
Integration of Single-Cell Analysis and Bulk RNA Sequencing Data Using Multi-Level Attention Graph Neural Network for Precise Prognostic Stratification in Thyroid Cancer

 and
and
Simple Summary
Abstract
1. Introduction
Study Hypothesis and Objective
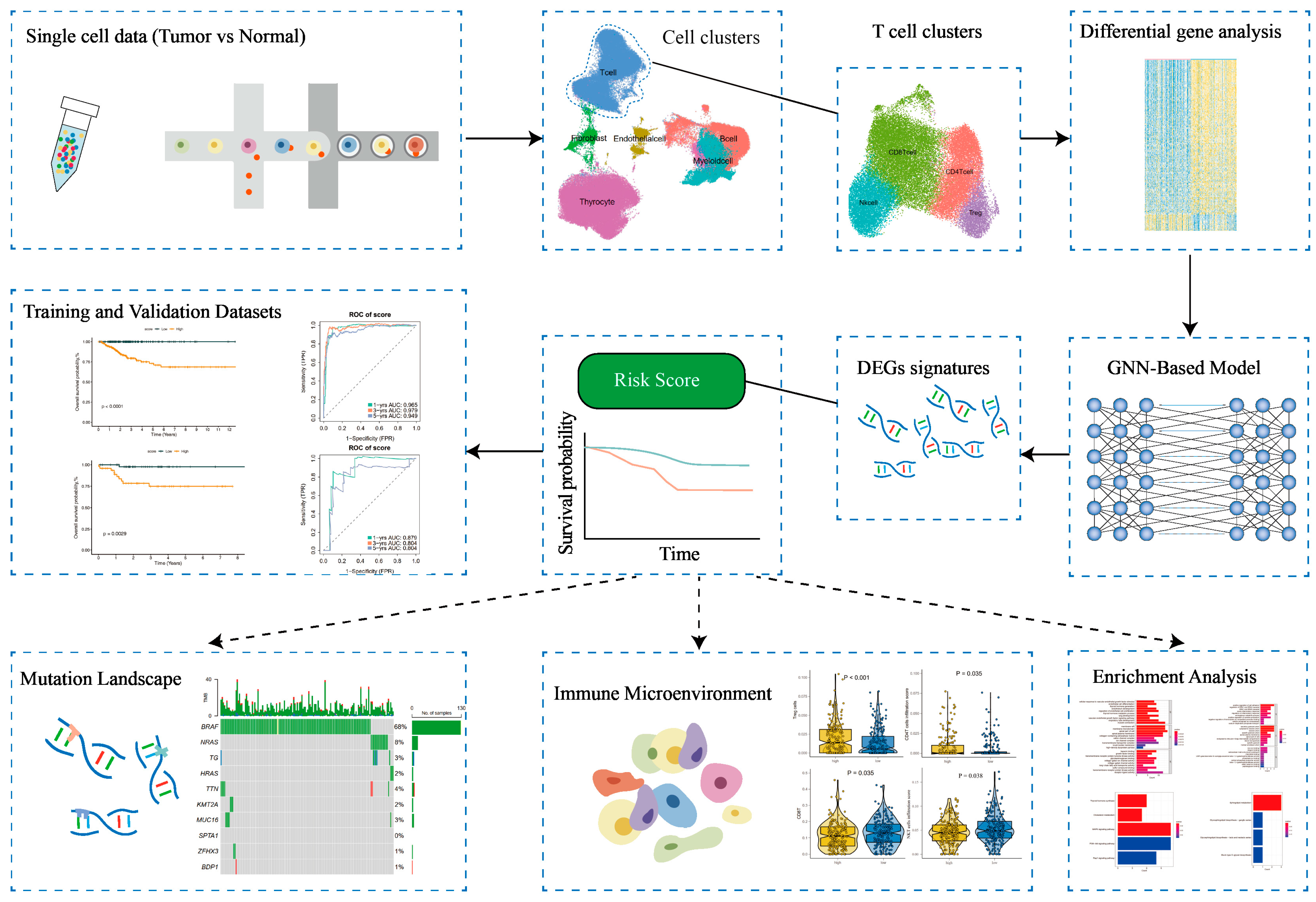
2. Materials and Methods
2.1. Acquisition of Raw Data
2.2. Processing of scRNA-Seq Data and Cell Annotation
2.3. Utilization of AUCell
2.4. Enhanced Analysis of Cell–Cell Communication
2.5. Gene Co-Expression Computation
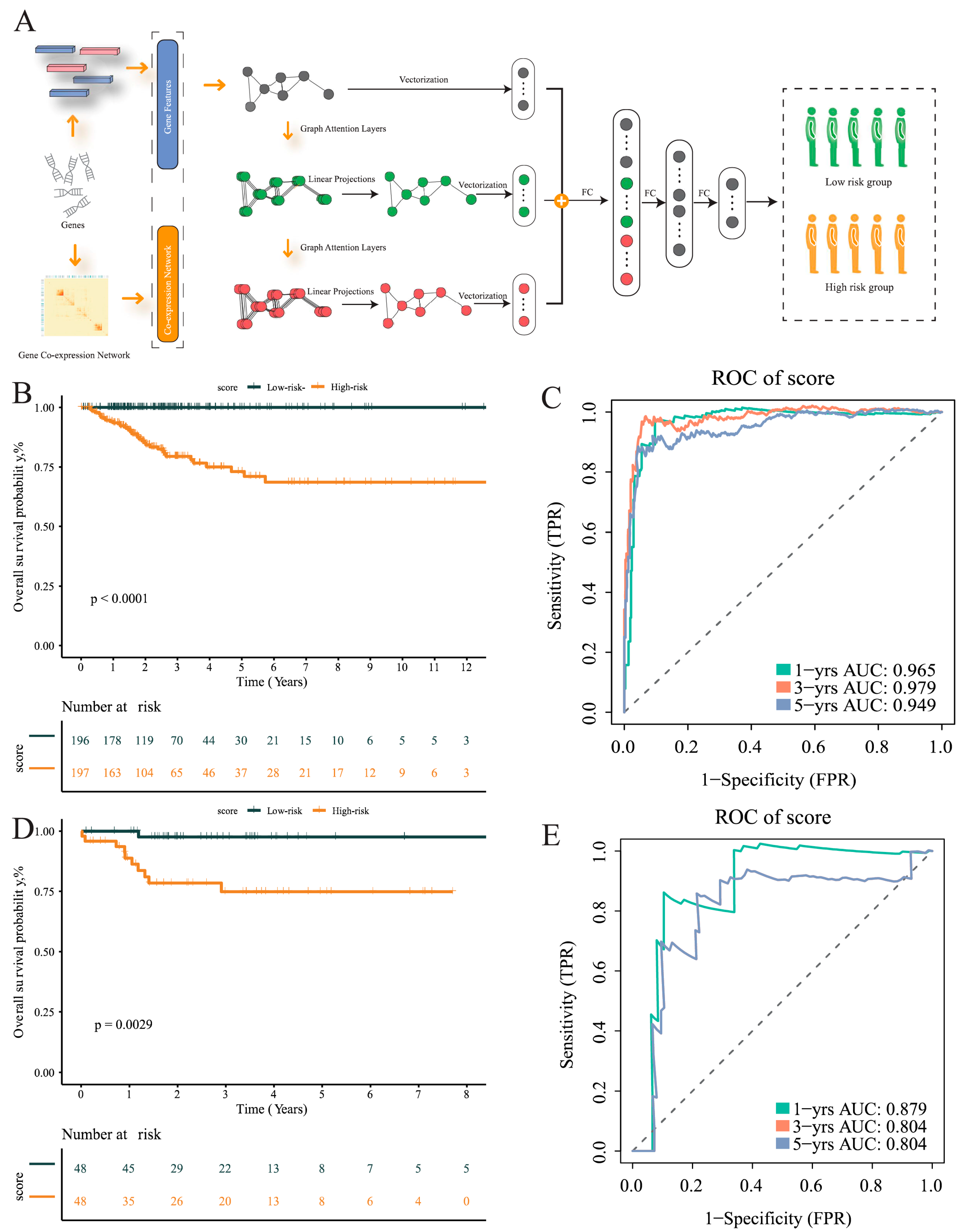
2.6. MLA-GNN Model
2.7. Prognosis Prediction Using MLA-GNN Model
2.8. Profiling of Immune Cell Subpopulations
2.9. Genetic Alterations and Tumorigenesis Through Mutation Analysis
2.10. Enrichment Analysis to Unveil Biological Significance in Genomic Data
2.11. Subtype Clinical Feature Analysis
3. Results
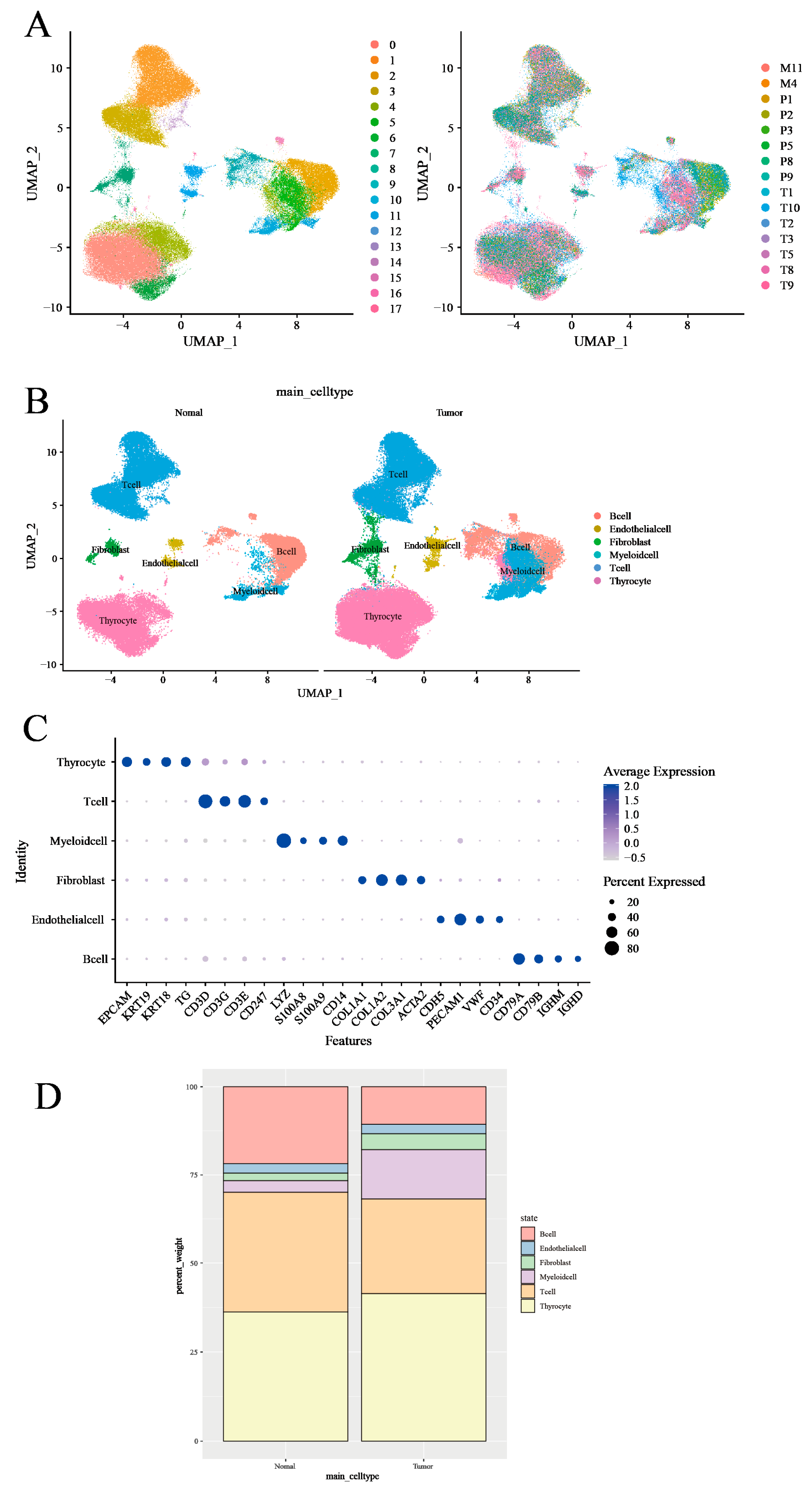
3.1. Characterization of the Tumor Microenvironment in Thyroid Tissues Through Identifying Main Clusters
3.2. T-Cell Heterogeneity and Metabolic Landscape in Thyroid Tumor Microenvironment Revealed by Single-Cell RNA Sequencing
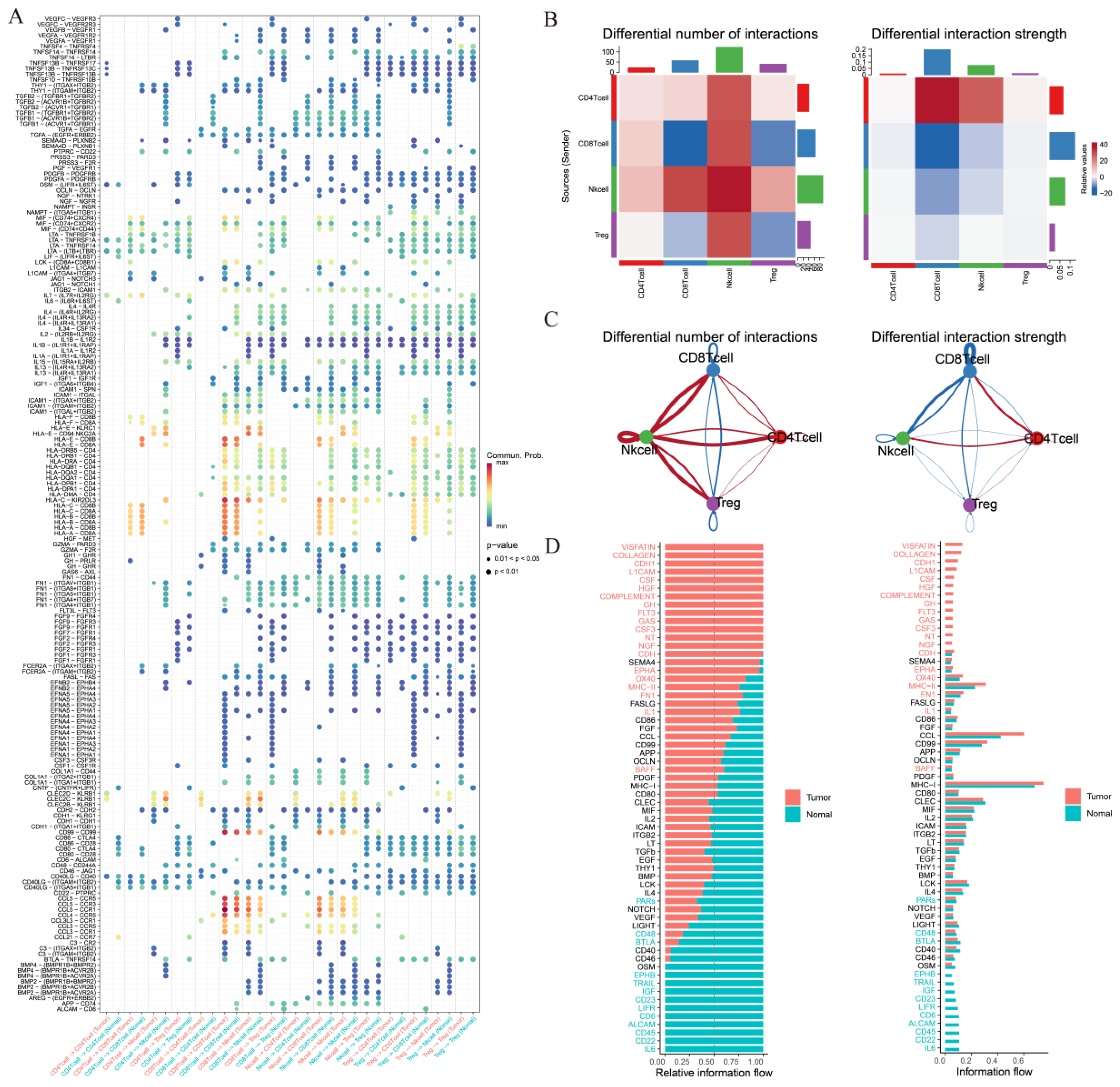
3.3. Comparative Intercellular Communication Analysis in Thyroid Cancer
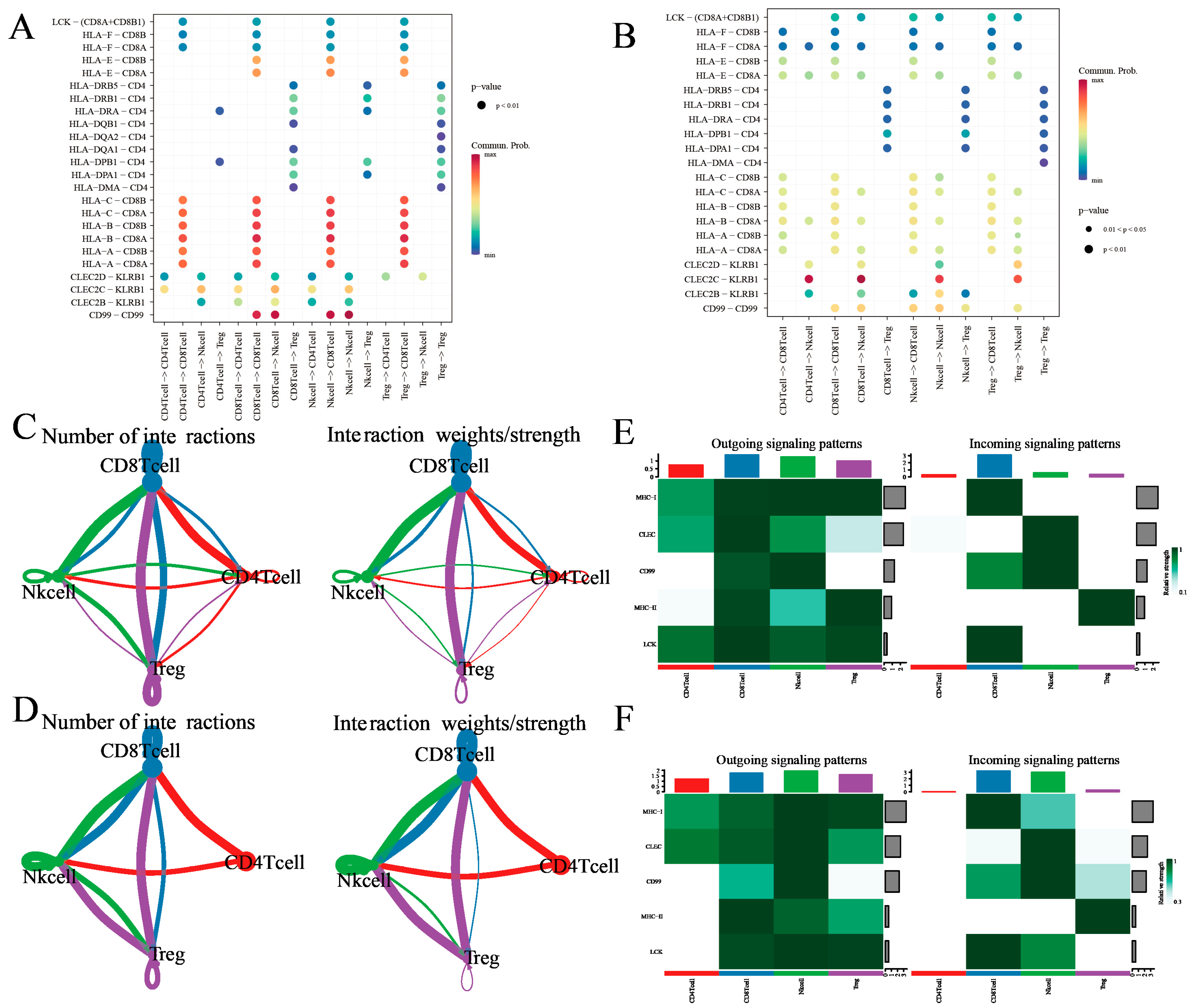
3.4. Treg Cell-Mediated Immune Communication Sheds Light on the Tumor Microenvironment: Insights from Cell Interactions
3.5. Development of a GNN Model Based on T-Cells’ Differential Genes for Risk Stratification and Prognosis Prediction
3.6. Differential Analysis of T-Cell Infiltration in High-Risk and Low-Risk Patient Groups
3.7. Differential Gene Expression and Pathway Analysis in Low-Risk Versus High-Risk Groups: A Comparative Study Involving Gene Ontology and KEGG Analyses
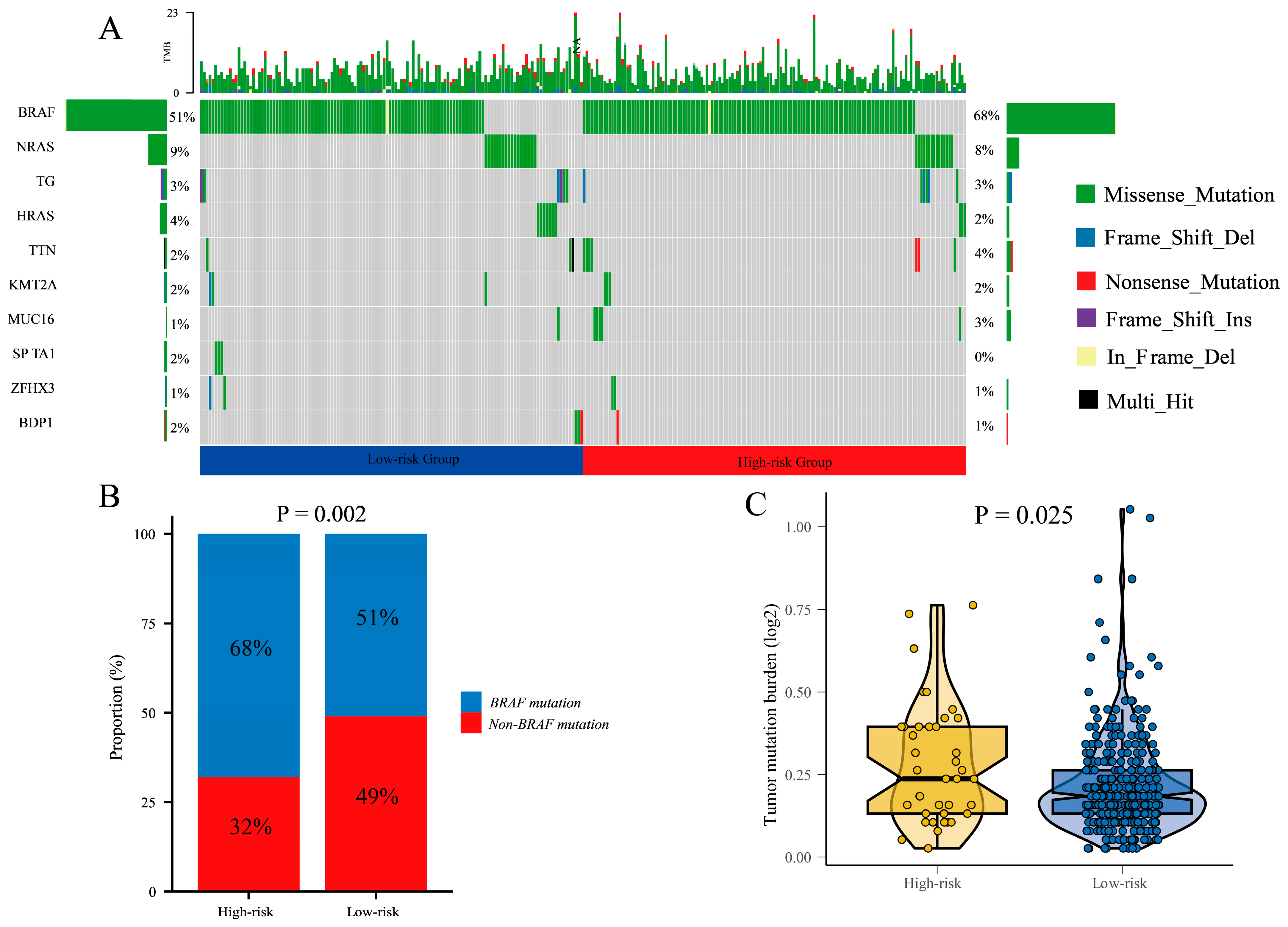
3.8. Differential Mutational Profiles and Tumor Mutational Burden in High-Risk and Low-Risk Groups: BRAF Mutations
3.9. Subgroup Analyses Reveal Improved Disease-Free Survival in Low-Risk Group: Age, Gender, and Stage-Specific Impact in Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Chen, D.W.; Lang, B.H.H.; McLeod, D.S.A.; Newbold, K.; Haymart, M.R. Thyroid cancer. Lancet 2023, 401, 1531–1544. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Pizzato, M.; Li, M.; Vignat, J.; Laversanne, M.; Singh, D.; La Vecchia, C.; Vaccarella, S. The epidemiological landscape of thyroid cancer worldwide: GLOBOCAN estimates for incidence and mortality rates in 2020. Lancet Diabetes Endocrinol. 2022, 10, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Brito, J.P.; Al Nofal, A.; Montori, V.M.; Hay, I.D.; Morris, J.C. The impact of subclinical disease and mechanism of detection on the rise in thyroid cancer incidence: A population-based study in Olmsted County, Minnesota during 1935 through 2012. Thyroid 2015, 25, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Haugen, B.R.; Alexander, E.K.; Bible, K.C.; Doherty, G.M.; Mandel, S.J.; Nikiforov, Y.E.; Pacini, F.; Randolph, G.W.; Sawka, A.M.; Schlumberger, M.; et al. 2015 American Thyroid Association management guidelines for adult patients with thyroid nodules and differentiated thyroid cancer: The American Thyroid Association guidelines task force on thyroid nodules and differentiated thyroid cancer. Thyroid 2016, 26, 1–133. [Google Scholar] [CrossRef]
- Brito, J.P.; Hay, I.D. Overdiagnosis of papillary carcinoma—Who benefits? Nat. Rev. Endocrinol. 2017, 13, 131–132. [Google Scholar] [CrossRef]
- Li, M.; Maso, L.D.; Vaccarella, S. Global trends in thyroid cancer incidence and the impact of overdiagnosis. Lancet Diabetes Endocrinol. 2020, 8, 468–470. [Google Scholar] [CrossRef]
- Wang, T.; Shi, J.; Li, L.; Zhou , X.; Zhang, H.; Zhang, X.; Wang, Y.; Liu, L.; Sheng, L. Single-Cell Transcriptome Analysis Reveals Inter-Tumor Heterogeneity in Bilateral Papillary Thyroid Carcinoma. Front Immunol. 2022, 13, 840811. [Google Scholar] [CrossRef]
- Pu, W.; Shi, X.; Yu, P.; Zhang, M.; Liu, Z.; Tan, L.; Han, P.; Ji, D.; Gan, H.; Wei, W.; et al. Single-cell transcriptomic analysis of the tumor ecosystems underlying initiation and progression of papillary thyroid carcinoma. Nat. Commun. 2021, 12, 6058. [Google Scholar] [CrossRef]
- Eraslan, G.; Avsec, Ž.; Gagneur, J.; Theis, F.J. Deep learning: New computational modelling techniques for genomics. Nat. Rev. Genet. 2019, 20, 389–403. [Google Scholar] [CrossRef]
- Wang, Z.; Han, Q.; Hu, X.; Wang, X.; Sun, R.; Huang, S.; Chen, W. Multi-omics clustering analysis carries out the molecular-specific subtypes of thyroid carcinoma: Implicating for the precise treatment strategies. Genes Immun. 2025, 26, 137–150. [Google Scholar] [CrossRef]
- Travaglini, K.J.; Nabhan, A.N.; Penland, L.; Sinha, R.; Krasnow, M.A. A molecular cell atlas of the human lung from single cell rna sequencing. Nature 2020, 587, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Fa, B.; Luo, C.; Tang, Z.; Yan, Y.; Zhang, Y.; Yu, Z. Pathway-based biomarker identification with crosstalk analysis for robust prognosis prediction in hepatocellular carcinoma. EBioMedicine 2019, 44, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Muzio, G.; O’Bray, L.; Borgwardt, K. Biological network analysis with deep learning. Brief Bioinform. 2021, 22, 1515–1530. [Google Scholar] [CrossRef]
- Wang, H.; Sham, P.; Tong, T.; Pang, H. Pathway-based single-cell RNA-Seq classification, clustering, and construction of gene–gene interactions networks using random forests. IEEE J. Biomed. Health Inform. 2020, 24, 1814–1822. [Google Scholar] [CrossRef]
- Xing, X.; Yang, F.; Li, H.; Zhang, J.; Zhao, Y.; Gao, M.; Huang, J.; Yao, J.; Birol, I. Multi-level attention graph neural network based on co-expression gene modules for disease diagnosis and prognosis. Bioinformatics 2022, 38, 2178–2186. [Google Scholar] [CrossRef]
- Bai, L.; Li, Z.; Tang, C.; Song, C.; Hu, F. Hypergraph-based analysis of weighted gene co-expression hypernetwork. Front Genet. 2025, 16, 1560841. [Google Scholar] [CrossRef]
- Lin, J.; Cai, B.; Lin, Q.; Lin, X.; Wang, B.; Chen, X. TLE4 downregulation identified by WGCNA and machine learning algorithm promotes papillary thyroid carcinoma progression via activating JAK/STAT pathway. J. Cancer 2024, 15, 4759–4776. [Google Scholar] [CrossRef]
- Zhao, H.; Zhang, S.; Shao, S.; Fang, H. Identification of a Prognostic 3-Gene Risk Prediction Model for Thyroid Cancer. Front Endocrinol. 2020, 11, 510. [Google Scholar] [CrossRef]
- Ruan, P.; Wang, S. DiSNEP: A Disease-Specific gene Network Enhancement to improve Prioritizing candidate disease genes. Brief Bioinform. 2021, 22, bbaa241. [Google Scholar] [CrossRef]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Chen, X.; Zhang, S.; Zhu, J.; Tang, B.; Wang, A.; Dong, L.; Zhang, Z.; Yu, C.; Sun, Y.; et al. The Genome Sequence Archive Family: Toward Explosive Data Growth and Diverse Data Types. Genom. Proteom. Bioinform. 2021, 19, 578–583. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Cong, S.; Wang, Y.; Ji, Q.; Liu, W.; Qu, N. Analysis of the key ligand receptor CADM1_CADM1 in the regulation of thyroid cancer based on scRNA-seq and bulk RNA-seq data. Front. Endocrinol. 2022, 13, 969914. [Google Scholar] [CrossRef]
- Gustavsson, E.K.; Zhang, D.; Reynolds, R.H.; Garcia-Ruiz, S.; Ryten, M. ggtranscript: An R package for the visualization and interpretation of transcript isoforms using ggplot2. Bioinformatics 2022, 38, 3844–3846. [Google Scholar] [CrossRef]
- Li, G.M.; Zhang, C.L.; Rui, R.P.; Sun, B.; Guo, W. Bioinformatics analysis of common differential genes of coronary artery disease and ischemic cardiomyopathy. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3553–3569. [Google Scholar]
- Madikizela, L.M.; Tutu, H.; Cukrowska, E.; Chimuka, L. Trends in Innovations and Recent Advances in Membrane Protected Extraction Techniques for Organics in Complex Samples. Crit. Rev. Anal. Chem. 2023, 53, 1197–1208. [Google Scholar] [CrossRef]
- Rocamora-Reverte, L.; Melzer, F.L.; Würzner, R.; Weinberger, B. The Complex Role of Regulatory T Cells in Immunity and Aging. Front. Immunol. 2021, 11, 616949. [Google Scholar] [CrossRef]
- Møller, S.H.; Hsueh, P.C.; Yu, Y.R.; Zhang, L.; Ho, P.C. Metabolic programs tailor T cell immunity in viral infection, cancer, and aging. Cell Metab. 2022, 34, 378–395. [Google Scholar] [CrossRef]
- Luo, Y.; Liang, H. Cancer Cells Employ the Most Prolific RNA Editors: A Closer Look at the Single-Cell Level. Cancer Res. 2023, 83, 351–353. [Google Scholar] [CrossRef]
- Multhoff, G.; Vaupel, P. Lactate-avid regulatory T cells: Metabolic plasticity controls immunosuppression in tumour microenvironment. Signal Transduct. Target. Ther. 2021, 6, 171. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Huang, Z.; Chen, Y.; Tian, H.; Chai, P.; Shen, Y.; Yao, Y.; Xu, S.; Ge, S.; Jia, R. Lactate and lactylation in cancer. Signal Transduct. Target. Ther. 2025, 10, 38. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.L.; Wu, M.; Zhang, Z.J.; Chang, S. The potential role of reprogrammed glucose metabolism: An emerging actionable codependent target in thyroid cancer. J. Transl. Med. 2023, 21, 735. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wang, C.; Zhang, W.; Tao, Y.; Guo, J.; Liu, Y.; Liu, Z.; Liu, D.; Mei, J.; Chen, F. Identification of biomarkers related to sepsis diagnosis based on bioinformatics and machine learning and experimental verification. Front. Immunol. 2023, 14, 1087691. [Google Scholar] [CrossRef]
- Murai, H.; Kodama, T.; Maesaka, K.; Tange, S.; Motooka, D.; Suzuki, Y.; Shigematsu, Y.; Inamura, K.; Mise, Y.; Saiura, A.; et al. Multiomics identifies the link between intratumor steatosis and the exhausted tumor immune microenvironment in hepatocellular carcinoma. Hepatology 2023, 77, 77–91. [Google Scholar] [CrossRef]
- Koll, F.J.; Banek, S.; Kluth, L.; Köllermann, J.; Bankov, K.; Chun, F.K.; Wild, P.J.; Weigert, A.; Reis, H. Tumor-associated macrophages and Tregs influence and represent immune cell infiltration of muscle-invasive bladder cancer and predict prognosis. J. Transl. Med. 2023, 21, 124. [Google Scholar] [CrossRef]
- Lixandru-Petre, I.O.; Dima, A.; Musat, M.; Dascalu, M.; Gradisteanu Pircalabioru, G.; Iliescu, F.S.; Iliescu, C. Machine Learning for Thyroid Cancer Detection, Presence of Metastasis, and Recurrence Predictions—A Scoping Review. Cancers 2025, 17, 1308. [Google Scholar] [CrossRef]
- Gong, J.; Jin, B.; Shang, L.; Liu, N. Characterization of the Immune Cell Infiltration Landscape of Thyroid Cancer for Improved Immunotherapy. Front. Mol. Biosci. 2021, 8, 714053. [Google Scholar] [CrossRef]
- Son, J.W.; Hong, J.Y.; Kim, Y.; Kim, W.J.; Shin, D.-Y.; Choi, H.-S.; Bak, S.H.; Moon, K.M. How Many Private Data Are Needed for Deep Learning in Lung Nodule Detection on CT Scans? A Retrospective Multicenter Study. Cancers 2022, 14, 3174. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Corhort | TCGA-THCA | GSE184362 |
|---|---|---|
| Number of patients | n = 489 | n = 11 |
| Age (Mean ± SD) | 46.53 ± 15.35 | - |
| Follow up time (Mean ± SD) (years) | 3.11 ± 2.65 | - |
| Follow up status | ||
| Alive | 442 (90.4%) | - |
| Dead | 47 (9.6%) | - |
| Gender | ||
| Male | 130 (26.6%) | - |
| Female | 359 (73.4%) | - |
| Clinical stage | ||
| Stage I | 281 (57.5%) | - |
| Stage II | 50 (10.2%) | - |
| Stage III | 105 (21.5%) | - |
| Stage IV | 51 (10.4%) | - |
| Unknown | 2 (0.4%) | - |
| T stage | ||
| T0 | - | 4 (36.4%) |
| T1 | 140 (28.6%) | 2 (18.2%) |
| T2 | 163 (33.3%) | - |
| T3 | 166 (34.0%) | - |
| T4 | 18 (3.7%) | 5 (45.4%) |
| Unknown | 2 (0.4%) | - |
| M stage | ||
| M0 | 273 (55.9%) | 7 (63.6%) |
| M1 | 8 (1.6%) | 4 (36.4%) |
| Unknown | 208 (42.5%) | - |
| N stage | ||
| N0 | 225 (46.0%) | 2 (18.2%) |
| N1 | 217 (44.4%) | 9 (81.8%) |
| Unknown | 47 (9.6%) | - |
| Histologic subtype | ||
| Classical | - | 8 (72.7%) |
| Follicular variant | - | 2 (18.2%) |
| Tall cell variant | - | 1 (9.1%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, L.; Huang, Z.; Chen, Y.; Wang, Z.; Lai, Z.; Peng, X.; Zhang, C.; Lin, R.; Ouyang, W.; Yu, Y.; et al. Integration of Single-Cell Analysis and Bulk RNA Sequencing Data Using Multi-Level Attention Graph Neural Network for Precise Prognostic Stratification in Thyroid Cancer. Cancers 2025, 17, 2411. https://doi.org/10.3390/cancers17142411
Tan L, Huang Z, Chen Y, Wang Z, Lai Z, Peng X, Zhang C, Lin R, Ouyang W, Yu Y, et al. Integration of Single-Cell Analysis and Bulk RNA Sequencing Data Using Multi-Level Attention Graph Neural Network for Precise Prognostic Stratification in Thyroid Cancer. Cancers. 2025; 17(14):2411. https://doi.org/10.3390/cancers17142411
Chicago/Turabian StyleTan, Langping, Zhenjun Huang, Yongjian Chen, Zehua Wang, Zijia Lai, Xinzhi Peng, Cheng Zhang, Ruichong Lin, Wenhao Ouyang, Yunfang Yu, and et al. 2025. "Integration of Single-Cell Analysis and Bulk RNA Sequencing Data Using Multi-Level Attention Graph Neural Network for Precise Prognostic Stratification in Thyroid Cancer" Cancers 17, no. 14: 2411. https://doi.org/10.3390/cancers17142411
APA StyleTan, L., Huang, Z., Chen, Y., Wang, Z., Lai, Z., Peng, X., Zhang, C., Lin, R., Ouyang, W., Yu, Y., & Long, M. (2025). Integration of Single-Cell Analysis and Bulk RNA Sequencing Data Using Multi-Level Attention Graph Neural Network for Precise Prognostic Stratification in Thyroid Cancer. Cancers, 17(14), 2411. https://doi.org/10.3390/cancers17142411