
Capacity for Compensatory Cyclin D2 Response Confers Trametinib Resistance in Canine Mucosal Melanoma


 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines, Cultures, and Cell Fate Assays
2.2. Western Blotting
2.3. Cell Cycle Flow Cytometry
2.4. siRNA Knockdown
2.5. RNA Extraction and Quantitative RT-PCR Analyses
2.6. EdU Incorporation
2.7. Colony Formation Assay
2.8. Reagents
2.9. Statistical Analysis
3. Results
3.1. Differential Intrinsic Resistance to Trametinib in a Series of MM Cell Lines
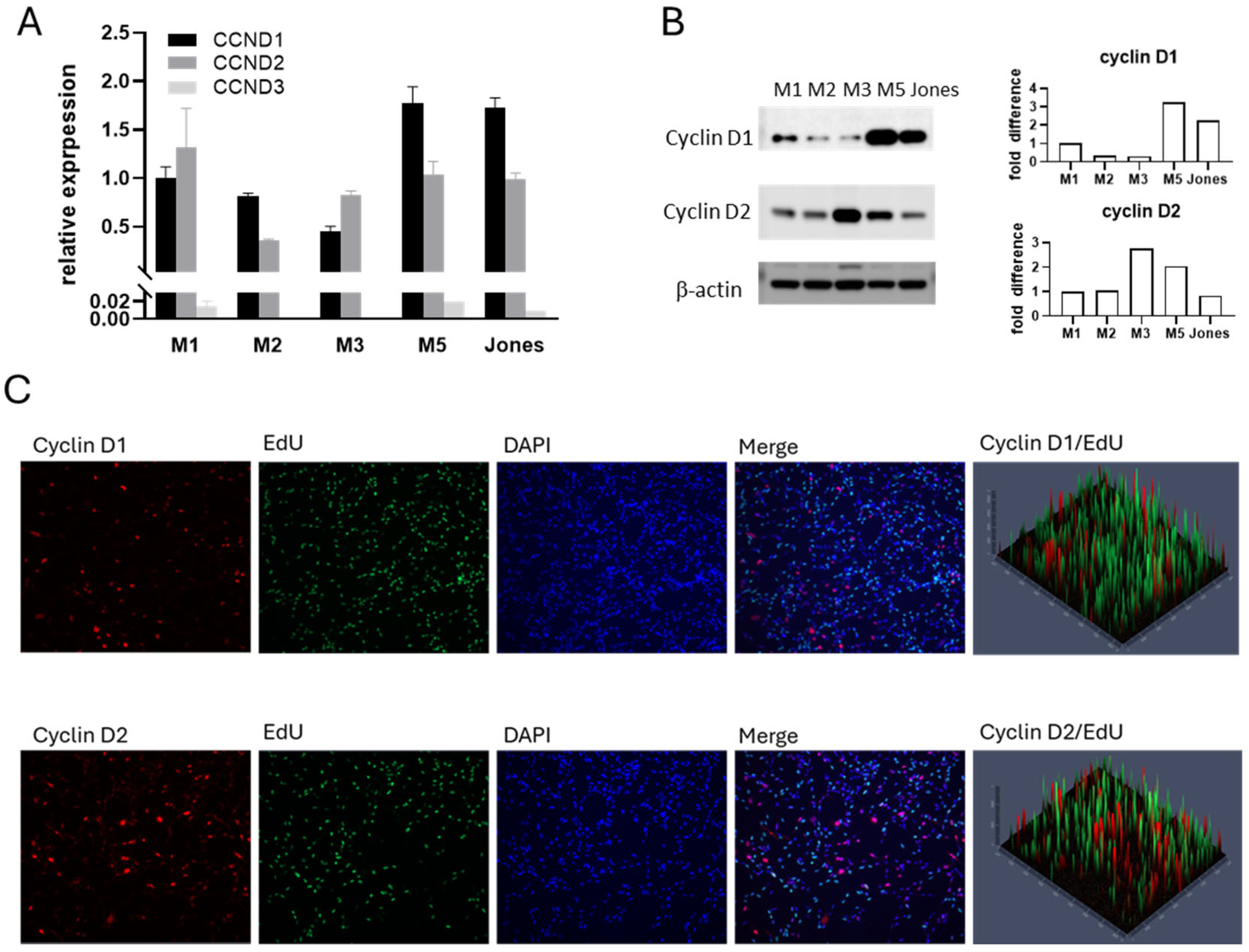
3.2. Cyclin D Expression Profiles in Canine MM Cells
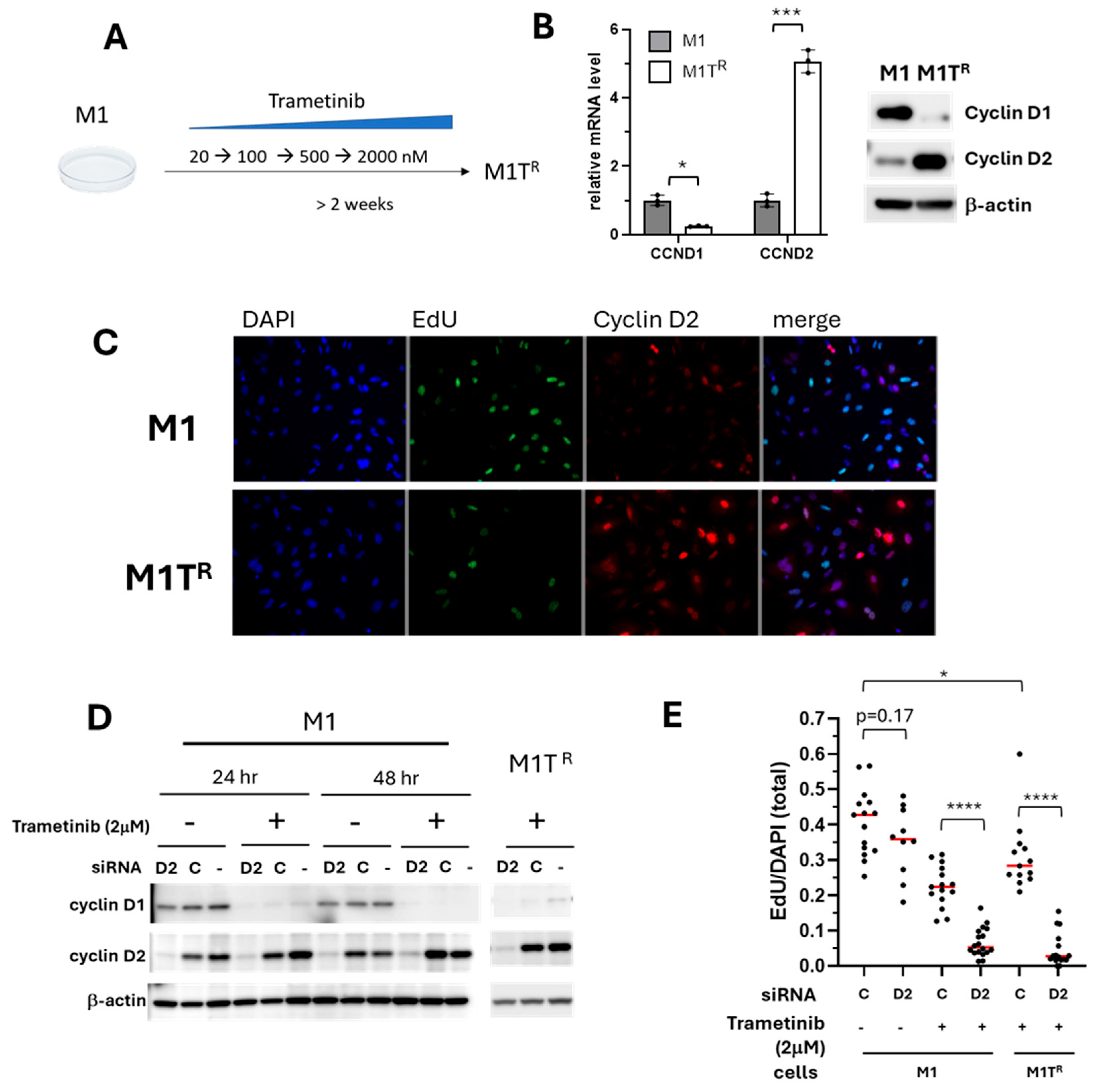
3.3. Trametinib-Resistance Projected by Compensatory Cyclin D2 Response
3.4. Inhibition of Compensatory Cyclin D2 Expression Conferred Sensitivity in Trametinib-Resistant MM
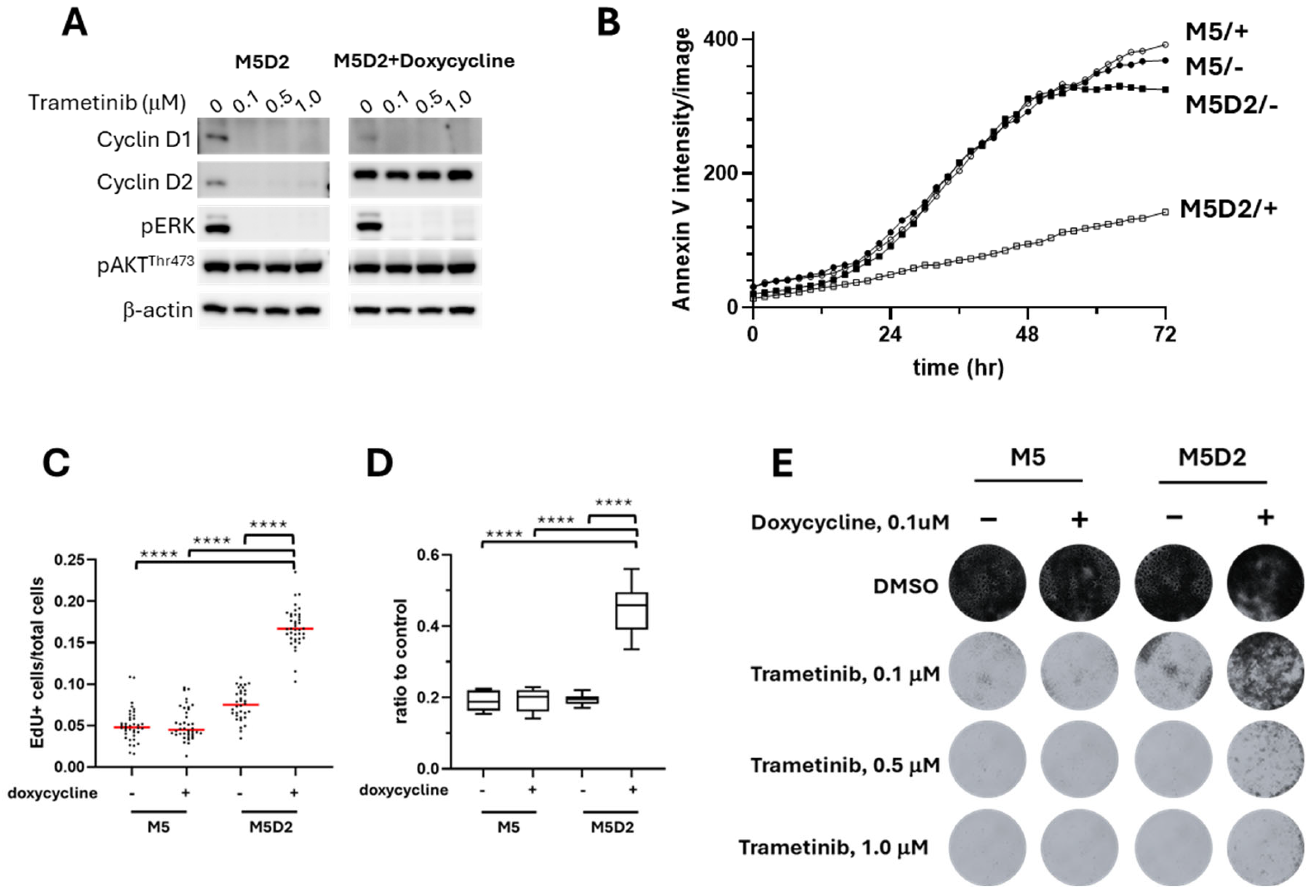
3.5. Induced Overexpression of Cyclin D2 in MM Confers Greater Trametinib Resistance
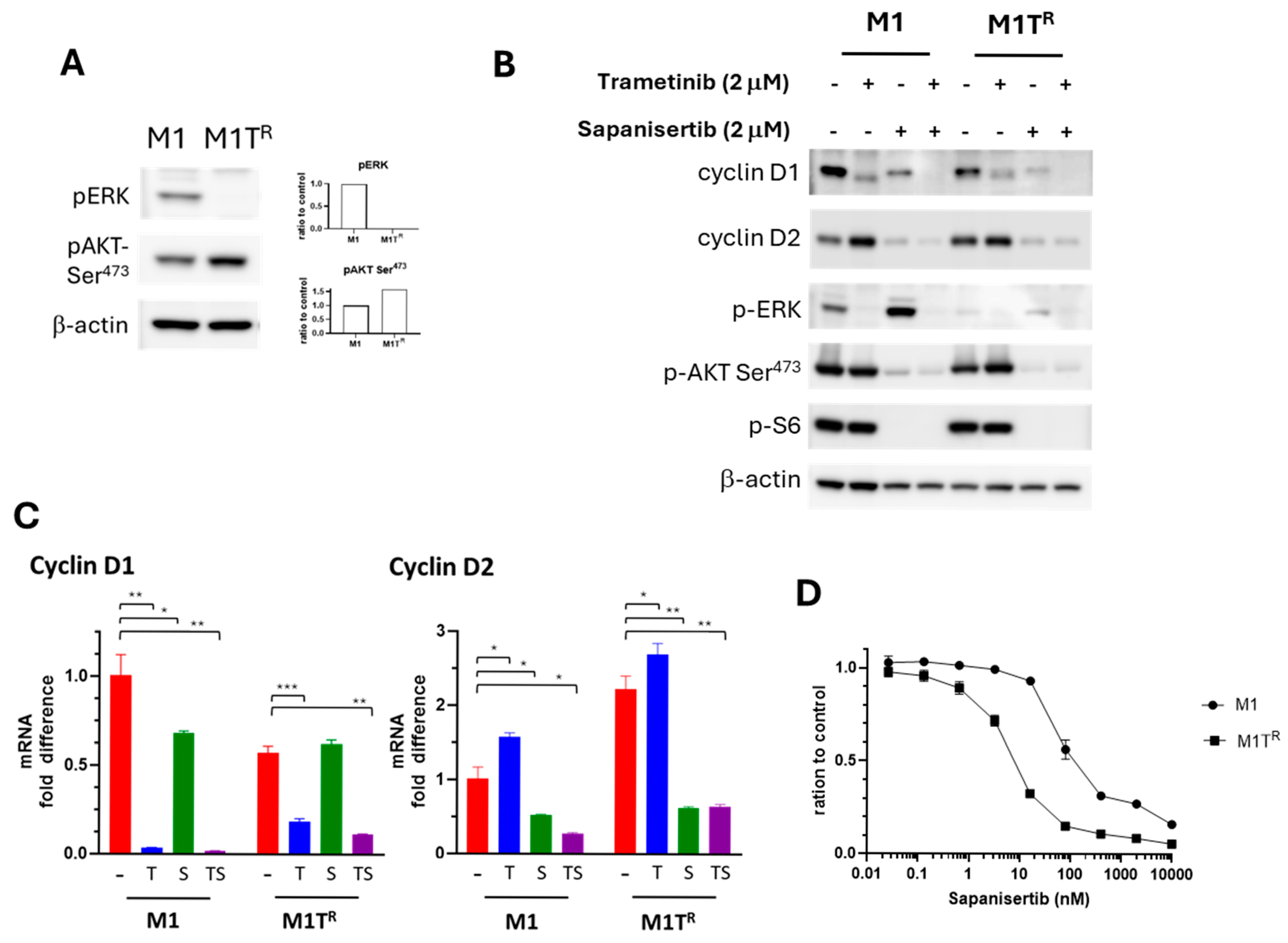
3.6. PI3K/AKT/mTOR Signal Transduction Pathway Inhibition SuppressedTrametinib-Stimulated Compensatory Cyclin D2 Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AV | Annexin V |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DMSO | Dimethyl Sulfoxide |
| EdU | 5-ethynyl-2′-deoxyuridine |
| GE | General Electric |
| HRP | Horseradish Peroxidase |
| KD | Knockdown |
| MM | Mucosal Melanoma |
| M5D2 | M5 Cells Overexpressing Cyclin D2 |
| M1TR | M1 Cells Conditionally Adapted to Trametinib |
| M2TR | M2 Cells Conditionally Adapted to Trametinib |
| MTS | 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium |
| PBS | Phosphate-Buffered Saline |
| PVDF | Polyvinylidene Difluoride |
References
- Vergara, I.A.; Wilmott, J.S.; Long, G.V.; Scolyer, R.A. Genetic drivers of non-cutaneous melanomas: Challenges and opportunities in a heterogeneous landscape. Exp. Dermatol. 2022, 31, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Tyrrell, H.; Payne, M. Combatting mucosal melanoma: Recent advances and future perspectives. Melanoma Manag. 2018, 5, Mmt11. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, R.D.; Antonescu, C.R.; Wolchok, J.D.; Chapman, P.B.; Roman, R.A.; Teitcher, J.; Panageas, K.S.; Busam, K.J.; Chmielowski, B.; Lutzky, J.; et al. KIT as a therapeutic target in metastatic melanoma. JAMA 2011, 305, 2327–2334. [Google Scholar] [CrossRef] [PubMed]
- Simpson, R.M.; Bastian, B.C.; Michael, H.T.; Webster, J.D.; Prasad, M.L.; Conway, C.M.; Prieto, V.M.; Gary, J.M.; Goldschmidt, M.H.; Esplin, D.G.; et al. Sporadic naturally occurring melanoma in dogs as a preclinical model for human melanoma. Pigment. Cell Melanoma Res. 2014, 27, 37–47. [Google Scholar] [CrossRef]
- Wong, K.; van der Weyden, L.; Schott, C.R.; Foote, A.; Constantino-Casas, F.; Smith, S.; Dobson, J.M.; Murchison, E.P.; Wu, H.; Yeh, I.; et al. Cross-species genomic landscape comparison of human mucosal melanoma with canine oral and equine melanoma. Nat. Commun. 2019, 10, 353. [Google Scholar] [CrossRef]
- Nishiya, A.T.; Massoco, C.O.; Felizzola, C.R.; Perlmann, E.; Batschinski, K.; Tedardi, M.V.; Garcia, J.S.; Mendonça, P.P.; Teixeira, T.F.; Zaidan Dagli, M.L. Comparative Aspects of Canine Melanoma. Vet. Sci. 2016, 3, 7. [Google Scholar] [CrossRef]
- Hernandez, B.; Adissu, H.A.; Wei, B.R.; Michael, H.T.; Merlino, G.; Simpson, R.M. Naturally Occurring Canine Melanoma as a Predictive Comparative Oncology Model for Human Mucosal and Other Triple Wild-Type Melanomas. Int. J. Mol. Sci. 2018, 19, 394. [Google Scholar] [CrossRef]
- Prouteau, A.; Andre, C. Canine Melanomas as Models for Human Melanomas: Clinical, Histological, and Genetic Comparison. Genes 2019, 10, 501. [Google Scholar] [CrossRef]
- Bosenberg, M.; Arnheiter, H.; Kelsh, R. Melanoma in mankind’s best friend. Pigment Cell Melanoma Res. 2014, 27, 1. [Google Scholar] [CrossRef]
- Stevenson, V.B.; Klahn, S.; LeRoith, T.; Huckle, W.R. Canine melanoma: A review of diagnostics and comparative mechanisms of disease and immunotolerance in the era of the immunotherapies. Front. Vet. Sci. 2023, 9, 1046636. [Google Scholar] [CrossRef]
- Gillard, M.; Cadieu, E.; De Brito, C.; Abadie, J.; Vergier, B.; Devauchelle, P.; Degorce, F.; Dreano, S.; Primot, A.; Dorso, L.; et al. Naturally occurring melanomas in dogs as models for non-UV pathways of human melanomas. Pigment. Cell Melanoma Res. 2014, 27, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Gardner, H.L.; Fenger, J.M.; London, C.A. Dogs as a Model for Cancer. Annu. Rev. Anim. Biosci. 2016, 4, 199–222. [Google Scholar] [CrossRef] [PubMed]
- Curtin, J.A.; Busam, K.; Pinkel, D.; Bastian, B.C. Somatic activation of KIT in distinct subtypes of melanoma. J. Clin. Oncol. 2006, 24, 4340–4346. [Google Scholar] [CrossRef] [PubMed]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef]
- Hintzsche, J.D.; Gorden, N.T.; Amato, C.M.; Kim, J.; Wuensch, K.E.; Robinson, S.E.; Applegate, A.J.; Couts, K.L.; Medina, T.M.; Wells, K.R.; et al. Whole-exome sequencing identifies recurrent SF3B1 R625 mutation and comutation of NF1 and KIT in mucosal melanoma. Melanoma Res. 2017, 27, 189–199. [Google Scholar] [CrossRef]
- Satzger, I.; Schaefer, T.; Kuettler, U.; Broecker, V.; Voelker, B.; Ostertag, H.; Kapp, A.; Gutzmer, R. Analysis of c-KIT expression and KIT gene mutation in human mucosal melanomas. Br. J. Cancer 2008, 99, 2065–2069. [Google Scholar] [CrossRef]
- Carlino, M.S.; Todd, J.R.; Rizos, H. Resistance to c-Kit inhibitors in melanoma: Insights for future therapies. Oncoscience 2014, 1, 423–426. [Google Scholar] [CrossRef]
- Smedley, R.C.; Thaiwong, T.; Deeth, L.E.; Kiupel, M. Correlation Between KIT Expression and c-Kit Mutations in 2 Subtypes of Canine Oral Melanocytic Neoplasms. Vet. Pathol. 2021, 58, 683–691. [Google Scholar] [CrossRef]
- Ito, K.; Kobayashi, M.; Kuroki, S.; Sasaki, Y.; Iwata, T.; Mori, K.; Kuroki, T.; Ozawa, Y.; Tetsuka, M.; Nakagawa, T.; et al. The proteasome inhibitor bortezomib inhibits the growth of canine malignant melanoma cells in vitro and in vivo. Vet. J. 2013, 198, 577–582. [Google Scholar] [CrossRef]
- Wack, K.; Drogowski, L.; Treloar, M.; Evans, A.; Ho, J.; Parwani, A.; Montalto, M.C. A multisite validation of whole slide imaging for primary diagnosis using standardized data collection and analysis. J. Pathol. Inform. 2016, 7, 49. [Google Scholar] [CrossRef]
- Dumaz, N.; Jouenne, F.; Delyon, J.; Mourah, S.; Bensussan, A.; Lebbé, C. Atypical BRAF and NRAS Mutations in Mucosal Melanoma. Cancers 2019, 11, 1133. [Google Scholar] [CrossRef] [PubMed]
- Hendricks, W.P.D.; Zismann, V.; Sivaprakasam, K.; Legendre, C.; Poorman, K.; Tembe, W.; Perdigones, N.; Kiefer, J.; Liang, W.; DeLuca, V.; et al. Somatic inactivating PTPRJ mutations and dysregulated pathways identified in canine malignant melanoma by integrated comparative genomic analysis. PLoS Genet. 2018, 14, e1007589. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Xia, R.; Ma, X.; Judson-Torres, R.L.; Zeng, H. Mucosal Melanoma: Pathological Evolution, Pathway Dependency and Targeted Therapy. Front. Oncol. 2021, 11, 702287. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.R.; Peer, C.J.; Richardson, W.J.; Hewitt, S.M.; Figg, W.D.; Simpson, R.M. Pharmacokinetics and tolerability of the dual TORC1/2 inhibitor sapanisertib in combination with the MEK inhibitor trametinib in dogs. Front. Vet. Sci. 2022, 9, 1056408. [Google Scholar] [CrossRef] [PubMed]
- Musgrove, E.A.; Caldon, C.E.; Barraclough, J.; Stone, A.; Sutherland, R.L. Cyclin D as a therapeutic target in cancer. Nat. Rev. Cancer 2011, 11, 558–572. [Google Scholar] [CrossRef]
- Lavoie, J.N.; L’Allemain, G.; Brunet, A.; Müller, R.; Pouysségur, J. Cyclin D1 Expression is Regulated Positively by the p42/p44MAPK and Negatively by the p38/HOGMAPK Pathway. J. Biol. Chem. 1996, 271, 20608–20616. [Google Scholar] [CrossRef]
- Chraybi, M.; Alsamad, I.A.; Copie-Bergman, C.; Baia, M.; André, J.; Dumaz, N.; Ortonne, N. Oncogene abnormalities in a series of primary melanomas of the sinonasal tract: NRAS mutations and cyclin D1 amplification are more frequent than KIT or BRAF mutations. Hum. Pathol. 2013, 44, 1902–1911. [Google Scholar] [CrossRef]
- Zamboni, C.; Brocca, G.; Ferraresso, S.; Ferro, S.; Sammarco, A.; Dal Corso, C.; Iussich, S.; de Andres, P.J.; Martìnez de Merlo, E.M.; Cavicchioli, L.; et al. Cyclin D1 immunohistochemical expression and somatic mutations in canine oral melanoma. Vet. Comp. Oncol. 2020, 18, 231–238. [Google Scholar] [CrossRef]
- Sewify, E.M.; Afifi, O.A.; Mosad, E.; Zaki, A.H.; El Gammal, S.A. Cyclin D1 amplification in multiple myeloma is associated with multidrug resistance expression. Clin. Lymphoma Myeloma Leuk. 2014, 14, 215–222. [Google Scholar] [CrossRef]
- Shi, Q.; Li, Y.; Li, S.; Jin, L.; Lai, H.; Wu, Y.; Cai, Z.; Zhu, M.; Li, Q.; Li, Y.; et al. LncRNA DILA1 inhibits Cyclin D1 degradation and contributes to tamoxifen resistance in breast cancer. Nat. Commun. 2020, 11, 5513. [Google Scholar] [CrossRef]
- Fuse, M.A.; Dinh, C.T.; Vitte, J.; Kirkpatrick, J.; Mindos, T.; Plati, S.K.; Young, J.I.; Huang, J.; Carlstedt, A.; Franco, M.C.; et al. Preclinical assessment of MEK1/2 inhibitors for neurofibromatosis type 2-associated schwannomas reveals differences in efficacy and drug resistance development. Neuro Oncol. 2019, 21, 486–497. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.R.; Michael, H.T.; Halsey, C.H.; Peer, C.J.; Adhikari, A.; Dwyer, J.E.; Hoover, S.B.; El Meskini, R.; Kozlov, S.; Weaver Ohler, Z.; et al. Synergistic targeted inhibition of MEK and dual PI3K/mTOR diminishes viability and inhibits tumor growth of canine melanoma underscoring its utility as a preclinical model for human mucosal melanoma. Pigment. Cell Melanoma Res. 2016, 29, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Del Pino, G.L.; Li, K.; Park, E.; Schmoker, A.M.; Ha, B.H.; Eck, M.J. Allosteric MEK inhibitors act on BRAF/MEK complexes to block MEK activation. Proc. Natl. Acad. Sci. USA 2021, 118, e2107207118. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, E.; Tsuchiya, A.; Imoto, M. Functions of cyclin D1 as an oncogene and regulation of cyclin D1 expression. Cancer Sci. 2007, 98, 629–635. [Google Scholar] [CrossRef]
- Chen, S.; Li, L. Degradation strategy of cyclin D1 in cancer cells and the potential clinical application. Front. Oncol. 2022, 12, 949688. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N. Engl. J. Med. 2012, 367, 107–114. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef]
- Long, G.V.; Fung, C.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Hyman, J.; Shahheydari, H.; Tembe, V.; Thompson, J.F.; Saw, R.P.; et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat. Commun. 2014, 5, 5694. [Google Scholar] [CrossRef]
- Garg, P.; Malhotra, J.; Kulkarni, P.; Horne, D.; Salgia, R.; Singhal, S.S. Emerging Therapeutic Strategies to Overcome Drug Resistance in Cancer Cells. Cancers 2024, 16, 2478. [Google Scholar] [CrossRef]
- Winder, M.; Virós, A. Mechanisms of Drug Resistance in Melanoma. In Mechanisms of Drug Resistance in Cancer Therapy; Mandalà, M., Romano, E., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 91–108. [Google Scholar] [CrossRef]
- He, X.; Gao, Y.; Deng, Y.; He, J.; Nolte, I.; Murua Escobar, H.; Yu, F. The Comparative Oncology of Canine Malignant Melanoma in Targeted Therapy: A Systematic Review of In Vitro Experiments and Animal Model Reports. Int. J. Mol. Sci. 2024, 25, 10387. [Google Scholar] [CrossRef]
- Aina, O.H.; Maeda, Y.; Harrison, M.; Zwingenberger, A.L.; Walker, N.J.; Lam, K.S.; Kent, M.S. Canine malignant melanoma alpha-3 integrin binding peptides. Vet. Immunol. Immunopathol. 2011, 143, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Fowles, J.S.; Dailey, D.D.; Gustafson, D.L.; Thamm, D.H.; Duval, D.L. The Flint Animal Cancer Center (FACC) Canine Tumour Cell Line Panel: A resource for veterinary drug discovery, comparative oncology and translational medicine. Vet. Comp. Oncol. 2017, 15, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Parekh, P.R.; Botting, G.M.; Thurber, D.B.; Boruszczak, M.; Murphy, W.; Bertenshaw, G.P. Predictive biomarkers for response to trametinib in non-small cell lung cancer. Tumour Biol. 2022, 44, 249–267. [Google Scholar] [CrossRef]
- Degan, S.; May, B.L.; Jin, Y.J.; Hammoda, M.B.; Sun, H.; Zhang, G.; Wang, Y.; Erdmann, D.; Warren, W.; Zhang, J.Y. Co-Treatment of Chloroquine and Trametinib Inhibits Melanoma Cell Proliferation and Decreases Immune Cell Infiltration. Front. Oncol. 2022, 12, 782877. [Google Scholar] [CrossRef]
- Atefi, M.; Titz, B.; Avramis, E.; Ng, C.; Wong, D.J.; Lassen, A.; Cerniglia, M.; Escuin-Ordinas, H.; Foulad, D.; Comin-Anduix, B.; et al. Combination of pan-RAF and MEK inhibitors in NRAS mutant melanoma. Mol. Cancer 2015, 14, 27. [Google Scholar] [CrossRef]
- Watanabe, M.; Sowa, Y.; Yogosawa, M.; Sakai, T. Novel MEK inhibitor trametinib and other retinoblastoma gene (RB)-reactivating agents enhance efficacy of 5-fluorouracil on human colon cancer cells. Cancer Sci. 2013, 104, 687–693. [Google Scholar] [CrossRef]
- Chen, B.; Pollard, J.W. Cyclin D2 Compensates for the Loss of Cyclin D1 in Estrogen-Induced Mouse Uterine Epithelial Cell Proliferation. Mol. Endocrinol. 2003, 17, 1368–1381. [Google Scholar] [CrossRef]
- Wei, B.R.; Hoover, S.B.; Peer, C.J.; Dwyer, J.E.; Adissu, H.A.; Shankarappa, P.; Yang, H.; Lee, M.; Peat, T.J.; Figg, W.D.; et al. Efficacy, Tolerability, and Pharmacokinetics of Combined Targeted MEK and Dual mTORC1/2 Inhibition in a Preclinical Model of Mucosal Melanoma. Mol. Cancer Ther. 2020, 19, 2308–2318. [Google Scholar] [CrossRef]
- Monti, M.; Benerini Gatta, L.; Bugatti, M.; Pezzali, I.; Picinoli, S.; Manfredi, M.; Lavazza, A.; Vanella, V.V.; De Giorgis, V.; Zanatta, L.; et al. Novel cellular systems unveil mucosal melanoma initiating cells and a role for PI3K/Akt/mTOR pathway in mucosal melanoma fitness. J. Transl. Med. 2024, 22, 35. [Google Scholar] [CrossRef]
- Saleban, M.; Harris, E.L.; Poulter, J.A. D-Type Cyclins in Development and Disease. Genes 2023, 14, 1445. [Google Scholar] [CrossRef]
- Tchakarska, G.; Sola, B. The double dealing of cyclin D1. Cell Cycle 2020, 19, 163–178. [Google Scholar] [CrossRef] [PubMed]
- González-Ruiz, L.; González-Moles, M.Á.; González-Ruiz, I.; Ruiz-Ávila, I.; Ayén, Á.; Ramos-García, P. An update on the implications of cyclin D1 in melanomas. Pigment. Cell Melanoma Res. 2020, 33, 788–805. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Wang, L.; Huang, S.; Heynen, G.J.J.E.; Prahallad, A.; Robert, C.; Haanen, J.; Blank, C.; Wesseling, J.; Willems, S.M.; et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 2014, 508, 118–122. [Google Scholar] [CrossRef]
- Vergani, E.; Busico, A.; Dugo, M.; Devecchi, A.; Valeri, B.; Cossa, M.; Di Guardo, L.; De Cecco, L.; Feltrin, E.; Valle, G.; et al. Genetic Layout of Melanoma Lesions Is Associated with BRAF/MEK-Targeted Therapy Resistance and Transcriptional Profiles. J. Investig. Dermatol. 2022, 142, 3030–3040.e3035. [Google Scholar] [CrossRef]
- Elnenaei, M.O.; Jadayel, D.M.; Matutes, E.; Morilla, R.; Owusu-Ankomah, K.; Atkinson, S.; Titley, I.; Mandala, E.M.; Catovsky, D. Cyclin D1 by flow cytometry as a useful tool in the diagnosis of B-cell malignancies. Leuk. Res. 2001, 25, 115–123. [Google Scholar] [CrossRef]
- Georgia, S.; Hinault, C.; Kawamori, D.; Hu, J.; Meyer, J.; Kanji, M.; Bhushan, A.; Kulkarni, R.N. Cyclin D2 is Essential for the Compensatory β-Cell Hyperplastic Response to Insulin Resistance in Rodents. Diabetes 2010, 59, 987–996. [Google Scholar] [CrossRef]
- Carthon, B.C.; Neumann, C.A.; Das, M.; Pawlyk, B.; Li, T.; Geng, Y.; Sicinski, P. Genetic replacement of cyclin D1 function in mouse development by cyclin D2. Mol. Cell. Biol. 2005, 25, 1081–1088. [Google Scholar] [CrossRef]
- Balcazar, N.; Sathyamurthy, A.; Elghazi, L.; Gould, A.; Weiss, A.; Shiojima, I.; Walsh, K.; Bernal-Mizrachi, E. mTORC1 activation regulates beta-cell mass and proliferation by modulation of cyclin D2 synthesis and stability. J. Biol. Chem. 2009, 284, 7832–7842. [Google Scholar] [CrossRef]
- Glassford, J.; Kassen, D.; Quinn, J.; Stengel, C.; Kallinikou, K.; Khwaja, A.; Yong, K.L. Inhibition of cell cycle progression by dual phosphatidylinositol-3-kinase and mTOR blockade in cyclin D2 positive multiple myeloma bearing IgH translocations. Blood Cancer J. 2012, 2, e50. [Google Scholar] [CrossRef]
- Hoshii, T.; Kasada, A.; Hatakeyama, T.; Ohtani, M.; Tadokoro, Y.; Naka, K.; Ikenoue, T.; Ikawa, T.; Kawamoto, H.; Fehling, H.J.; et al. Loss of mTOR complex 1 induces developmental blockage in early T-lymphopoiesis and eradicates T-cell acute lymphoblastic leukemia cells. Proc. Natl. Acad. Sci. USA 2014, 111, 3805–3810. [Google Scholar] [CrossRef]
- Rodrik-Outmezguine, V.S.; Chandarlapaty, S.; Pagano, N.C.; Poulikakos, P.I.; Scaltriti, M.; Moskatel, E.; Baselga, J.; Guichard, S.; Rosen, N. mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer Discov. 2011, 1, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Kida, A.; Kakihana, K.; Kotani, S.; Kurosu, T.; Miura, O. Glycogen synthase kinase-3β and p38 phosphorylate cyclin D2 on Thr280 to trigger its ubiquitin/proteasome-dependent degradation in hematopoietic cells. Oncogene 2007, 26, 6630–6640. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Gavert, N.; Brabletz, T.; Ben-Ze’ev, A. A Necessary Role for Cyclin D2 Induction During Colon Cancer Progression Mediated by L1. Cells 2024, 13, 1810. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, R.; Hunter, I.A.; Rajaganeshan, R.; Perry, S.L.; Guillou, P.; Jayne, D.G. Expression of cyclin D2 is an independent predictor of the development of hepatic metastasis in colorectal cancer. Color. Dis. 2010, 12, 316–323. [Google Scholar] [CrossRef]
- Koyama-Nasu, R.; Nasu-Nishimura, Y.; Todo, T.; Ino, Y.; Saito, N.; Aburatani, H.; Funato, K.; Echizen, K.; Sugano, H.; Haruta, R.; et al. The critical role of cyclin D2 in cell cycle progression and tumorigenicity of glioblastoma stem cells. Oncogene 2013, 32, 3840–3845. [Google Scholar] [CrossRef]
- Matsubayashi, H.; Sato, N.; Fukushima, N.; Yeo, C.J.; Walter, K.M.; Brune, K.; Sahin, F.; Hruban, R.H.; Goggins, M. Methylation of Cyclin D2 is Observed Frequently in Pancreatic Cancer but is Also an Age-related Phenomenon in Gastrointestinal Tissues. Clin. Cancer Res. 2003, 9, 1446–1452. [Google Scholar]
- Evron, E.; Umbricht, C.B.; Korz, D.; Raman, V.; Loeb, D.M.; Niranjan, B.; Buluwela, L.; Weitzman, S.A.; Marks, J.; Sukumar, S. Loss of Cyclin D2 Expression in the Majority of Breast Cancers Is Associated with Promoter Hypermethylation. Cancer Res. 2001, 61, 2782–2787. [Google Scholar]
- Jardim, D.L.; Millis, S.Z.; Ross, J.S.; Woo, M.S.; Ali, S.M.; Kurzrock, R. Cyclin Pathway Genomic Alterations Across 190,247 Solid Tumors: Leveraging Large-Scale Data to Inform Therapeutic Directions. Oncologist 2021, 26, e78–e89. [Google Scholar] [CrossRef]
- Ding, Z.Y.; Li, R.; Zhang, Q.J.; Wang, Y.; Jiang, Y.; Meng, Q.Y.; Xi, Q.L.; Wu, G.H. Prognostic role of cyclin D2/D3 in multiple human malignant neoplasms: A systematic review and meta-analysis. Cancer Med. 2019, 8, 2717–2729. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, B.-R.; Verdi, V.; Zhang, S.; Mock, B.A.; Shive, H.R.; Simpson, R.M. Capacity for Compensatory Cyclin D2 Response Confers Trametinib Resistance in Canine Mucosal Melanoma. Cancers 2025, 17, 2357. https://doi.org/10.3390/cancers17142357
Wei B-R, Verdi V, Zhang S, Mock BA, Shive HR, Simpson RM. Capacity for Compensatory Cyclin D2 Response Confers Trametinib Resistance in Canine Mucosal Melanoma. Cancers. 2025; 17(14):2357. https://doi.org/10.3390/cancers17142357
Chicago/Turabian StyleWei, Bih-Rong, Vincenzo Verdi, Shuling Zhang, Beverly A. Mock, Heather R. Shive, and R. Mark Simpson. 2025. "Capacity for Compensatory Cyclin D2 Response Confers Trametinib Resistance in Canine Mucosal Melanoma" Cancers 17, no. 14: 2357. https://doi.org/10.3390/cancers17142357
APA StyleWei, B.-R., Verdi, V., Zhang, S., Mock, B. A., Shive, H. R., & Simpson, R. M. (2025). Capacity for Compensatory Cyclin D2 Response Confers Trametinib Resistance in Canine Mucosal Melanoma. Cancers, 17(14), 2357. https://doi.org/10.3390/cancers17142357