossRef]
- León-Letelier, R.A.; Katayama, H.; Hanash, S. Mining the Immunopeptidome for Antigenic Peptides in Cancer. Cancers 2022, 14, 4968. [Google Scholar] [CrossRef]
- Duan, Z.; Shi, R.; Gao, B.; Cai, J. N-linked glycosylation of PD-L1/PD-1: An emerging target for cancer diagnosis and treatment. J. Transl. Med. 2024, 22, 705. [Google Scholar] [CrossRef]
- Rajesh, C.; Cummings, R.D.; Radhakrishnan, P. Unraveling the glyco-immunity nexus in pancreatic cancer. Mol. Cancer 2025, 24, 211. [Google Scholar] [CrossRef]
- Karamitopoulou, E. Tumour microenvironment of pancreatic cancer: Immune landscape is dictated by molecular and histopathological features. Br. J. Cancer 2019, 121, 5–14. [Google Scholar] [CrossRef]
- Estephan, H.; Tailor, A.; Parker, R.; Kreamer, M.; Papandreou, I.; Campo, L.; Easton, A.; Moon, E.J.; Denko, N.C.; Ternette, N.; et al. Hypoxia promotes tumor immune evasion by suppressing MHC-I expression and antigen presentation. EMBO J. 2025, 44, 903–922. [Google Scholar] [CrossRef]
- De Meo, M.L.; Spicer, J.D. The role of neutrophil extracellular traps in cancer progression and metastasis. In Seminars in Immunology; Academic Press: Cambridge, MA, USA, 2021; Volume 57. [Google Scholar] [CrossRef]
- Mitsis, M.; Drosou, P.; Tatsis, V.; Markopoulos, G.S. Neutrophil Extracellular Traps and Pancreatic Cancer Development: A Vicious Cycle. Cancers 2022, 14, 3339. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, A.; Hashimoto, S. Plasticity and Tumor Microenvironment in Pancreatic Cancer: Genetic, Metabolic, and Immune Perspectives. Cancers 2024, 16, 4094. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, C.M.; Lynch, T.P.; Sodi, V.L.; Falcone, J.N.; Schwab, L.P.; Peacock, D.L.; Vocadlo, D.J.; Seagroves, T.N.; Reginato, M.J. O-GlcNAcylation Regulates Cancer Metabolism and Survival Stress Signaling via Regulation of the HIF-1 Pathway. Mol. Cell 2014, 54, 820–831. [Google Scholar] [CrossRef] [PubMed]
- Olivier-Van Stichelen, S.; Dehennaut, V.; Buzy, A.; Zachayus, J.-L.; Guinez, C.; Mir, A.-M.; El Yazidi-Belkoura, I.; Copin, M.-C.; Boureme, D.; Loyaux, D.; et al. O-GlcNAcylation stabilizes β-catenin through direct competition with phosphorylation at threonine 41. FASEB J. 2014, 28, 3325–3338. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Jin, X.; Zhang, D.; Li, D.; Hao, F.; Feng, Y.; Gu, S.; Meng, F.; Tian, M.; et al. O- GlcNAcylation destabilizes the active tetrameric PKM2 to promote the Warburg effect. Proc. Natl. Acad. Sci. USA 2017, 114, 13732–13737. [Google Scholar] [CrossRef]
- Lu, Q.; Zhang, X.; Liang, T.; Bai, X. O-GlcNAcylation: An important post-translational modification and a potential therapeutic target for cancer therapy. Mol. Med. 2022, 28, 115. [Google Scholar] [CrossRef]
- Lumibao, J.C.; Tremblay, J.R.; Hsu, J.; Engle, D.D. Altered glycosylation in pancreatic cancer and beyond. J. Exp. Med. 2022, 219, e20211505. [Google Scholar] [CrossRef]
- Backe, S.J.; Sager, R.A.; Woodford, M.R.; Makedon, A.M.; Mollapour, M. Post-translational modifications of Hsp90 and translating the chaperone code. J. Biol. Chem. 2020, 295, 11099–11117. [Google Scholar] [CrossRef] [PubMed]
- Bali, P.; Pranpat, M.; Bradner, J.; Balasis, M.; Fiskus, W.; Guo, F.; Rocha, K.; Kumaraswamy, S.; Boyapalle, S.; Atadja, P.; et al. Inhibition of Histone Deacetylase 6 Acetylates and Disrupts the Chaperone Function of Heat Shock Protein 90. J. Biol. Chem. 2005, 280, 26729–26734. [Google Scholar] [CrossRef] [PubMed]
- Scroggins, B.T.; Robzyk, K.; Wang, D.; Marcu, M.G.; Tsutsumi, S.; Beebe, K.; Cotter, R.J.; Felts, S.; Toft, D.; Karnitz, L.; et al. An Acetylation Site in the Middle Domain of Hsp90 Regulates Chaperone Function. Mol. Cell 2007, 25, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Menezes, S.; Okail, M.H.; Jalil, S.M.A.; Kocher, H.M.; Cameron, A.J.M. Cancer-associated fibroblasts in pancreatic cancer: New subtypes, new markers, new targets. J. Pathol. 2022, 257, 526–544. [Google Scholar] [CrossRef]
- Morgan, A.; Griffin, M.; Kameni, L.; Wan, D.C.; Longaker, M.T.; Norton, J.A. Medical Biology of Cancer-Associated Fibroblasts in Pancreatic Cancer. Biology 2023, 12, 1044. [Google Scholar] [CrossRef]
- Zou, S.; Tong, Q.; Liu, B.; Huang, W.; Tian, Y.; Fu, X. Targeting STAT3 in Cancer Immunotherapy. Mol. Cancer 2020, 19, 145. [Google Scholar] [CrossRef]
- Demel, U.M.; Böger, M.; Yousefian, S.; Grunert, C.; Zhang, L.; Hotz, P.W.; Gottschlich, A.; Köse, H.; Isaakidis, K.; Vonficht, D.; et al. Activated SUMOylation restricts MHC class I antigen presentation to confer immune evasion in cancer. J. Clin. Investig. 2022, 132, e152383. [Google Scholar] [CrossRef]
- Miller-Ocuin, J.L.; Liang, X.; Boone, B.A.; Doerfler, W.R.; Singhi, A.D.; Tang, D.; Kang, R.; Lotze, M.T.; Zeh, H.J., III. DNA released from neutrophil extracellular traps (NETs) activates pancreatic stellate cells and enhances pancreatic tumor growth. OncoImmunology 2019, 8, e1605822. [Google Scholar] [CrossRef]
- Deng, H.; Lin, C.; Garcia-Gerique, L.; Fu, S.; Cruz, Z.; Bonner, E.E.; Rosenwasser, M.; Rajagopal, S.; Sadhu, M.N.; Gajendran, C.; et al. A Novel Selective Inhibitor JBI-589 Targets PAD4-Mediated Neutrophil Migration to Suppress Tumor Progression. Cancer Res. 2022, 82, 3561–3572. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, K.; Li, J.; Wang, C.; Li, P.; Du, L. Neutrophil extracellular traps in cancer: From mechanisms to treatments. Clin. Transl. Med. 2025, 15, e70368. [Google Scholar] [CrossRef]
- Ortega-Batista, A.; Jaén-Alvarado, Y.; Moreno-Labrador, D.; Gómez, N.; García, G.; Guerrero, E.N. Single-Cell Sequencing: Genomic and Transcriptomic Approaches in Cancer Cell Biology. Int. J. Mol. Sci. 2025, 26, 2074. [Google Scholar] [CrossRef]
- Liu, J.; Xu, X.; Zhong, H.; Yu, M.; Abuduaini, N.; Zhang, S.; Yang, X.; Feng, B. Glycosylation and Its Role in Immune Checkpoint Proteins: From Molecular Mechanisms to Clinical Implications. Biomedicines 2024, 12, 1446. [Google Scholar] [CrossRef] [PubMed]
- Kufe, D.W. MUC1-C oncoprotein as a target in breast cancer: Activation of signaling pathways and therapeutic approaches. Oncogene 2012, 32, 1073–1081. [Google Scholar] [CrossRef]
- Zhu, D.; Lu, Y.; Wang, Y.; Wang, Y. PAD4 and Its Inhibitors in Cancer Progression and Prognosis. Pharmaceutics 2022, 14, 2414. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Yang, G.; Zhou, W.; Qiu, J.; Chen, G.; Luo, W.; Zhao, F.; You, L.; Zheng, L.; Zhang, T.; et al. Targeting hypoxic tumor microenvironment in pancreatic cancer. J. Hematol. Oncol. 2021, 14, 14. [Google Scholar] [CrossRef]
- Du, L.; Lee, J.-H.; Jiang, H.; Wang, C.; Wang, S.; Zheng, Z.; Shao, F.; Xu, D.; Xia, Y.; Li, J.; et al. β-Catenin induces transcriptional expression of PD-L1 to promote glioblastoma immune evasion. J. Exp. Med. 2020, 217, e20191115. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Jenkins, R.W.; Sullivan, R.J. Mechanisms of Resistance to Immune Checkpoint Blockade. Am. J. Clin. Dermatol. 2018, 20, 41–54. [Google Scholar] [CrossRef]
- Kwee, S.A.; Tiirikainen, M. Beta-catenin activation and immunotherapy resistance in hepatocellular carcinoma: Mechanisms and biomarkers. Hepatoma Res. 2021, 2021, 8. [Google Scholar] [CrossRef]
- Griesemer, A.D.; Sorenson, E.C.; Hardy, M.A. The Role of the Thymus in Tolerance. Transplantation 2010, 90, 465–474. [Google Scholar] [CrossRef]
- Zhai, J.; Gu, X.; Liu, Y.; Hu, Y.; Jiang, Y.; Zhang, Z. Chemotherapeutic and targeted drugs-induced immunogenic cell death in cancer models and antitumor therapy: An update review. Front. Pharmacol. 2023, 14, 1152934. [Google Scholar] [CrossRef]
- Borrello, M.G.; Degl’innocenti, D.; Pierotti, M.A. Inflammation and cancer: The oncogene-driven connection. Cancer Lett. 2008, 267, 262–270. [Google Scholar] [CrossRef]
- Peng, H.; James, C.A.; Cullinan, D.R.; Hogg, G.D.; Mudd, J.L.; Zuo, C.; Takchi, R.; Caldwell, K.E.; Liu, J.; DeNardo, D.G.; et al. Neoadjuvant FOLFIRINOX Therapy Is Associated with Increased Effector T Cells and Reduced Suppressor Cells in Patients with Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 6761–6771. [Google Scholar] [CrossRef]
- Mowen, K.A.; David, M. Unconventional post-translational modifications in immunological signaling. Nat. Immunol. 2014, 15, 512–520. [Google Scholar] [CrossRef] [PubMed]
- De Sanctis, F.; Sandri, S.; Ferrarini, G.; Pagliarello, I.; Sartoris, S.; Ugel, S.; Marigo, I.; Molon, B.; Bronte, V. The Emerging Immunological Role of Post-Translational Modifications by Reactive Nitrogen Species in Cancer Microenvironment. Front. Immunol. 2014, 5, 69. [Google Scholar] [CrossRef] [PubMed]
- Brentville, V.A.; Metheringham, R.L.; Daniels, I.; Atabani, S.; Symonds, P.; Cook, K.W.; Vankemmelbeke, M.; Choudhury, R.; Vaghela, P.; Gijon, M.; et al. Combination vaccine based on citrullinated vimentin and enolase peptides induces potent CD4-mediated anti-tumor responses. J. Immunother. Cancer 2020, 8, e000560. [Google Scholar] [CrossRef]
- Imamura, T.; Ashida, R.; Ohshima, K.; Uesaka, K.; Sugiura, T.; Ohgi, K.; Yamada, M.; Otsuka, S.; Hatakeyama, K.; Nagashima, T.; et al. Characterization of pancreatic cancer with ultra-low tumor mutational burden. Sci. Rep. 2023, 13, 4359. [Google Scholar] [CrossRef] [PubMed]
- Lui, I.; Schaefer, K.; Kirkemo, L.L.; Zhou, J.; Perera, R.M.; Leung, K.K.; Wells, J.A. Hypoxia Induces Extensive Protein and Proteolytic Remodeling of the Cell Surface in Pancreatic Adenocarcinoma (PDAC) Cell Lines. J. Proteome Res. 2025, 24, 2791–2800. [Google Scholar] [CrossRef]
- Li, K.; Yuan, J.; Trafton, D.; Wang, J.; Niu, N.; Yuan, C.; Liu, X.; Zheng, L. Pancreatic ductal adenocarcinoma immune microenvironment and immunotherapy prospects. Chronic Dis. Transl. Med. 2020, 6, 6–17. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic Cell Death in Cancer Therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.N.; Heiden, M.G.V. Metabolism in the Tumor Microenvironment. Annu. Rev. Cancer Biol. 2020, 4, 17–40. [Google Scholar] [CrossRef]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras Maintains Pancreatic Tumors through Regulation of Anabolic Glucose Metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef]
- Guillaumond, F.; Leca, J.; Olivares, O.; Lavaut, M.-N.; Vidal, N.; Berthezène, P.; Dusetti, N.J.; Loncle, C.; Calvo, E.; Turrini, O.; et al. Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, 3919–3924. [Google Scholar] [CrossRef] [PubMed]
- Rohrbach, A.S.; Slade, D.J.; Thompson, P.R.; Mowen, K.A. Activation of PAD4 in NET formation. Front. Immunol. 2012, 3, 360. [Google Scholar] [CrossRef]
- Karamitopoulou, E.; Wenning, A.S.; Acharjee, A.; Aeschbacher, P.; Marinoni, I.; Zlobec, I.; Gloor, B.; Perren, A. Spatial Heterogeneity of Immune Regulators Drives Dynamic Changes in Local Immune Responses, Affecting Disease Outcomes in Pancreatic Cancer. Clin. Cancer Res. 2024, 30, 4215–4226. [Google Scholar] [CrossRef]
- Xia, Y.; Ma, J.; Yang, X.; Liu, D.; Zhu, Y.; Zhao, Y.; Fei, X.; Xu, D.; Dai, J. Identifying the Spatial Architecture That Restricts the Proximity of CD8+ T Cells to Tumor Cells in Pancreatic Ductal Adenocarcinoma. Cancers 2024, 16, 1434. [Google Scholar] [CrossRef]
- Sethumadhavan, S.; Silva, M.; Philbrook, P.; Nguyen, T.; Hatfield, S.M.; Ohta, A.; Sitkovsky, M.V. Hypoxia and hypoxia-inducible factor (HIF) downregulate antigen-presenting MHC class I molecules limiting tumor cell recognition by T cells. PLoS ONE 2017, 12, e0187314. [Google Scholar] [CrossRef]
- Lyman, M.R.; Mitchell, J.T.; Raghavan, S.; Kagohara, L.T.; Huff, A.L.; Haldar, S.D.; Shin, S.M.; Guinn, S.; Barrett, B.; Longway, G.; et al. Spatial Proteomics and Transcriptomics Reveal Early Immune Cell Organization in Pancreatic Intraepithelial Neoplasia. J. Clin. Investig. 2025, 10, e191595. [Google Scholar] [CrossRef]
- Zhu, D.; Zhang, Y.; Wang, S. Histone citrullination: A new target for tumors. Mol. Cancer 2021, 20, 90. [Google Scholar] [CrossRef]
- Rappu, P.; Suwal, U.; Siljamäki, E.; Heino, J. Inflammation-related citrullination of matrisome proteins in human cancer. Front. Oncol. 2022, 12, 1035188. [Google Scholar] [CrossRef]
- Zhu, T.; Yang, Q.; Qian, X.; Wu, X.; Fang, J.; Lin, Y.; Feng, Y.; Gao, J.; Xia, Q. GPRC5A/CXCL8/NLRP3-mediated neutrophil extracellular traps drive gemcitabine-nab-paclitaxel resistance in pancreatic adenocarcinoma. Cancer Biol. Med. 2025, 22, 832–853. [Google Scholar] [CrossRef]
- Boelaars, K.; Rodriguez, E.; Huinen, Z.R.; Liu, C.; Wang, D.; Springer, B.O.; Olesek, K.; Goossens-Kruijssen, L.; van Ee, T.; Lindijer, D.; et al. Pancreatic cancer-associated fibroblasts modulate macrophage differentiation via sialic acid-Siglec interactions. Commun. Biol. 2024, 7, 430. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.-Y.; Qi, L.-L.; Kang, F.-B.; Wang, L. The intriguing roles of Siglec family members in the tumor microenvironment. Biomark. Res. 2022, 10, 22. [Google Scholar] [CrossRef]
- van Houtum, E.J.H.; Büll, C.; Cornelissen, L.A.M.; Adema, G.J. Siglec Signaling in the Tumor Microenvironment. Front. Immunol. 2021, 12, 790317. [Google Scholar] [CrossRef]
- Peng, L.; Wang, D.; Han, Y.; Huang, T.; He, X.; Wang, J.; Ou, C. Emerging Role of Cancer-Associated Fibroblasts-Derived Exosomes in Tumorigenesis. Front. Immunol. 2022, 12, 795372. [Google Scholar] [CrossRef]
- Nedaeinia, R.; Najafgholian, S.; Salehi, R.; Goli, M.; Ranjbar, M.; Nickho, H.; Javanmard, S.H.; Ferns, G.A.; Manian, M. The role of cancer-associated fibroblasts and exosomal miRNAs-mediated intercellular communication in the tumor microenvironment and the biology of carcinogenesis: A systematic review. Cell Death Discov. 2024, 10, 380. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.D.; Hwang, J.; Marciel, M.P.; Bellis, S.L. The pro-inflammatory cytokines IL-1β and IL-6 promote upregulation of the ST6GAL1 sialyltransferase in pancreatic cancer cells. J. Biol. Chem. 2024, 300, 107752. [Google Scholar] [CrossRef]
- Berglund, A.K.; Hinson, A.L.; Schnabel, L.V. TGF-β downregulates antigen processing and presentation genes and MHC I surface expression through a Smad3-dependent mechanism. bioRxiv 2023. [Google Scholar] [CrossRef]
- Gu, Y.; Fang, Y.; Wu, X.; Xu, T.; Hu, T.; Xu, Y.; Ma, P.; Wang, Q.; Shu, Y. The emerging roles of SUMOylation in the tumor microenvironment and therapeutic implications. Exp. Hematol. Oncol. 2023, 12, 58. [Google Scholar] [CrossRef]
- Park, S.; Paszek, M. Abstract 1549: Mucins form a nanoscale material barrier against immune cell attack. J. Biol. Chem. 2023, 299, S390. [Google Scholar] [CrossRef]
- Lustig, M.; Chan, C.; Jansen, J.H.M.; Bräutigam, M.; Kölling, M.A.; Gehlert, C.L.; Baumann, N.; Mester, S.; Foss, S.; Andersen, J.T.; et al. Disruption of the sialic acid/Siglec-9 axis improves antibody-mediated neutrophil cytotoxicity towards tumor cells. Front. Immunol. 2023, 14, 1178817. [Google Scholar] [CrossRef]
- Dobie, C.; Skropeta, D. Insights into the role of sialylation in cancer progression and metastasis. Br. J. Cancer 2020, 124, 76–90. [Google Scholar] [CrossRef] [PubMed]
- Boelaars, K.; van Kooyk, Y. Targeting myeloid cells for cancer immunotherapy: Siglec-7/9/10/15 and their ligands. Trends Cancer 2023, 10, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Patskovsky, Y.; Natarajan, A.; Patskovska, L.; Nyovanie, S.; Joshi, B.; Morin, B.; Brittsan, C.; Huber, O.; Gordon, S.; Michelet, X.; et al. Molecular mechanism of phosphopeptide neoantigen immunogenicity. Nat. Commun. 2023, 14, 3763. [Google Scholar] [CrossRef] [PubMed]
- Apoorvi, T.; Yury, P.; Iryna, V.; Michelle, K. Phosphopeptide Neoantigens as Emerging Targets in Cancer Immunotherapy. J. Cancer Immunol. 2024, 6, 135–147. [Google Scholar] [CrossRef]
- Koning, T.; Cordova, F.; Aguilar, G.; Sarmiento, J.; Mardones, G.A.; Boric, M.; Varas-Godoy, M.; Lladser, A.; Duran, W.N.; Ehrenfeld, P.; et al. S-Nitrosylation in endothelial cells contributes to tumor cell adhesion and extravasation during breast cancer metastasis. Biol. Res. 2023, 56, 51. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Lin, Y.-S.; Chen, C.-H.; Chen, Y.-J. Annexin A2-mediated cancer progression and therapeutic resistance in nasopharyngeal carcinoma. J. Biomed. Sci. 2018, 25, 30. [Google Scholar] [CrossRef]
- Shi, H.; Han, X.; Sun, Y.; Shang, C.; Wei, M.; Ba, X.; Zeng, X. Chemokine (C-X-C motif) ligand 1 and CXCL2 produced by tumor promote the generation of monocytic myeloid-derived suppressor cells. Cancer Sci. 2018, 109, 3826–3839. [Google Scholar] [CrossRef]
- Smith, L.E.; Rogowska-Wrzesinska, A. The challenge of detecting modifications on proteins. Essays Biochem. 2020, 64, 135–153. [Google Scholar] [CrossRef]
- Pinkse, M.; Evaristo, G.; Pieterse, M.; Yu, Y.; Verhaert, P. MS approaches to select peptides with post-translational modifications from amphibian defense secretions prior to full sequence elucidation. EuPA Open Proteom. 2014, 5, 32–40. [Google Scholar] [CrossRef]
- Geiszler, D.J.; Polasky, D.A.; Yu, F.; Nesvizhskii, A.I. Detecting diagnostic features in MS/MS spectra of post-translationally modified peptides. Nat. Commun. 2023, 14, 4132. [Google Scholar] [CrossRef]
- Bons, J.; Hunter, C.L.; Chupalov, R.; Causon, J.; Antonoplis, A.; Rose, J.; MacLean, B.; Schilling, B. Localization and Quantification of Post-Translational Modifications of Proteins Using Electron Activated Dissociation Fragmentation on a Fast-Acquisition Time-of-Flight Mass Spectrometer. J. Am. Soc. Mass Spectrom. 2023, 34, 2199–2210. [Google Scholar] [CrossRef]
- Ma, J.; Hart, G.W. O-GlcNAc profiling: From proteins to proteomes. Clin. Proteom. 2014, 11, 8. [Google Scholar] [CrossRef]
- Udeshi, N.D.; Hart, G.W.; Slawson, C. From Fringe to the Mainstream: How ETD MS Brought O-GlcNAc to the Masses. Mol. Cell. Proteom. 2024, 23, 100859. [Google Scholar] [CrossRef] [PubMed]
- Burt, R.A.; Alghusen, I.M.; Ephrame, S.J.; Villar, M.T.; Artigues, A.; Slawson, C. Mapping the O-GlcNAc Modified Proteome: Applications for Health and Disease. Front. Mol. Biosci. 2022, 9, 920727. [Google Scholar] [CrossRef]
- Mechref, Y.; Madera, M.; Novotny, M.V. Glycoprotein enrichment through lectin affinity techniques. Methods Mol. Biol. 2008, 424, 373–396. [Google Scholar] [CrossRef]
- Wu, F.; Li, W.; Lu, H.; Li, L. Recent Advances in Mass Spectrometry-Based Studies of Post-Translational Modifications in Alzheimer’s Disease. Mol. Cell. Proteom. 2025, 24, 101003. [Google Scholar] [CrossRef]
- Morgenstern, D.; Wolf-Levy, H.; Tickotsky-Moskovitz, N.; Cooper, I.; Buchman, A.S.; Bennett, D.A.; Beeri, M.S.; Levin, Y. Optimized Glycopeptide Enrichment Method–It Is All About the Sauce. Anal. Chem. 2022, 94, 10308–10313. [Google Scholar] [CrossRef]
- Flender, D.; Vilenne, F.; Adams, C.; Boonen, K.; Valkenborg, D.; Baggerman, G. Exploring the dynamic landscape of immunopeptidomics: Unravelling posttranslational modifications and navigating bioinformatics terrain. Mass Spectrom. Rev. 2024, 44, 599–629. [Google Scholar] [CrossRef]
- Arshad, S.; Cameron, B.; Joglekar, A.V. Immunopeptidomics for autoimmunity: Unlocking the chamber of immune secrets. npj Syst. Biol. Appl. 2025, 11, 10. [Google Scholar] [CrossRef] [PubMed]
- Pongcharoen, S.; Kaewsringam, N.; Somaparn, P.; Roytrakul, S.; Maneerat, Y.; Pintha, K.; Topanurak, S. Immunopeptidomics in the cancer immunotherapy era. Explor. Target. Anti Tumor Ther. 2024, 5, 801–817. [Google Scholar] [CrossRef]
- Brown, R.; Stuart, S.S.; Houel, S.; Ahn, N.G.; Old, W.M. Large-Scale Examination of Factors Influencing Phosphopeptide Neutral Loss during Collision Induced Dissociation. J. Am. Soc. Mass Spectrom. 2015, 26, 1128–1142. [Google Scholar] [CrossRef]
- Potel, C.M.; Lemeer, S.; Heck, A.J.R. Phosphopeptide Fragmentation and Site Localization by Mass Spec-trometry: An Update. Anal. Chem. 2018, 91, 126–141. [Google Scholar]
- Myers, S.A.; Daou, S.; Affar, E.B.; Burlingame, A. Electron transfer dissociation (ETD): The mass spectrometric breakthrough essential for O-GlcNAc protein site assignments—A study of the O-GlcNAcylated protein Host Cell Factor C1. Proteomics 2013, 13, 982–991. [Google Scholar] [CrossRef]
- Riley, N.M.; Malaker, S.A.; Driessen, M.D.; Bertozzi, C.R. Optimal Dissociation Methods Differ for N- and O-Glycopeptides. J. Proteome Res. 2020, 19, 3286–3301. [Google Scholar] [CrossRef] [PubMed]
- Riley, N.M.; Sikora, J.W.; Seckler, H.S.; Greer, J.B.; Fellers, R.T.; LeDuc, R.D.; Westphall, M.S.; Thomas, P.M.; Kelleher, N.L.; Coon, J.J. The Value of Activated Ion Electron Transfer Dissociation for High-Throughput Top-Down Characterization of Intact Proteins. Anal. Chem. 2018, 90, 8553–8560. [Google Scholar] [CrossRef]
- Kessler, A.L.; Fort, K.L.; Resemann, H.C.; Krüger, P.; Wang, C.; Koch, H.; Hauschild, J.-P.; Marino, F.; Heck, A.J. Increased EThcD Efficiency on the Hybrid Orbitrap Excedion Pro Mass Analyzer Extends the Depth in Identification and Sequence Coverage of HLA Class I Immunopeptidomes. Mol. Cell. Proteom. 2025, 24, 101049. [Google Scholar] [CrossRef]
- Qi, Y.A.; Maity, T.K.; Cultraro, C.M.; Misra, V.; Zhang, X.; Ade, C.; Gao, S.; Milewski, D.; Nguyen, K.D.; Ebrahimabadi, M.H.; et al. Proteogenomic Analysis Unveils the HLA Class I-Presented Immunopeptidome in Melanoma and EGFR-Mutant Lung Adenocarcinoma. Mol. Cell. Proteom. 2021, 20, 100136. [Google Scholar] [CrossRef]
- Yang, K.L.; Yu, F.; Teo, G.C.; Li, K.; Demichev, V.; Ralser, M.; Nesvizhskii, A.I. MSBooster: Improving peptide identification rates using deep learning-based features. Nat. Commun. 2023, 14, 4539. [Google Scholar] [CrossRef]
- Sandalova, T.; Sala, B.M.; Achour, A. Structural aspects of chemical modifications in the MHC-restricted immunopeptidome; Implications for immune recognition. Front. Chem. 2022, 10, 861609. [Google Scholar] [CrossRef]
- Purcell, A.W.; van Driel, I.R.; Gleeson, P.A. Impact of glycans on T-cell tolerance to glycosylated self-antigens. Immunol. Cell Biol. 2008, 86, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Schnaar, R.L. Glycans and glycan-binding proteins in immune regulation: A concise introduction to glycobiology for the allergist. J. Allergy Clin. Immunol. 2015, 135, 609–615. [Google Scholar] [CrossRef] [PubMed]
- van Kooyk, Y.; Rabinovich, G.A. Protein-glycan interactions in the control of innate and adaptive immune responses. Nat. Immunol. 2008, 9, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Wang, B.; Chen, Z.; Urabe, G.; Glover, M.S.; Shi, X.; Guo, L.-W.; Kent, K.C.; Li, L. Electron-Transfer/Higher-Energy Collision Dissociation (EThcD)-Enabled Intact Glycopeptide/Glycoproteome Characterization. J. Am. Soc. Mass Spectrom. 2017, 28, 1751–1764. [Google Scholar] [CrossRef]
- Baliban, R.C.; DiMaggio, P.A.; Plazas-Mayorca, M.D.; Young, N.L.; Garcia, B.A.; Floudas, C.A. A Novel Approach for Untargeted Post-translational Modification Identification Using Integer Linear Optimization and Tandem Mass Spectrometry. Mol. Cell. Proteom. 2010, 9, 764–779. [Google Scholar] [CrossRef]
- Steckel, A.; Uray, K.; Schlosser, G. Detection of protein posttranslational modifications by mass spectrometry. In Amino Acids, Peptides and Proteins; Ryadnov, M., Hudecz, F., Eds.; The Royal Society of Chemistry: London, UK, 2020; Volume 44, pp. 140–170. [Google Scholar]
- Rebak, A.S.; Hendriks, I.A.; Nielsen, M.L. Characterizing citrullination by mass spectrometry-based proteomics. Philos. Trans. R. Soc. B Biol. Sci. 2023, 378, 20220237. [Google Scholar] [CrossRef] [PubMed]
- Kacen, A.; Javitt, A.; Kramer, M.P.; Morgenstern, D.; Tsaban, T.; Shmueli, M.D.; Teo, G.C.; Leprevost, F.d.V.; Barnea, E.; Yu, F.; et al. Post-translational modifications reshape the antigenic landscape of the MHC I immunopeptidome in tumors. Nat. Biotechnol. 2022, 41, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, J.B.; Kaabinejadian, S.; Yari, H.; Peters, B.; Barra, C.; Gragert, L.; Hildebrand, W.; Nielsen, M. Machine learning reveals limited contribution of trans-only encoded variants to the HLA-DQ immunopeptidome. Commun. Biol. 2023, 6, 442. [Google Scholar] [CrossRef]
- Wilhelm, M.; Zolg, D.P.; Graber, M.; Gessulat, S.; Schmidt, T.; Schnatbaum, K.; Schwencke-Westphal, C.; Seifert, P.; Krätzig, N.d.A.; Zerweck, J.; et al. Deep learning boosts sensitivity of mass spectrometry-based immunopeptidomics. Nat. Commun. 2021, 12, 3346. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Liao, Y.; Wen, B.; Li, K.; Dou, Y.; Savage, S.R.; Zhang, B. caAtlas: An immunopeptidome atlas of human cancer. iScience 2021, 24, 103107. [Google Scholar] [CrossRef]
- Wen, B.; Wang, C.; Li, K.; Han, P.; Holt, M.V.; Savage, S.R.; Lei, J.T.; Dou, Y.; Shi, Z.; Li, Y.; et al. DeepMVP: Deep learning models trained on high-quality data accurately predict PTM sites and variant-induced alterations. Nat. Methods 2025, 22, 1857–1867. [Google Scholar] [CrossRef]
- Wang, J.; Tao, X.; Zhu, J.; Dai, Z.; Du, Y.; Xie, Y.; Chu, X.; Fu, G.; Lei, Z. Tumor organoid-immune co-culture models: Exploring a new perspective of tumor immunity. Cell Death Discov. 2025, 11, 195. [Google Scholar] [CrossRef] [PubMed]
- Dijkstra, K.K.; Cattaneo, C.M.; Weeber, F.; Chalabi, M.; Van De Haar, J.; Fanchi, L.F.; Slagter, M.; Van Der Velden, D.L.; Kaing, S.; Kelderman, S.; et al. Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 2018, 174, 1586–1598.e12. [Google Scholar] [CrossRef]
- Moravec, Z.; Zhao, Y.; Voogd, R.; Cook, D.R.; Kinrot, S.; Capra, B.; Yang, H.; Raud, B.; Ou, J.; Xuan, J.; et al. Discovery of tumor-reactive T cell receptors by massively parallel library synthesis and screening. Nat. Biotechnol. 2024, 43, 214–222. [Google Scholar] [CrossRef]
- Wang, X.; Song, X.; Li, Y.; Ding, Y.; Yin, C.; Ren, T.; Zhang, W. Integrated system for screening tumor-specific TCRs, epitopes, and HLA subtypes using single-cell sequencing data. J. Immunother. Cancer 2025, 13, e012029. [Google Scholar] [CrossRef]
- Kuilman, T.; Schrikkema, D.S.; Gadiot, J.; Gomez-Eerland, R.; Bies, L.; Walker, J.; Spaapen, R.M.; Kok, H.; Houg, D.; Viyacheva, M.; et al. Enabling next-generation engineered TCR-T therapies based on high-throughput TCR discovery from diagnostic tumor biopsies. Nat. Commun. 2025, 16, 649. [Google Scholar] [CrossRef]
- Ramanathan, R.K.; Lee, K.M.; McKolanis, J.; Hitbold, E.; Schraut, W.; Moser, A.J.; Warnick, E.; Whiteside, T.; Osborne, J.; Kim, H.; et al. Phase I study of a MUC1 vaccine composed of different doses of MUC1 peptide with SB-AS2 adjuvant in resected and locally advanced pancreatic cancer. Cancer Immunol. Immunother. 2004, 54, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Engelhard, V.H.; Obeng, R.C.; Cummings, K.L.; Petroni, G.R.; Ambakhutwala, A.L.; Chianese-Bullock, K.A.; Smith, K.T.; Lulu, A.; Varhegyi, N.; Smolkin, M.E.; et al. MHC-restricted phosphopeptide antigens: Preclinical validation and first-in-humans clinical trial in participants with high-risk melanoma. J. Immunother. Cancer 2020, 8, e000262. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.; Hamid, O.; Shum, E.; Wise, D.R.; Balar, A.V.; Weber, J.S.; LoRusso, P.; Shafi, S.; Rimm, D.L.; Tolcher, A.W.; et al. Trial in progress: A phase I/II, open-label, dose-escalation, safety and tolerability study of NC318 in subjects with advanced or metastatic solid tumors. J. Clin. Oncol. 2020, 38, TPS3166. [Google Scholar] [CrossRef]
- Assouline, S.E.; Mehta, A.; Hanel, W.; Doucet, S.; Johnston, P.B.; Danilov, A.; Cooper, B.W.; Chudnovsky, A.; Ding, J.; Long, T.; et al. Phase I/II Study of Subasumstat (TAK-981) in Combination With Rituximab in Relapsed/Refractory Non-Hodgkin Lymphoma. Clin. Lymphoma Myeloma Leuk. 2025, 25, 788–799.e11. [Google Scholar] [CrossRef] [PubMed]
- Ottensmeier, C.H.; Pinato, D.J.J.; Armstrong, A.C.; Symeonides, S.N.; Patel, P.M.; Danson, S.; Korolewicz, J.; Cameron, D.; Miller, R.M.; Master, F.; et al. Modi-1, anti-citrullinated neoepitope vaccine, alone and combined with checkpoint inhibitors in patients with head and neck, breast, renal, and ovarian cancers: ModiFy phase I/II basket clinical trial—Report after completion of monotherapy dose-finding. J. Clin. Oncol. 2023, 41, 2566. [Google Scholar] [CrossRef]
- Durrant, L.G.; Masters, F.; Paston, S.; Miller, R.; Pinato, D.J.; Herbertson, R.; Armstrong, A.; Symeonides, S.; Ottensmeier, C. Abstract CT256: Modi-1, anti-citrullinated neoepitope vaccine alone and combined with checkpoint inhibitors in patients with head and neck, breast, renal and ovarian carcinoma: Protocol for the ModiFY phase I/II basket clinical Ttial. Cancer Res. 2023, 83, CT256. [Google Scholar] [CrossRef]
- Penny, S.A.; Abelin, J.G.; Malaker, S.A.; Myers, P.T.; Saeed, A.Z.; Steadman, L.G.; Bai, D.L.; Ward, S.T.; Shabanowitz, J.; Hunt, D.F.; et al. Tumor Infiltrating Lymphocytes Target HLA-I Phosphopeptides Derived From Cancer Signaling in Colorectal Cancer. Front. Immunol. 2021, 12, 723566. [Google Scholar] [CrossRef]
- Lepisto, A.J.; Moser, A.J.; Zeh, H.; Lee, K.; Bartlett, D.; McKolanis, J.R.; Geller, B.A.; Schmotzer, A.; Potter, D.P.; Whiteside, T.; et al. A phase I/II study of a MUC1 peptide pulsed autologous dendritic cell vaccine as adjuvant therapy in patients with resected pancreatic and biliary tumors. Cancer Ther. 2008, 6, 955–964. [Google Scholar]
- Ota, S.; Miyashita, M.; Yamagishi, Y.; Ogasawara, M. Baseline immunity predicts prognosis of pancreatic cancer patients treated with WT1 and/or MUC1 peptide-loaded dendritic cell vaccination and a standard chemotherapy. Hum. Vaccines Immunother. 2021, 17, 5563–5572. [Google Scholar] [CrossRef]
- McGinty, J.W.; Marré, M.L.; Bajzik, V.; Piganelli, J.D.; James, E.A. T Cell Epitopes and Post-Translationally Modified Epitopes in Type 1 Diabetes. Curr. Diabetes Rep. 2015, 15, 90. [Google Scholar] [CrossRef] [PubMed]
- Gabba, A.; Attariya, R.; Behren, S.; Pett, C.; van der Horst, J.C.; Yurugi, H.; Yu, J.; Urschbach, M.; Sabin, J.; Birrane, G.; et al. MUC1 Glycopeptide Vaccine Modified with a GalNAc Glycocluster Targets the Macrophage Galactose C-type Lectin on Dendritic Cells to Elicit an Improved Humoral Response. J. Am. Chem. Soc. 2023, 145, 13027–13037. [Google Scholar] [CrossRef]
- Gao, T.; Cen, Q.; Lei, H. A review on development of MUC1-based cancer vaccine. Biomed. Pharmacother. 2020, 132, 110888. [Google Scholar] [CrossRef]
- Lakshminarayanan, V.; Supekar, N.T.; Wei, J.; McCurry, D.B.; Dueck, A.C.; Kosiorek, H.E.; Trivedi, P.P.; Bradley, J.M.; Madsen, C.S.; Pathangey, L.B.; et al. MUC1 Vaccines, Comprised of Glycosylated or Non-Glycosylated Peptides or Tumor-Derived MUC1, Can Circumvent Immunoediting to Control Tumor Growth in MUC1 Transgenic Mice. PLoS ONE 2016, 11, e0145920. [Google Scholar] [CrossRef]
- McDonald, D.M.; Byrne, S.N.; Payne, R.J. Synthetic self-adjuvanting glycopeptide cancer vaccines. Front. Chem. 2015, 3, 60. [Google Scholar] [CrossRef] [PubMed]
- Katayama, H.; Kobayashi, M.; Irajizad, E.; Sevillano, A.M.; Patel, N.; Mao, X.; Rusling, L.; Vykoukal, J.; Cai, Y.; Hsiao, F.; et al. Protein citrullination as a source of cancer neoantigens. J. Immunother. Cancer 2021, 9, e002549. [Google Scholar] [CrossRef]
- Loh, T.J.; Lim, J.J.; Jones, C.M.; Dao, H.T.; Tran, M.T.; Baker, D.G.; La Gruta, N.L.; Reid, H.H.; Rossjohn, J. The molecular basis underlying T cell specificity towards citrullinated epitopes presented by HLA-DR4. Nat. Commun. 2024, 15, 6201. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.; Daniels, I.; Symonds, P.; Pitt, T.; Gijon, M.; Xue, W.; Metheringham, R.; Durrant, L.; Brentville, V. Citrullinated α-enolase is an effective target for anti-cancer immunity. OncoImmunology 2017, 7, e1390642. [Google Scholar] [CrossRef]
- Tang, S.; Tang, R.; Chen, G.; Zhang, D.; Lin, K.; Yang, H.; Fu, J.; Guo, Y.; Lin, F.; Dong, X.; et al. Personalized neoantigen hydrogel vaccine combined with PD-1 and CTLA-4 double blockade elicits antitumor response in liver metastases by activating intratumoral CD8+CD69+T cells. J. Immunother. Cancer 2024, 12, e009543. [Google Scholar] [CrossRef]
- Hu, Q.; Shi, Y.; Wang, H.; Bing, L.; Xu, Z. Post-translational modifications of immune checkpoints: Unlocking new potentials in cancer immunotherapy. Exp. Hematol. Oncol. 2025, 14, 37. [Google Scholar] [CrossRef]
- Liu, Z.; Lv, J.; Dang, Q.; Liu, L.; Weng, S.; Wang, L.; Zhou, Z.; Kong, Y.; Li, H.; Han, Y.; et al. Engineering neoantigen vaccines to improve cancer personalized immunotherapy. Int. J. Biol. Sci. 2022, 18, 5607–5623. [Google Scholar] [CrossRef]
- Liao, J.-Y.; Zhang, S. Safety and Efficacy of Personalized Cancer Vaccines in Combination With Immune Checkpoint Inhibitors in Cancer Treatment. Front. Oncol. 2021, 11, 663264. [Google Scholar] [CrossRef]
- Nair, R.; Somasundaram, V.; Kuriakose, A.; Krishn, S.R.; Raben, D.; Salazar, R.; Nair, P. Deciphering T-cell exhaustion in the tumor microenvironment: Paving the way for innovative solid tumor therapies. Front. Immunol. 2025, 16, 1548234. [Google Scholar] [CrossRef]
- Zhu, T.; Wu, X.; Liao, Y.; Yan, Y.; Yu, M.; Wang, L.; Xia, Q. The role of innate immune cells as modulators of the tumor microenvironment in the metastasis and treatment of pancreatic cancer. Clin. Cancer Bull. 2023, 2, 2. [Google Scholar] [CrossRef]
- Jin, L.; Kim, H.; Shi, J. Neutrophil in the Pancreatic Tumor Microenvironment. Biomolecules 2021, 11, 1170. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Jia, R.; Taledaohan, A.; Wang, Y.; Wang, Y. Structure–Activity Relationship of PAD4 Inhibitors and Their Role in Tumor Immunotherapy. Pharmaceutics 2024, 16, 335. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhao, B.; Dong, H.; Li, T.; Cheng, X.; Gong, W.; Wang, J.; Zhang, J.; Xin, G.; Yu, Y.; et al. Inhibition of O-GlcNAc transferase activates type I interferon-dependent antitumor immunity by bridging cGAS-STING pathway. eLife 2024, 13, RP94849. [Google Scholar] [CrossRef]
- Al Saoud, R.; Hamrouni, A.; Idris, A.; Mousa, W.K.; Abu Izneid, T. Recent advances in the development of sialyltransferase inhibitors to control cancer metastasis: A comprehensive review. Biomed. Pharmacother. 2023, 165, 115091. [Google Scholar] [CrossRef]
- Lin, S.-Y.; Schmidt, E.N.; Takahashi-Yamashiro, K.; Macauley, M.S. Roles for Siglec-glycan interactions in regulating immune cells. Semin. Immunol. 2024, 77, 101925. [Google Scholar] [CrossRef]
- Egan, H.; Treacy, O.; Lynch, K.; Leonard, N.A.; O’malley, G.; Reidy, E.; O’neill, A.; Corry, S.M.; De Veirman, K.; Vanderkerken, K.; et al. Targeting stromal cell sialylation reverses T cell-mediated immunosuppression in the tumor microenvironment. Cell Rep. 2023, 42, 112475. [Google Scholar] [CrossRef]
- Rao, T.C.; Beggs, R.R.; Ankenbauer, K.E.; Hwang, J.; Ma, V.P.-Y.; Salaita, K.; Bellis, S.L.; Mattheyses, A.L. ST6Gal-I–mediated sialylation of the epidermal growth factor receptor modulates cell mechanics and enhances invasion. J. Biol. Chem. 2022, 298, 101726. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Zhou, Y.; Guo, L.; Feng, S. Biological function of sialic acid and sialylation in human health and disease. Cell Death Discov. 2024, 10, 415. [Google Scholar] [CrossRef]
- Lv, D.; Fei, Y.; Chen, H.; Wang, J.; Han, W.; Cui, B.; Feng, Y.; Zhang, P.; Chen, J. Crosstalk between T lymphocyte and extracellular matrix in tumor microenvironment. Front. Immunol. 2024, 15, 1340702. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, L.; Wan, D.; Zhou, L.; Zheng, S.; Lin, S.; Qiao, Y. Extracellular matrix and its therapeutic potential for cancer treatment. Signal Transduct. Target. Ther. 2021, 6, 153. [Google Scholar] [CrossRef]
- Mai, Z.; Lin, Y.; Lin, P.; Zhao, X.; Cui, L. Modulating extracellular matrix stiffness: A strategic approach to boost cancer immunotherapy. Cell Death Dis. 2024, 15, 307. [Google Scholar] [CrossRef]
- Lundberg, K.; Nijenhuis, S.; Vossenaar, E.R.; Palmblad, K.; van Venrooij, W.J.; Klareskog, L.; Zendman, A.; Harris, H.E. Citrullinated proteins have increased immunogenicity and arthritogenicity and their presence in arthritic joints correlates with disease severity. Arthritis Res. Ther. 2005, 7, R458–67. [Google Scholar] [CrossRef]
- Postow, M.A.; Sidlow, R.; Hellmann, M.D. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. N. Engl. J. Med. 2018, 378, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Lewis, H.D.; Liddle, J.; Coote, J.E.; Atkinson, S.J.; Barker, M.D.; Bax, B.D.; Bicker, K.L.; Bingham, R.P.; Campbell, M.; Chen, Y.H.; et al. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat. Chem. Biol. 2015, 11, 189–191. [Google Scholar] [CrossRef]
- Ferrara, B.; Pignatelli, C.; Cossutta, M.; Citro, A.; Courty, J.; Piemonti, L. The Extracellular Matrix in Pancreatic Cancer: Description of a Complex Network and Promising Therapeutic Options. Cancers 2021, 13, 4442. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Li, Y.; Zhang, S.; Wang, X.; Dou, H.; Yu, X.; Zhang, Z.; Yang, S.; Xiao, M. Extracellular matrix remodeling in tumor progression and immune escape: From mechanisms to treatments. Mol. Cancer 2023, 22, 48. [Google Scholar] [CrossRef] [PubMed]
- Nicolas-Boluda, A.; Vaquero, J.; Vimeux, L.; Guilbert, T.; Barrin, S.; Kantari-Mimoun, C.; Ponzo, M.; Renault, G.; Deptula, P.; Pogoda, K.; et al. Tumor stiffening reversion through collagen crosslinking inhibition improves T cell migration and anti-PD-1 treatment. eLife 2021, 10, e58688. [Google Scholar] [CrossRef]
- Jiang, C.; Wang, M.; Yao, W.; Lv, G.; Liu, X.; Wang, G. Comprehensive Analysis on Prognosis and Immune Infiltration of Lysyl Oxidase Family Members in Pancreatic Adenocarcinoma With Experimental Verification. Front. Mol. Biosci. 2022, 9, 778857. [Google Scholar] [CrossRef]
- Miller, B.W.; Morton, J.P.; Pinese, M.; Saturno, G.; Jamieson, N.B.; McGhee, E.; Timpson, P.; Leach, J.; McGarry, L.; Shanks, E.; et al. Targeting the LOX / hypoxia axis reverses many of the features that make pancreatic cancer deadly: Inhibition of LOX abrogates metastasis and enhances drug efficacy. EMBO Mol. Med. 2015, 7, 1063–1076. [Google Scholar] [CrossRef]
- Mommen, G.P.M.; Frese, C.K.; Meiring, H.D.; Brink, J.v.G.-V.D.; de Jong, A.P.J.M.; van Els, C.A.C.M.; Heck, A.J.R. Expanding the detectable HLA peptide repertoire using electron-transfer/higher-energy collision dissociation (EThcD). Proc. Natl. Acad. Sci. USA 2014, 111, 4507–4512. [Google Scholar] [CrossRef]
- Riley, N.M.; Westphall, M.S.; Coon, J.J. Activated Ion-Electron Transfer Dissociation Enables Comprehensive Top-Down Protein Fragmentation. J. Proteome Res. 2017, 16, 2653–2659. [Google Scholar] [CrossRef]
- Palacios, P.A.; Flores, I.; Cereceda, L.; Otero, F.F.; Müller, M.; Brebi, P.; Contreras, H.R.; Carreño, L.J. Patient-Derived Organoid Models for NKT Cell-Based Cancer Immunotherapy. Cancers 2025, 17, 406. [Google Scholar] [CrossRef] [PubMed]
- Magré, L.; Verstegen, M.M.A.; Buschow, S.; van der Laan, L.J.W.; Peppelenbosch, M.; Desai, J. Emerging organoid-immune co-culture models for cancer research: From oncoimmunology to personalized immunotherapies. J. Immunother. Cancer 2023, 11, e006290. [Google Scholar] [CrossRef] [PubMed]
- Esposito, A.; Agostini, A.; Quero, G.; Piro, G.; Priori, L.; Caggiano, A.; Scaglione, G.; Battaglia, A.; Calegari, M.A.; Salvatore, L.; et al. Colorectal cancer patients-derived immunity-organoid platform unveils cancer-specific tissue markers associated with immunotherapy resistance. Cell Death Dis. 2024, 15, 878. [Google Scholar] [CrossRef]
- Tsai, S.; McOlash, L.; Palen, K.; Johnson, B.; Duris, C.; Yang, Q.; Dwinell, M.B.; Hunt, B.; Evans, D.B.; Gershan, J.; et al. Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3D tumor microenvironment models. BMC Cancer 2018, 18, 335. [Google Scholar] [CrossRef]
- Caron, E.; Aebersold, R.; Banaei-Esfahani, A.; Chong, C.; Bassani-Sternberg, M. A Case for a Human Immuno-Peptidome Project Consortium. Immunity 2017, 47, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Vizcaíno, J.A.; Kubiniok, P.; Kovalchik, K.A.; Ma, Q.; Duquette, J.D.; Mongrain, I.; Deutsch, E.W.; Peters, B.; Sette, A.; Sirois, I.; et al. The Human Immunopeptidome Project: A Roadmap to Predict and Treat Immune Diseases. Mol. Cell. Proteom. 2020, 19, 31–49. [Google Scholar] [CrossRef]
- Kraemer, A.I.; Chong, C.; Huber, F.; Pak, H.; Stevenson, B.J.; Müller, M.; Michaux, J.; Altimiras, E.R.; Rusakiewicz, S.; Simó-Riudalbas, L.; et al. The immunopeptidome landscape associated with T cell infiltration, inflammation and immune editing in lung cancer. Nat. Cancer 2023, 4, 608–628. [Google Scholar] [CrossRef]
- Xu, H.; Hu, R.; Dong, X.; Kuang, L.; Zhang, W.; Tu, C.; Li, Z.; Zhao, Z. ImmuneApp for HLA-I epitope prediction and immunopeptidome analysis. Nat. Commun. 2024, 15, 8926. [Google Scholar] [CrossRef]
- Schneeweis, C.; Hassan, Z.; Schick, M.; Keller, U.; Schneider, G. The SUMO pathway in pancreatic cancer: Insights and inhibition. Br. J. Cancer 2020, 124, 531–538. [Google Scholar] [CrossRef]
- Kumar, S.; Schoonderwoerd, M.J.A.; Kroonen, J.S.; de Graaf, I.J.; Sluijter, M.; Ruano, D.; González-Prieto, R.; Vries, M.V.-D.; Rip, J.; Arens, R.; et al. Targeting pancreatic cancer by TAK-981: A SUMOylation inhibitor that activates the immune system and blocks cancer cell cycle progression in a preclinical model. Gut 2022, 71, 2266–2283. [Google Scholar] [CrossRef]
- Luo, Y.; Li, Z.; Kong, Y.; He, W.; Zheng, H.; An, M.; Lin, Y.; Zhang, D.; Yang, J.; Zhao, Y.; et al. KRAS mutant–driven SUMOylation controls extracellular vesicle transmission to trigger lymphangiogenesis in pancreatic cancer. J. Clin. Investig. 2022, 132, e157644. [Google Scholar] [CrossRef]
- de la Torre Medina, J.; Joshi, U.; Sonowal, H.; Kuang, Y.; Ren, T.; Chen, D.H.; Tharuka, M.D.N.; Nguyen-Ta, K.; Gros, H.; Mikulski, Z.; et al. Immunomodulation of Pancreatic Cancer via Inhibition of SUMOylation and CD155/TIGIT Pathway. bioRxiv 2025. [Google Scholar] [CrossRef]
- Zheng, Y.; Sun, W.; Shan, C.; Li, B.; Liu, J.; Xing, H.; Xu, Q.; Cui, B.; Zhu, W.; Chen, J.; et al. β-hydroxybutyrate inhibits ferroptosis-mediated pancreatic damage in acute liver failure through the increase of H3K9bhb. Cell Rep. 2022, 41, 111847. [Google Scholar] [CrossRef]
- Liang, X.; Tian, R.; Li, T.; Wang, H.; Qin, Y.; Qian, M.; Fan, J.; Wang, D.; Cui, H.-Y.; Jiang, J. Integrative insights into the role of CAV1 in ketogenic diet and ferroptosis in pancreatic cancer. Cell Death Discov. 2025, 11, 139. [Google Scholar] [CrossRef] [PubMed]
- Ku, B.; Eisenbarth, D.; Baek, S.; Jeong, T.-K.; Kang, J.-G.; Hwang, D.; Noh, M.-G.; Choi, C.; Choi, S.; Seol, T.; et al. PRMT1 promotes pancreatic cancer development and resistance to chemotherapy. Cell Rep. Med. 2024, 5, 101461. [Google Scholar] [CrossRef] [PubMed]
- Baretti, M.; Ahuja, N.; Azad, N.S. Targeting the epigenome of pancreatic cancer for therapy: Challenges and opportunities. Ann. Pancreat. Cancer 2019, 2, 18. [Google Scholar] [CrossRef]
- Chen, G.; Guo, G.; Zhou, X.; Chen, H. Potential mechanism of ferroptosis in pancreatic cancer (Review). Oncol. Lett. 2019, 19, 579–587. [Google Scholar] [CrossRef]
- Shen, X.; Chen, Y.; Tang, Y.; Lu, P.; Liu, M.; Mao, T.; Weng, Y.; Yu, F.; Liu, Y.; Tang, Y.; et al. Targeting pancreatic cancer glutamine dependency confers vulnerability to GPX4-dependent ferroptosis. Cell Rep. Med. 2025, 6, 101928. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).