
Engineering Innate Immunity: Recent Advances and Future Directions for CAR-NK and CAR–Macrophage Therapies in Solid Tumors
 , , , ,
, , , ,
Simple Summary
Abstract
1. Introduction
2. CAR-NK Cells for Solid Tumors
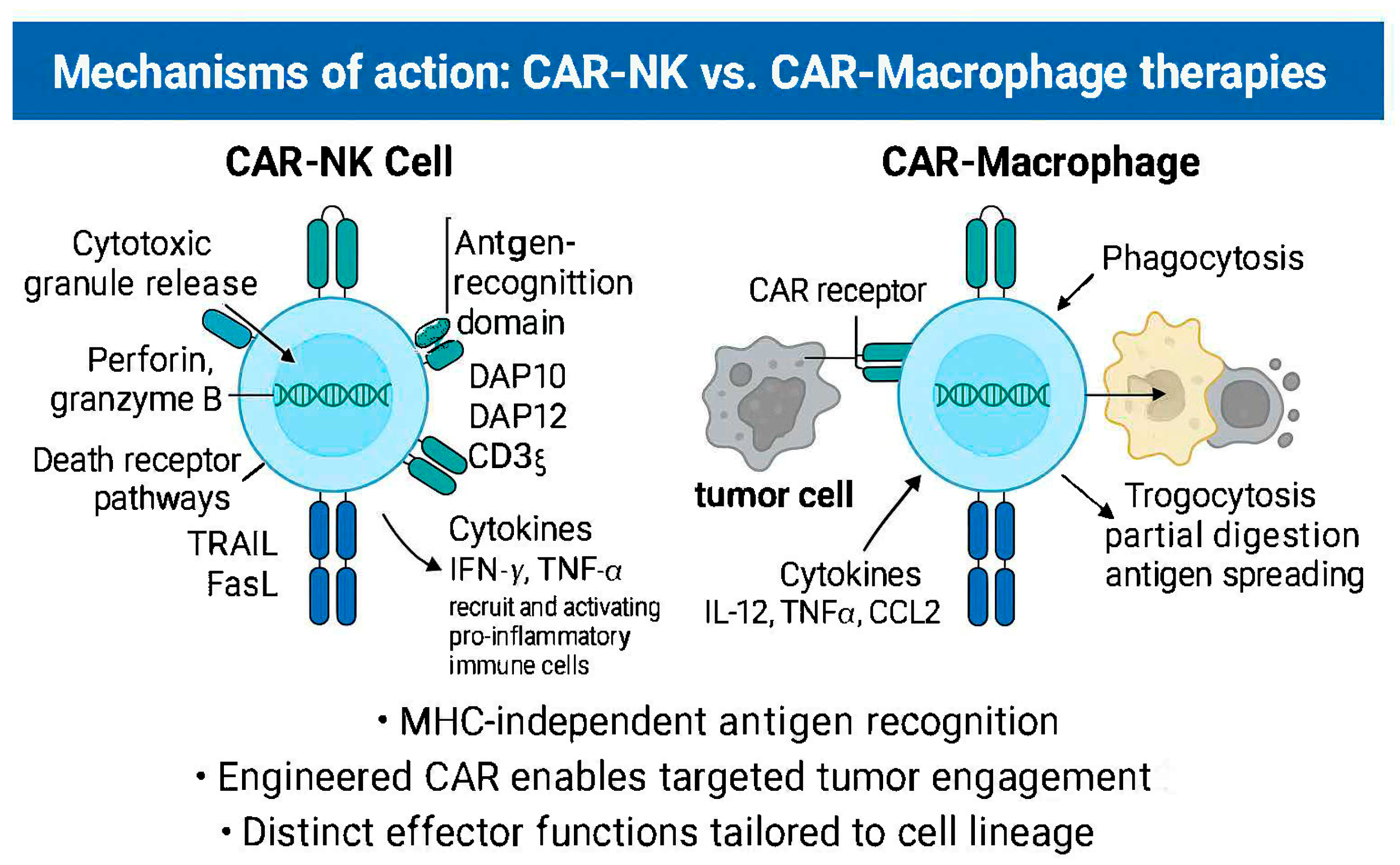
2.1. Mechanisms of Action
2.2. Recent Advances: 2023–2025
2.3. Preclinical Data
2.4. Limitations
2.5. Future Directions
3. CAR–Macrophages (CAR-MΦ) for Solid Tumors
3.1. Mechanisms of Action
3.2. CAR-MΦ Design Features
3.3. Recent Advances: 2023–2025
3.4. Limitations
3.5. Future Directions
4. Dueling Perspectives on CAR-MΦ and CAR-NK Cells
4.1. Comparative Perspective: CAR-MΦ Versus CAR-NK
4.2. Future Opportunities and Combinatorial Strategies
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Beavis, P.A.; Slaney, C.Y.; Kershaw, M.H.; Gyorki, D.E.; Neeson, P.J.; Darcy, P.K. Reprogramming the tumor microenvironment to enhance adoptive cellular therapy. Semin. Immunol. 2016, 28, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumor response to PD-L1 blockade by contributing to the exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Majzner, R.G.; Mackall, C.L. Tumor antigen escape from CAR T-cell therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Newick, K.; O’Brien, S.; Moon, E.; Albelda, S.M. CAR T cell therapy for solid tumors. Annu. Rev. Med. 2017, 68, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—Assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Blache, U.; Popp, G.; Dünkel, A.; Koehl, U.; Fricke, S. Potential solutions for the manufacture of CAR T cells in cancer immunotherapy. Nat. Commun. 2022, 13, 5225. [Google Scholar] [CrossRef]
- Daher, M.; Rezvani, K. Next generation natural killer cells for cancer immunotherapy: The promise of genetic engineering. Curr. Opin. Immunol. 2018, 51, 146–153. [Google Scholar] [CrossRef]
- Tsiverioti, C.A.; Gottschlich, A.; Trefny, M.; Theurich, S.; Anders, H.-J.; Kroiss, M.; Kobold, S. Beyond CAR T cells: Exploring alternative cell sources for CAR-like cellular therapies. Biol. Chem. 2024, 405, 485–515. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, M.A.; Williamson, A.P.; Steinbach, A.M.; Roberts, E.W.; Kern, N.; Headley, M.B.; Vale, R.D. Chimeric antigen receptors that trigger phagocytosis. eLife 2018, 7, e36688. [Google Scholar] [CrossRef]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef]
- Chiossone, L.; Dumas, P.-Y.; Vienne, M.; Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 2018, 18, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Prager, I.; Watzl, C. Mechanisms of natural killer cell-mediated cellular cytotoxicity. J. Leukoc. Biol. 2019, 105, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Romee, R.; Leong, J.W.; Fehniger, T.A. Utilizing cytokines to function-enable human NK cells for the immunotherapy of cancer. Scientifica 2014, 2014, 205796. [Google Scholar] [CrossRef]
- Böttcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Sousa, C.R.E. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell 2018, 172, 1022–1037.e14. [Google Scholar] [CrossRef]
- Marcus, A.; Gowen, B.G.; Thompson, T.W.; Iannello, A.; Ardolino, M.; Deng, W.; Wang, L.; Shifrin, N.; Raulet, D.H. Recognition of tumors by the innate immune system and natural killer cells. Adv. Immunol. 2014, 122, 91–128. [Google Scholar]
- Chen, Y.; Wang, M.; Huang, S.; Han, L.; Cai, Y.; Xu, X.; Sun, S.; Chen, Z.; Chen, J.; Yu, J.; et al. Ectopic expression of NKG7 enhances CAR-T function and improves the therapeutic efficacy in liquid and solid tumors. Pharmacol. Res. 2024, 210, 107506. [Google Scholar] [CrossRef]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell 2018, 23, 181–192.e5. [Google Scholar] [CrossRef] [PubMed]
- Altvater, B.; Landmeier, S.; Pscherer, S.; Temme, J.; Schweer, K.; Kailayangiri, S.; Campana, D.; Juergens, H.; Pule, M.; Rossig, C. 2B4 (CD244) signaling by recombinant antigen-specific chimeric receptors costimulates natural killer cell activation to leukemia and neuroblastoma cells. Clin. Cancer Res. 2009, 15, 4857–4866. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zeng, J.; Liu, T.; Xu, Q. 2B4 costimulatory domain enhancing cytotoxic ability of anti-CD5 CAR-NK cells against T cell malignancies. J. Hematol. Oncol. 2020, 13, 49. [Google Scholar]
- Zhu, H.; Kaufman, D.S. An iPSC perspective on NK cells as cancer immunotherapy. Trends Mol. Med. 2019, 25, 956–968. [Google Scholar]
- Cichocki, F.; van der Stegen, S.J.C.; Miller, J.S. Engineered and banked iPSCs for advanced NK- and T-cell immunotherapies. Blood 2023, 141, 846–855. [Google Scholar] [CrossRef] [PubMed]
- Strecker, M.I.; Wlotzka, K.; Strassheimer, F.; Roller, B.; Ludmirski, G.; König, S.; Röder, J.; Opitz, C.; Alekseeva, T.; Reul, J.; et al. AAV-mediated gene transfer of a checkpoint inhibitor in combination with HER2-targeted CAR-NK cells as an experimental therapy for glioblastoma. Oncoimmunology 2022, 11, 2127508. [Google Scholar] [CrossRef]
- Robbins, Y.; Greene, S.; Friedman, J.; Clavijo, P.E.; Van Waes, C.; Fabian, K.P.; Padget, M.R.; Sater, H.A.; Lee, J.H.; Soon-Shiong, P.; et al. Tumor control via targeting PD-L1 with chimeric antigen receptor-modified NK cells. eLife 2020, 9, e54854. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Kerbauy, L.N.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Du, Z.; Ng, Y.Y.; Zha, S.; Wang, S. piggyBac system to co-express NKG2D CAR and IL-15 to augment the in vivo persistence and anti-AML activity of human peripheral blood NK cells. Mol. Ther. Methods Clin. Dev. 2021, 23, 582–596. [Google Scholar] [CrossRef]
- Tian, J.; Tong, D.; Li, Z.; Wang, E.; Yu, Y.; Lv, H.; Hu, Z.; Sun, F.; Wang, G.; He, M.; et al. Mage transposon: A novel gene delivery system for mammalian cells. Nucleic Acids Res. 2024, 52, 2724–2739. [Google Scholar] [CrossRef]
- Hsu, L.J.; Liu, C.L.; Kuo, M.L.; Shen, C.N.; Shen, C.R. An alternative cell therapy for cancers: Induced pluripotent stem cell (iPSC)-Derived natural killer cells. Biomedicines 2021, 9, 1323. [Google Scholar] [CrossRef]
- Goldenson, B.H.; Hor, P.; Kaufman, D.S. iPSC-derived natural killer cell therapies—Expansion and targeting. Front. Immunol. 2022, 13, 841107. [Google Scholar] [CrossRef] [PubMed]
- Pomeroy, E.J.; Hunzeker, J.T.; Kluesner, M.G.; Lahr, W.S.; Smeester, B.A.; Crosby, M.R.; Lonetree, C.-L.; Yamamoto, K.; Bendzick, L.; Miller, J.S.; et al. A genetically engineered primary human natural killer cell platform for cancer immunotherapy. Mol. Ther. 2020, 28, 52–63. [Google Scholar] [CrossRef]
- Kremer, V.; Ligtenberg, M.A.; Zendehdel, R.; Seitz, C.; Duivenvoorden, A.; Wennerberg, E.; Colón, E.; Scherman-Plogell, A.-H.; Lundqvist, A. Genetic engineering of human NK cells to express CXCR2 improves migration to renal cell carcinoma. J. Immunother. Cancer 2017, 5, 73. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Chu, J.; Chan, W.K.; Zhang, J.; Wang, Y.; Cohen, J.B.; Victor, A.; Meisen, W.H.; Kim, S.-H.; Grandi, P.; et al. CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Sci. Rep. 2015, 5, 11483. [Google Scholar] [CrossRef]
- Arcangeli, S.; Rotiroti, M.C.; Bardelli, M.; Simonelli, L.; Magnani, C.F.; Biondi, A.; Biagi, E.; Tettamanti, S.; Varani, L. Balance of anti-CD123 chimeric antigen receptor binding affinity and density for the targeting of acute myeloid leukemia. Mol. Ther. 2021, 29, 1229–1241. [Google Scholar]
- Chanmee, T.; Ontong, P.; Konno, K.; Itano, N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers 2014, 6, 1670–1690. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Hadiloo, K.; Taremi, S.; Heidari, M.; Esmaeilzadeh, A. The CAR macrophage cells, a novel generation of chimeric antigen-based approach against solid tumors. Biomark. Res. 2023, 11, 103. [Google Scholar] [CrossRef]
- Li, J.; Chen, P.; Ma, W. The next frontier in immunotherapy: Potential and challenges of CAR-macrophages. Exp. Hematol. Oncol. 2024, 13, 76. [Google Scholar] [CrossRef] [PubMed]
- Paasch, D.; Meyer, J.; Stamopoulou, A.; Lenz, D.; Kuehle, J.; Kloos, D.; Buchegger, T.; Holzinger, A.; Falk, C.S.; Kloth, C.; et al. Ex vivo generation of CAR macrophages from hematopoietic stem and progenitor cells for use in cancer therapy. Cells 2022, 11, 994. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Han, G.; Gu, J.; Chen, Z.; Wu, J. Role of tumor-associated macrophages in hepatocellular carcinoma: Impact, mechanism, and therapy. Front. Immunol. 2024, 15, 1429812. [Google Scholar] [CrossRef]
- Reiss, K.A.; Yuan, Y.; Barton, D.; Cushing, D.; Ronczka, A.; Klichinsky, M.; Dees, E.C. A phase 1, first-in-human (FIH) study of adenovirally transduced autologous macrophages engineered to contain an anti-HER2 chimeric antigen receptor (CAR) in subjects with HER2 overexpressing solid tumors. J. Clin. Oncol. 2022, 40 (Suppl. S4), TPS668. Available online: https://ascopubs.org/doi/10.1200/JCO.2022.40.4_suppl.TPS668 (accessed on 1 May 2025). [CrossRef]
- Shah, Z.; Tian, L.; Li, Z.; Jin, L.; Zhang, J.; Li, Z.; Barr, T.; Tang, H.; Feng, M.; Caligiuri, M.A.; et al. Human anti-PSCA CAR macrophages possess potent antitumor activity against pancreatic cancer. Cell Stem Cell 2024, 31, 803–817. [Google Scholar] [CrossRef]
- Lu, J.; Ma, Y.; Li, Q.; Xu, Y.; Xue, Y.; Xu, S. CAR Macrophages: A promising novel immunotherapy for solid tumors and beyond. Biomark. Res. 2024, 12, 86. [Google Scholar] [CrossRef]
- Chen, S.; Saeed, A.F.; Liu, Q.; Jiang, Q.; Xu, H.; Xiao, G.G.; Rao, L.; Duo, Y. Macrophages in immunoregulation and therapeutics. Signal Transduct. Target. Ther. 2023, 8, 207. [Google Scholar] [CrossRef]
- Gargett, T.; Brown, M.P. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front. Pharmacol. 2014, 5, 235. [Google Scholar] [CrossRef]
- Shi, Z.D.; Tchao, J.; Wu, L.; Carman, A.J. Precision installation of a highly efficient suicide gene safety switch in human induced pluripotent stem cells. Stem Cells Transl. Med. 2020, 9, 1378–1388. [Google Scholar] [CrossRef] [PubMed]
- Kosti, P.; Opzoomer, J.W.; Larios-Martinez, K.I.; Henley-Smith, R.; Scudamore, C.L.; Okesola, M.; Taher, M.Y.M.; Davies, D.M.; Muliaditan, T.; Larcombe-Young, D.; et al. Hypoxia-sensing CAR-T cells provide safety and efficacy in treating solid tumors. Cell Rep. Med. 2021, 2, 100227. [Google Scholar] [CrossRef]
- He, H.; Liao, Q.; Zhao, C.; Zhu, C.; Feng, M.; Liu, Z.; Jiang, L.; Zhang, L.; Ding, X.; Yuan, M.; et al. Conditioned CAR-T cells by hypoxia-inducible transcription amplification system significantly enhance systemic safety and retain antitumor efficacy. J. Immunother. Cancer 2021, 9, e002755. [Google Scholar] [CrossRef]
- Chen, S.; Li, L.; Yuan, H.; Gui, H.; Wan, Q.; Wang, M.; Lv, H.; Wang, C.; Zhu, L.; Nie, Y.; et al. Intratumoral injection of R848 and poly(I:C) synergistically promoted antitumor immune responses by reprogramming macrophage polarization and activating DCs in lung cancer. Clin. Exp. Immunol. 2025, 219, uxae110. [Google Scholar] [CrossRef] [PubMed]
- Huo, Y.; Zhang, H.; Sa, L.; Zheng, W.; He, Y.; Lyu, H.; Sun, M.; Zhang, L.; Shan, L.; Yang, A.; et al. M1 polarization enhances the antitumor activity of chimeric antigen receptor macrophages in solid tumors. J. Transl. Med. 2023, 21, 225. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, X.; Zhang, X.; Aziz, A.U.; Wang, D. CAR-NK cell therapy: A transformative approach to overcoming oncological challenges. Biomolecules 2024, 14, 1035. [Google Scholar] [CrossRef]
- Page, A.; Chuvin, N.; Valladeau-Guilemond, J.; Depil, S. Development of NK cell-based cancer immunotherapies through receptor engineering. Cell Mol. Immunol. 2024, 21, 315–331. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Liu, J. Emerging roles of CAR-NK cell therapies in tumor immunotherapy: Current status and future directions. Cell Death Discov. 2024, 10, 318. [Google Scholar] [CrossRef]
- Khawar, M.B.; Sun, H. CAR-NK cells: From natural basis to design for kill. Front. Immunol. 2021, 12, 707542. [Google Scholar] [CrossRef]
- Cifaldi, L.; Melaiu, O.; Giovannoni, R.; Benvenuto, M.; Focaccetti, C.; Nardozi, D.; Barillari, G.; Bei, R. DNAM-1 chimeric receptor-engineered NK cells: A new frontier for CAR-NK cell-based immunotherapy. Front. Immunol. 2023, 14, 1197053. [Google Scholar] [CrossRef]
- Look, T.; Sankowski, R.; Bouzereau, M.; Fazio, S.; Sun, M.; Buck, A.; Binder, N.; Mastall, M.; Prisco, F.; Seehusen, F.; et al. CAR T cells, CAR NK cells, and CAR macrophages exhibit distinct traits in glioma models but are similarly enhanced when combined with cytokines. Cell Rep. Med. 2025, 6, 101234. [Google Scholar] [CrossRef]
- Strassheimer, F.; Elleringmann, P.; Ludmirski, G.; Roller, B.; Macas, J.; Alekseeva, T.; Cakmak, P.; Aliraj, B.; Krenzlin, H.; Demes, M.C.; et al. CAR-NK cell therapy combined with checkpoint inhibition induces an NKT cell response in glioblastoma. Br. J. Cancer 2025, 132, 849–860. [Google Scholar] [CrossRef]
- Peng, L.; Sferruzza, G.; Yang, L.; Zhou, L.; Chen, S. CAR-T and CAR-NK as cellular cancer immunotherapy for solid tumors. Cell Mol. Immunol. 2024, 21, 1089–1108. [Google Scholar] [CrossRef]
- Zadorozhna, M.; Mangieri, D. Mechanisms of Chemopreventive and Therapeutic Proprieties of Ginger Extracts in Cancer. Int. J. Mol. Sci. 2021, 22, 6599. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Isola, S.; Gammeri, L.; Furci, F.; Gangemi, S.; Pioggia, G.; Allegra, A. Vitamin C Supplementation in the Treatment of Autoimmune and Onco-Hematological Diseases: From Prophylaxis to Adjuvant Therapy. Int. J. Mol. Sci. 2024, 25, 7284. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kwon, W.A.; Lee, M.K. Evolving Treatment Landscape of Frontline Therapy for Metastatic Urothelial Carcinoma: Current Insights and Future Perspectives. Cancers 2024, 16, 4078. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]

{kind=link}
| Feature | CAR-NK Cells | CAR–Macrophages (CAR-MΦ) |
|---|---|---|
| Lineage | Innate lymphoid lineage | Myeloid lineage |
| Mechanism of Cytotoxicity | Granule exocytosis (perforin, granzyme B), death receptor signaling (TRAIL, FasL), cytokine release | Phagocytosis, trogocytosis, antibody-dependent cellular phagocytosis (ADCP) |
| Tumor Infiltration | Limited natural infiltration; enhanced via chemokine receptor engineering (e.g., CXCR2, CCR5) [34,54] | Naturally efficient stromal penetration via chemotaxis and TME responsiveness [45] |
| CAR Signaling Domains | CD3ζ, DAP10, DAP12, 2B4, DNAM-1 [22,55,56,57] | FcRγ, DAP12, Megf10; Syk-/PI3K-dependent activation [45] |
| Secretory Profile | IFN-γ, TNF-α, GM-CSF; promotes recruitment and priming of adaptive immunity [53,55] | IL-12, TNF-α, CCL2, CXCL9; induces M1 polarization and immune recruitment [45,52] |
| Immune Recruitment Capacity | Indirect via cytokines and cross-talk with DCs and T cells [17,18,19] | Direct recruitment of CD8+ T cells, NK cells, DCs via chemokines [39,40] |
| Antigen Presentation | Limited | Active antigen presentation with potential for T cell priming and antigen spreading [41] |
| Clinical Stage | Phase I/II trials for hematologic malignancies and emerging solid tumor targets (e.g., mesothelin, HER2) [28] | Phase I (e.g., CT-0508 for HER2+ tumors); preclinical models for mesothelin, PSCA [43,44] |
| In Vivo Persistence | Transient (days to weeks), often supported by IL-15- or iPSC-derived platforms [25,28] | Very limited; no expansion, terminal differentiation; being enhanced with IL-15- and iPSC-based strategies [45,52] |
| Manufacturing Source | Peripheral blood, cord blood, iPSC-derived platforms under development [24,25] | Autologous monocytes; iPSC-derived macrophages emerging for off-the-shelf production [45] |
| Key Limitations | Short persistence, low transduction efficiency, inter-donor variability | Low durability, complex GMP manufacturing, cytokine-related toxicity risk |
| Trial Name/ID | Cell Type | Target Antigen | Tumor Type | Study Phase | Key Findings |
|---|---|---|---|---|---|
| CT-0508 (NCT04660929) | CAR–Macrophage | HER2 | HER2+ solid tumors | Phase I (clinical) | 44% SD in HER2 3+ tumors; TME remodeling with CD8+ T cell expansion |
| PD-L1 CAR-NK Trial (preclinical) | CAR-NK | PD-L1 | Head and neck squamous cell carcinoma | Preclinical | PD-L1 targeting enhanced tumor cytotoxicity and control in xenografts |
| iPSC-derived CAR-NK (preclinical) | CAR-NK | CD19 (model antigen) | Various solid tumors | Preclinical | Stable phenotype and cytotoxicity; potential for off-the-shelf use |
| Anti-PSCA CAR-MΦ (preclinical) | CAR–Macrophage | PSCA | Pancreatic cancer | Preclinical | Robust antitumor activity; required repeated dosing |
| HER2/Mesothelin CAR-MΦ (preclinical) | CAR–Macrophage | HER2/Mesothelin | Ovarian, breast, pancreatic | Preclinical | M1 polarization; enhanced phagocytosis and immune recruitment |
| Glioma CAR-NK/MΦ combo (preclinical) | CAR-NK and CAR–Macrophage | EGFRvIII, HER2 | Glioblastoma | Preclinical | Distinct activity profiles; combination enhanced by cytokines |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amoozgar, B.; Bangolo, A.; Mansour, C.; Elias, D.; Mohamed, A.; Thor, D.C.; Ehsanullah, S.U.; Tran, H.H.-V.; Aguilar, I.K.; Weissman, S. Engineering Innate Immunity: Recent Advances and Future Directions for CAR-NK and CAR–Macrophage Therapies in Solid Tumors. Cancers 2025, 17, 2397. https://doi.org/10.3390/cancers17142397
Amoozgar B, Bangolo A, Mansour C, Elias D, Mohamed A, Thor DC, Ehsanullah SU, Tran HH-V, Aguilar IK, Weissman S. Engineering Innate Immunity: Recent Advances and Future Directions for CAR-NK and CAR–Macrophage Therapies in Solid Tumors. Cancers. 2025; 17(14):2397. https://doi.org/10.3390/cancers17142397
Chicago/Turabian StyleAmoozgar, Behzad, Ayrton Bangolo, Charlene Mansour, Daniel Elias, Abdifitah Mohamed, Danielle C. Thor, Syed Usman Ehsanullah, Hadrian Hoang-Vu Tran, Izage Kianifar Aguilar, and Simcha Weissman. 2025. "Engineering Innate Immunity: Recent Advances and Future Directions for CAR-NK and CAR–Macrophage Therapies in Solid Tumors" Cancers 17, no. 14: 2397. https://doi.org/10.3390/cancers17142397
APA StyleAmoozgar, B., Bangolo, A., Mansour, C., Elias, D., Mohamed, A., Thor, D. C., Ehsanullah, S. U., Tran, H. H.-V., Aguilar, I. K., & Weissman, S. (2025). Engineering Innate Immunity: Recent Advances and Future Directions for CAR-NK and CAR–Macrophage Therapies in Solid Tumors. Cancers, 17(14), 2397. https://doi.org/10.3390/cancers17142397