
Blood-Epigenetic Biomarker Associations with Tumor Immunophenotype in Patients with Urothelial Carcinoma from JAVELIN Bladder 100

 , , , ,
, , , ,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Description of Specimens Collected for Analysis
2.2. Identification of Candidate CCMs Using the EpiSwitch Platform
2.2.1. Preparation of 3D Genomic Templates
2.2.2. Array Design
2.2.3. Translation of Array-Based 3D Genomic Markers to PCR Readouts
2.2.4. Identification of Top 25 CCMs Associated with Tumor JAV-Immuno Scores
2.3. Prioritization of Candidate CCMs Based on Estimated Interactions with TMB and OS
Model Development
2.4. Assessment of Potential CCM Gene Expression
2.5. Visium Data Processing
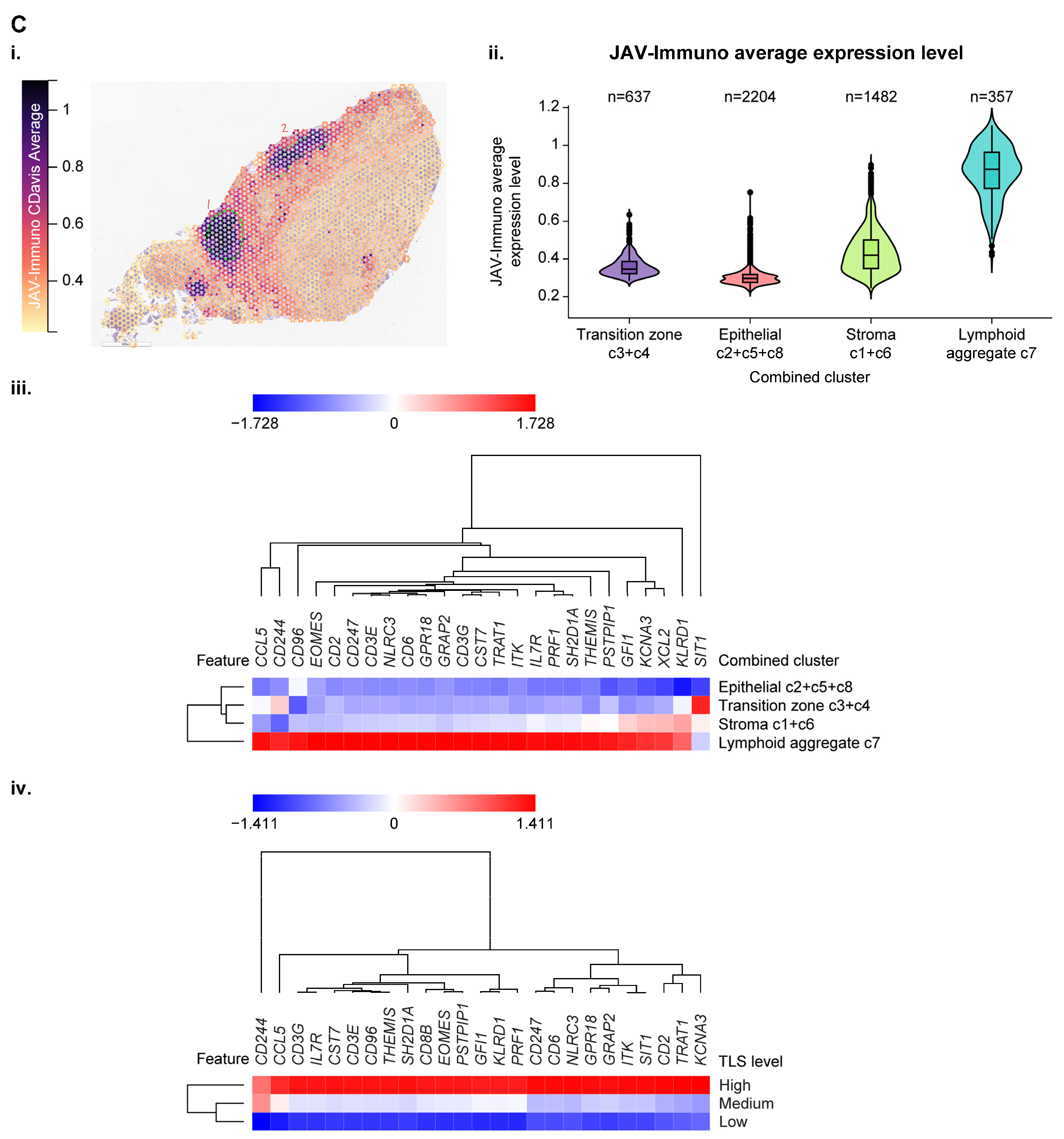
3. Results
3.1. Selection of a Tumor Immune Gene Expression Signature to Screen for Candidate CCMs
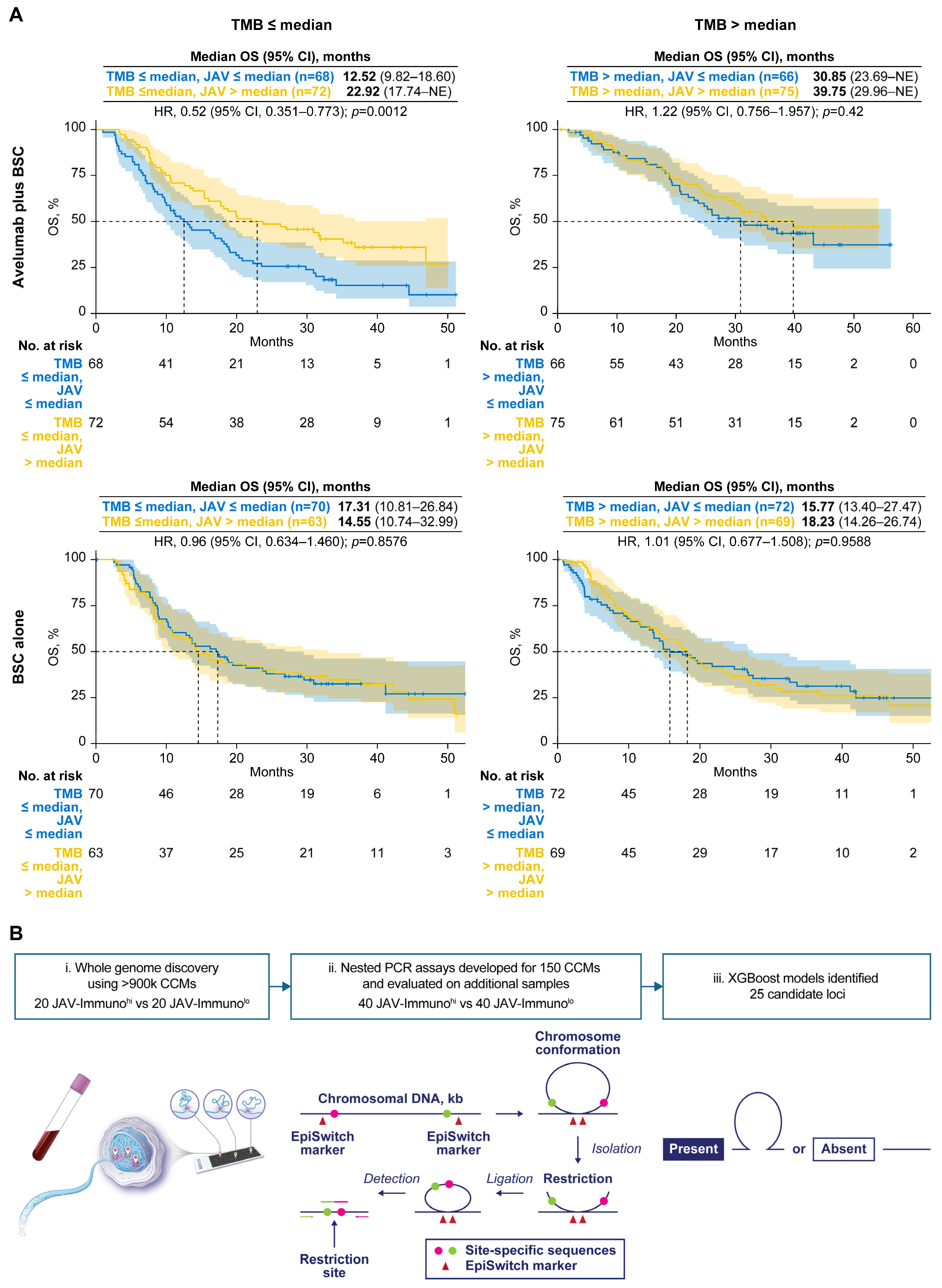
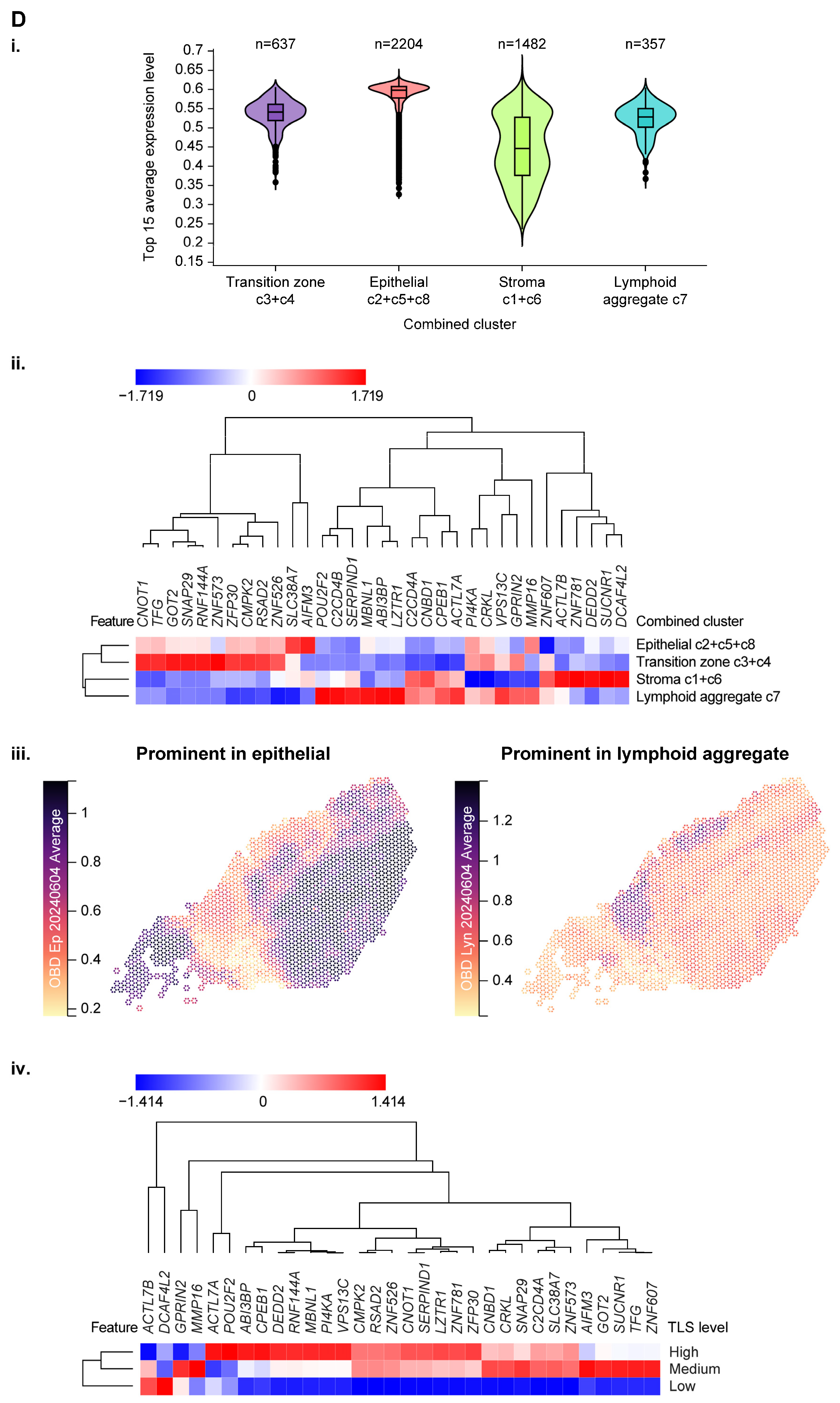
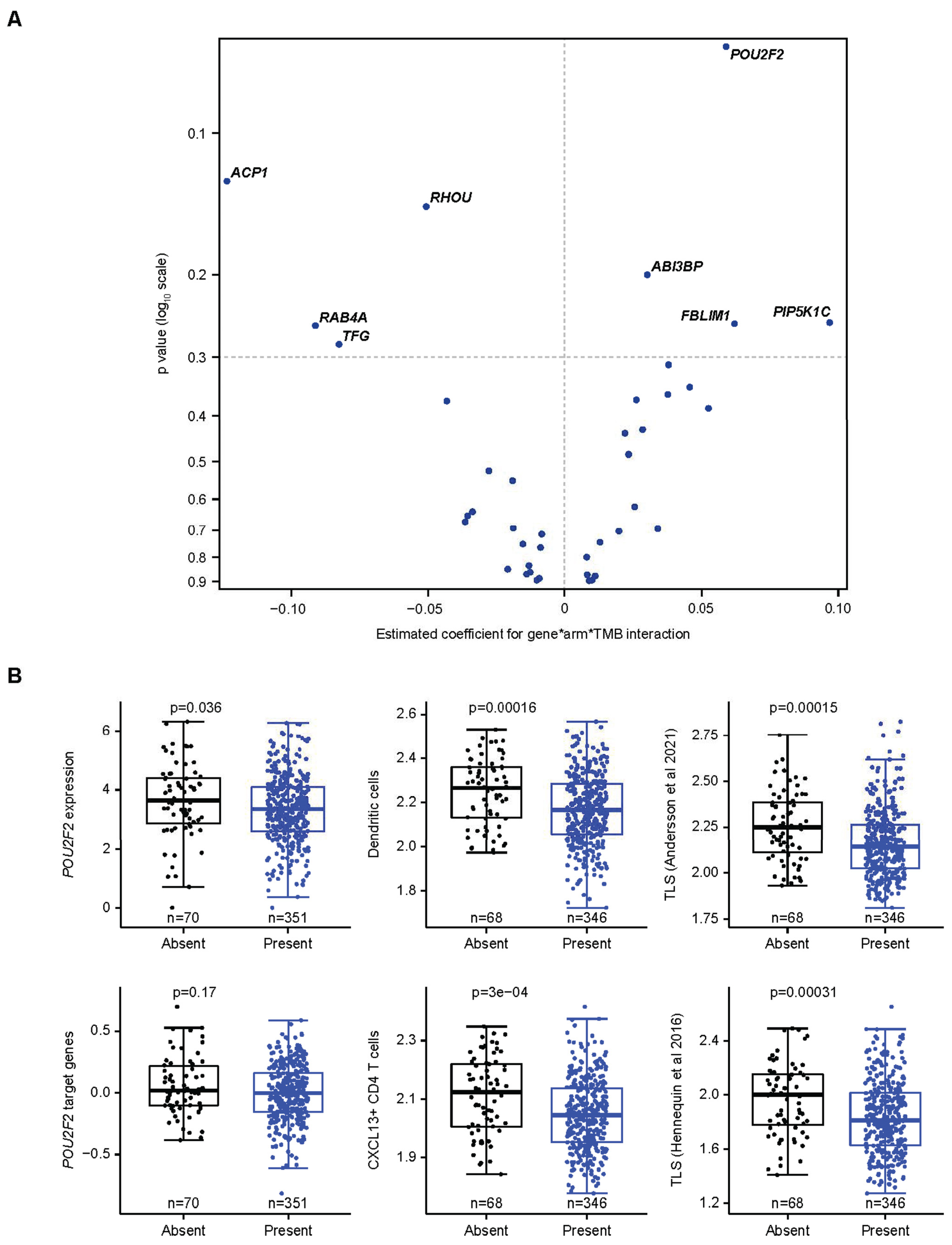
3.2. Screening and Selection of Candidate CCMs Through Assessment of Potential Interactions with Treatment Effect and TMB as Well as Tumor Phenotype
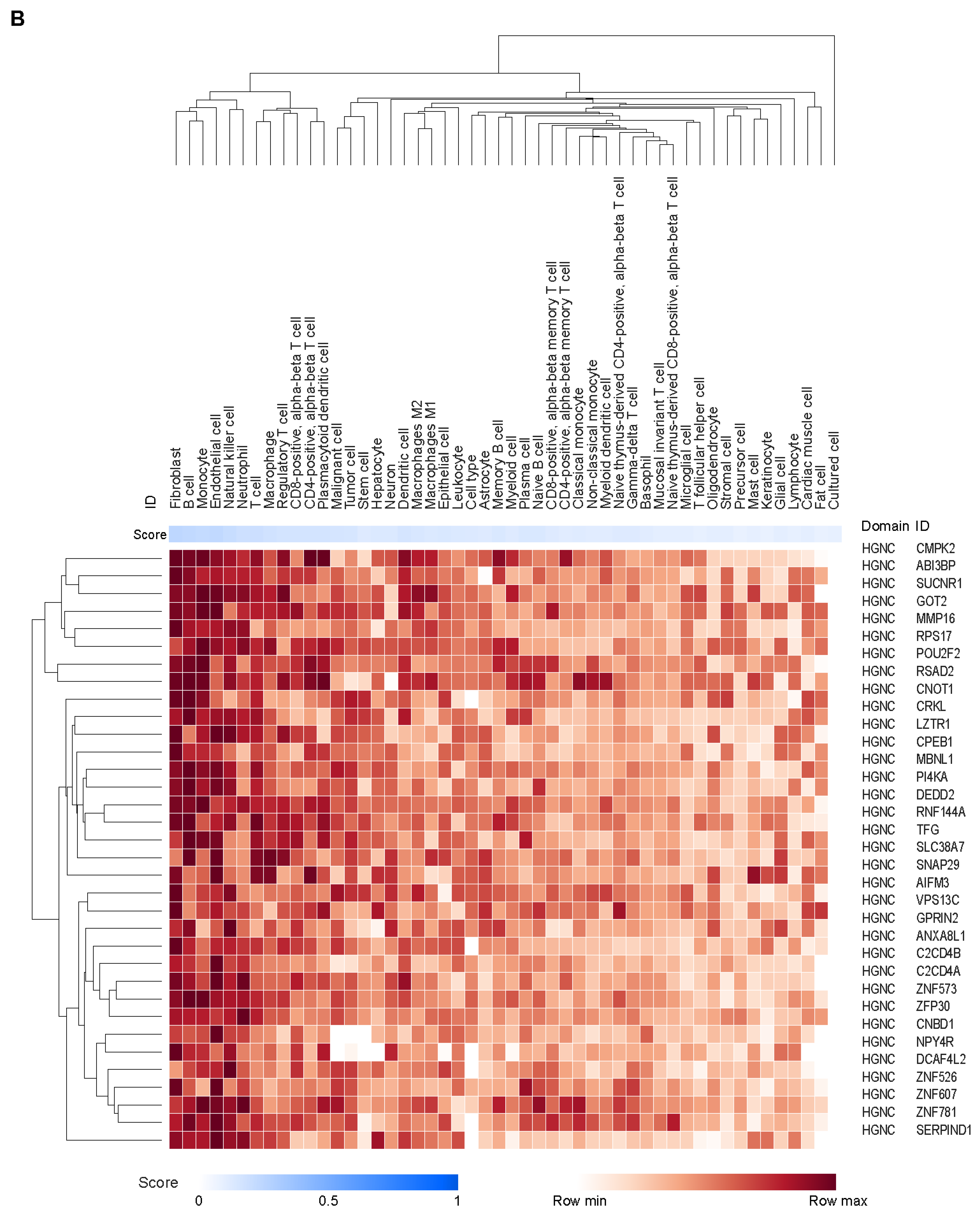
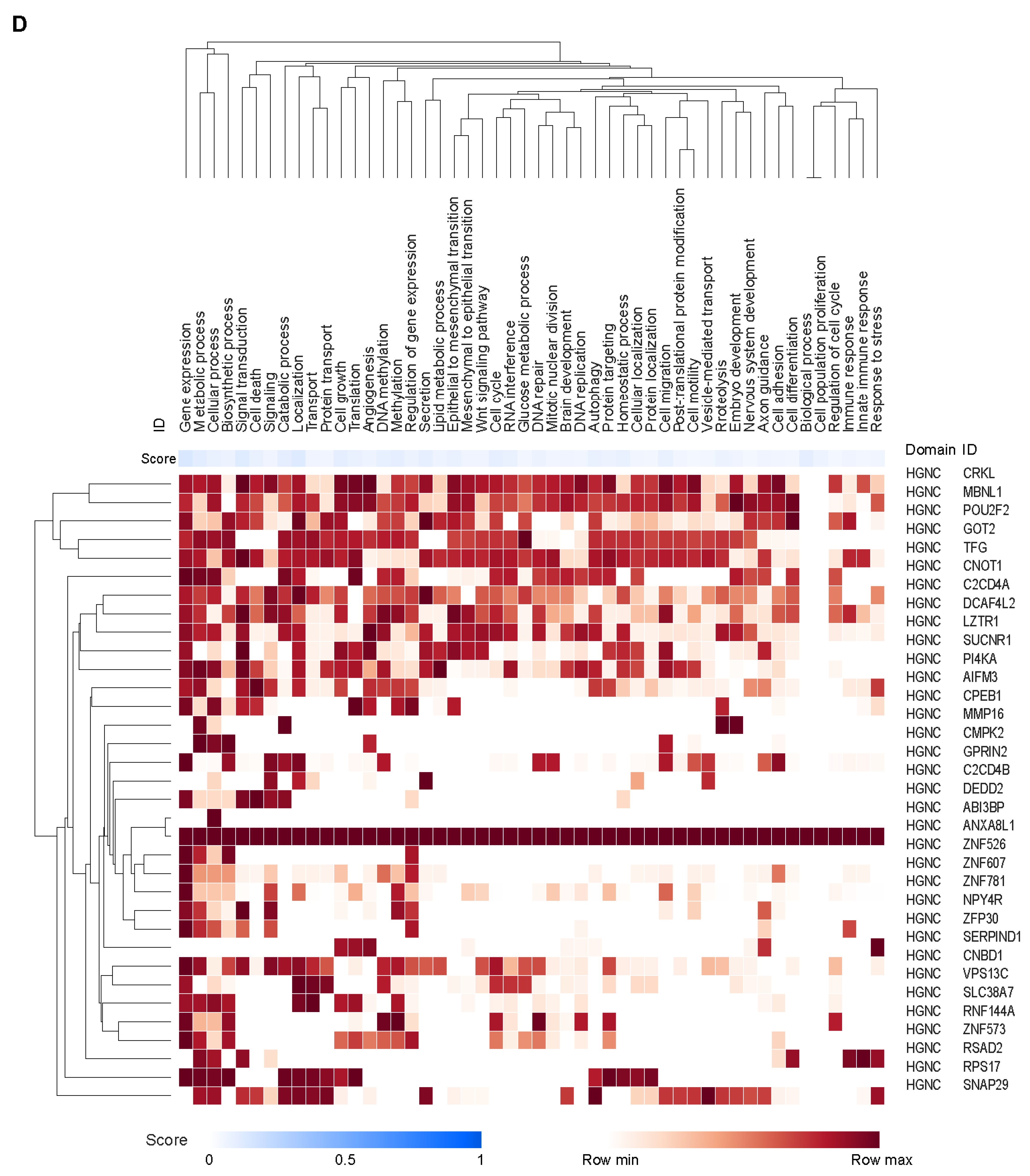
3.3. Potential Cell Types and Pathways Represented by the Genes Covered by the CCMs
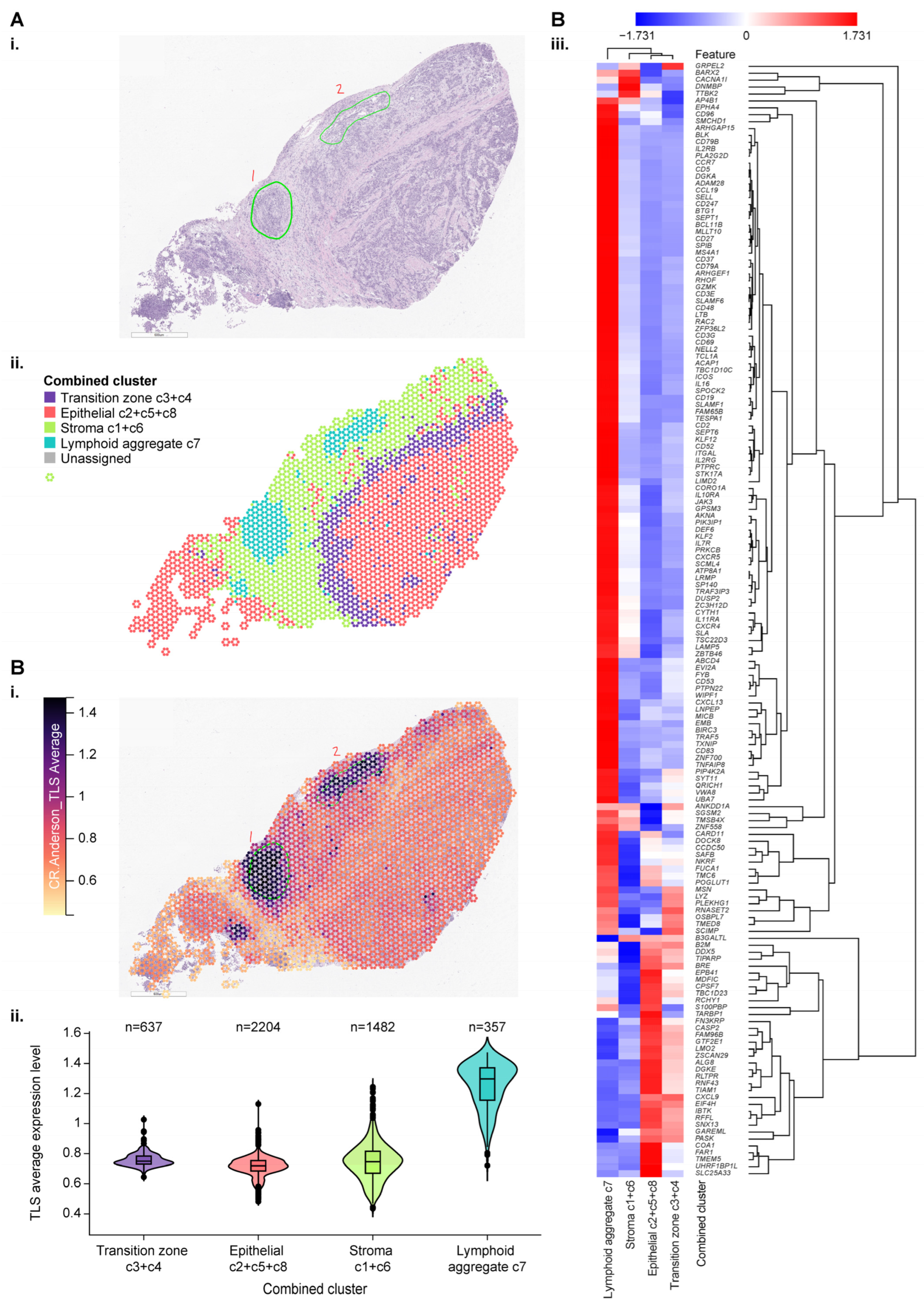
3.4. Use of Spatial Profiling to Locate CCM Genes Within the TME
3.5. Specific Assessment of Associations Between a CCM-Covered Gene, TMB, and Avelumab Treatment Effect
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Banchereau, R.; Leng, N.; Zill, O.; Sokol, E.; Liu, G.; Pavlick, D.; Maund, S.; Liu, L.-F.; Kadel, E.; Baldwin, N.; et al. Molecular determinants of response to PD-L1 blockade across tumor types. Nat. Commun. 2021, 12, 3969. [Google Scholar] [CrossRef]
- Powles, T.; Sridhar, S.S.; Loriot, Y.; Bellmunt, J.; Mu, X.J.; Ching, K.A.; Pu, J.; Sternberg, C.N.; Petrylak, D.P.; Tambaro, R.; et al. Avelumab maintenance in advanced urothelial carcinoma: Biomarker analysis of the phase 3 JAVELIN Bladder 100 trial. Nat. Med. 2021, 27, 2200–2211. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Park, S.H.; Voog, E.; Caserta, C.; Valderrama, B.P.; Gurney, H.; Kalofonos, H.; Radulović, S.; Demey, W.; Ullén, A.; et al. Avelumab maintenance therapy for advanced or metastatic urothelial carcinoma. N. Engl. J. Med. 2020, 383, 1218–1230. [Google Scholar] [CrossRef]
- Powles, T.; Bellmunt, J.; Comperat, E.; De Santis, M.; Huddart, R.; Loriot, Y.; Necchi, A.; Valderrama, B.P.; Ravaud, A.; Shariat, S.F.; et al. ESMO Clinical Practice Guideline interim update on first-line therapy in advanced urothelial carcinoma. Ann. Oncol. 2024, 35, 485–490. [Google Scholar] [CrossRef]
- Grivas, P.; Grande, E.; Davis, I.D.; Moon, H.H.; Grimm, M.-O.; Gupta, S.; Barthélémy, P.; Thibault, C.; Guenther, S.; Hanson, S.; et al. Avelumab first-line maintenance treatment for advanced urothelial carcinoma: Review of evidence to guide clinical practice. ESMO Open 2023, 8, 102050. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Chen, D.S.; Powles, T.; Turley, S.J. The cancer-immunity cycle: Indication, genotype, and immunotype. Immunity 2023, 56, 2188–2205. [Google Scholar] [CrossRef]
- Khan, Z.; Di Nucci, F.; Kwan, A.; Hammer, C.; Mariathasan, S.; Rouilly, V.; Carroll, J.; Fontes, M.; Acosta, S.L.; Guardino, E.; et al. Polygenic risk for skin autoimmunity impacts immune checkpoint blockade in bladder cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 12288–12294. [Google Scholar] [CrossRef]
- Dizman, N.; Meza, L.; Bergerot, P.; Alcantara, M.; Dorff, T.; Lyou, Y.; Frankel, P.; Cui, Y.; Mira, V.; Llamas, M.; et al. Nivolumab plus ipilimumab with or without live bacterial supplementation in metastatic renal cell carcinoma: A randomized phase 1 trial. Nat. Med. 2022, 28, 704–712. [Google Scholar] [CrossRef]
- Cuartero, S.; Stik, G.; Stadhouders, R. Three-dimensional genome organization in immune cell fate and function. Nat. Rev. Immunol. 2022, 23, 206–221. [Google Scholar] [CrossRef] [PubMed]
- Johanson, T.M.; Chan, W.F.; Keenan, C.R.; Allan, R.S. Genome organization in immune cells: Unique challenges. Nat. Rev. Immunol. 2019, 19, 448–456. [Google Scholar] [CrossRef]
- Tordini, F.; Aldinucci, M.; Milanesi, L.; Liò, P.; Merelli, I. The genome conformation as an integrator of multi-omic data: The example of damage spreading in cancer. Front. Genet. 2016, 7, 194. [Google Scholar] [CrossRef]
- Bonev, B.; Cavalli, G. Organization and function of the 3D genome. Nat. Rev. Genet. 2016, 17, 661–678. [Google Scholar] [CrossRef]
- Tien, F.-M.; Lu, H.-H.; Lin, S.-Y.; Tsai, H.-C. Epigenetic remodeling of the immune landscape in cancer: Therapeutic hurdles and opportunities. J. Biomed. Sci. 2023, 30, 3. [Google Scholar] [CrossRef]
- Kempfer, R.; Pombo, A. Methods for mapping 3D chromosome architecture. Nat. Rev. Genet. 2019, 21, 207–226. [Google Scholar] [CrossRef]
- Dekker, J.; Rippe, K.; Dekker, M.; Kleckner, N. Capturing chromosome conformation. Science 2002, 295, 1306–1311. [Google Scholar] [CrossRef]
- Jakub, J.W.; Grotz, T.E.; Jordan, P.; Hunter, E.; Pittelkow, M.; Ramadass, A.; Akoulitchev, A.; Markovic, S. A pilot study of chromosomal aberrations and epigenetic changes in peripheral blood samples to identify patients with melanoma. Melanoma Res. 2015, 25, 406–411. [Google Scholar] [CrossRef]
- Carini, C.; Hunter, E.; Ramadass, A.S.; Green, J.; Akoulitchev, A.; McInnes, I.B.; Goodyear, C.S. Chromosome conformation signatures define predictive markers of inadequate response to methotrexate in early rheumatoid arthritis. J. Transl. Med. 2018, 16, 18. [Google Scholar] [CrossRef]
- Alshaker, H.; Mills, R.; Hunter, E.; Salter, M.; Ramadass, A.; Skinner, B.M.; Westra, W.; Green, J.; Akoulitchev, A.; Winkler, M.; et al. Chromatin conformation changes in peripheral blood can detect prostate cancer and stratify disease risk groups. J. Transl. Med. 2021, 19, 46. [Google Scholar] [CrossRef]
- Pchejetski, D.; Hunter, E.; Dezfouli, M.; Salter, M.; Powell, R.; Green, J.; Naithani, T.; Koutsothanasi, C.; Alshaker, H.; Jaipuria, J.; et al. Circulating chromosome conformation signatures significantly enhance PSA positive predicting value and overall accuracy for prostate cancer detection. Cancers 2023, 15, 821. [Google Scholar] [CrossRef] [PubMed]
- Hunter, E.; Salter, M.; Powell, R.; Dring, A.; Naithani, T.; Chatziioannou, M.E.; Gebregzabhar, A.; Issa, M.; Green, J.; Ng, S.; et al. Development and validation of blood-based predictive biomarkers for response to PD-1/PD-L1 checkpoint inhibitors: Evidence of a universal systemic core of 3D immunogenetic profiling across multiple oncological indications. Cancers 2023, 15, 2696. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R. BEDTools: The Swiss-army tool for genome feature analysis. Curr. Protoc. Bioinform. 2014, 47, 11–12. [Google Scholar] [CrossRef]
- Chen, T.; Guestrin, C. XGBoost: A scalable tree boosting system. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; Association for Computing Machinery: San Francisco, CA, USA, 2016; pp. 785–794. [Google Scholar]
- Biecek, P.; Burzykowski, T. Shapley. Additive Explanations (SHAP) for Average Attributions. In Explanatory Model Analysis, 1st ed.; Chapman and Hall/CRC: Boca Raton, FL, USA, 2021; pp. 95–106. [Google Scholar]
- Salter, M.; Corfield, E.; Ramadass, A.; Grand, F.; Green, J.; Westra, J.; Lim, C.R.; Farrimond, L.; Feneberg, E.; Scaber, J.; et al. Initial identification of a blood-based chromosome conformation signature for aiding in the diagnosis of amyotrophic lateral sclerosis. EBioMedicine 2018, 33, 169–184. [Google Scholar] [CrossRef]
- Zou, H.; Hastie, T. Regularization and variable selection via the elastic net. J. R. Stat. Soc. Stat. Methodol. Ser. B 2005, 67, 301–320. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression shrinkage and selection via the Lasso. J. R. Stat. Soc. Ser. B Stat. Methodol. 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Hoerl, A.E.; Kennard, R.W. Ridge regression: Applications to nonorthogonal problems. Technometrics 1970, 12, 69–82. [Google Scholar] [CrossRef]
- Tiddi, I.; Schlobach, S. Knowledge graphs as tools for explainable machine learning: A survey. Artif. Intell. 2022, 302, 103627. [Google Scholar] [CrossRef]
- Zhao, E.; Stone, M.R.; Ren, X.; Guenthoer, J.; Smythe, K.S.; Pulliam, T.; Williams, S.R.; Uytingco, C.R.; Taylor, S.E.B.; Nghiem, P.; et al. Spatial transcriptomics at subspot resolution with BayesSpace. Nat. Biotechnol. 2021, 39, 1375–1384. [Google Scholar] [CrossRef]
- Hu-Lieskovan, S.; Braiteh, F.; Grilley-Olson, J.E.; Wang, X.; Forgie, A.; Bonato, V.; Jacobs, I.A.; Chou, J.; Johnson, M.L. Association of tumor mutational burden and immune gene expression with response to PD-1 blockade by sasanlimab across tumor types and routes of administration. Target. Oncol. 2021, 16, 773–787. [Google Scholar] [CrossRef]
- Motzer, R.J.; Robbins, P.B.; Powles, T.; Albiges, L.; Haanen, J.B.; Larkin, J.; Mu, X.J.; Ching, K.A.; Uemura, M.; Pal, S.K.; et al. Avelumab plus axitinib versus sunitinib in advanced renal cell carcinoma: Biomarker analysis of the phase 3 JAVELIN Renal 101 trial. Nat. Med. 2020, 26, 1733–1741. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Hunter, E.; Akoulitchev, A.; Park, P.; Winchester, D.J.; A Moo-Young, T.; A Prinz, R. Epigenetic chromatin conformation changes in peripheral blood can detect thyroid cancer. Surgery 2018, 165, 44–49. [Google Scholar] [CrossRef]
- Karlsson, M.; Zhang, C.; Méar, L.; Zhong, W.; Digre, A.; Katona, B.; Sjöstedt, E.; Butler, L.; Odeberg, J.; Dusart, P.; et al. A single-cell type transcriptomics map of human tissues. Sci. Adv. 2021, 7, eabh2169. [Google Scholar] [CrossRef]
- Tabula Sapiens Consortium; Jones, R.C.; Karkanias, J.; Krasnow, M.A.; Pisco, A.O.; Quake, S.R.; Salzman, J.; Yosef, N.; Bulthaup, B.; Brown, P.; et al. The Tabula Sapiens: A multiple-organ, single-cell transcriptomic atlas of humans. Science 2022, 376, eabl4896. [Google Scholar]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef]
- Higgs, B.W.; Morehouse, C.A.; Streicher, K.; Brohawn, P.Z.; Pilataxi, F.; Gupta, A.; Ranade, K. Interferon gamma messenger RNA signature in tumor biopsies predicts outcomes in patients with non-small cell lung carcinoma or urothelial cancer treated with durvalumab. Clin. Cancer Res. 2018, 24, 3857–3866. [Google Scholar] [CrossRef]
- Fehrenbacher, L.; Spira, A.; Ballinger, M.; Kowanetz, M.; Vansteenkiste, J.; Mazieres, J.; Park, K.; Smith, D.; Artal-Cortes, A.; Lewanski, C.; et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): A multicentre, open-label, phase 2 randomised controlled trial. Lancet 2016, 387, 1837–1846. [Google Scholar] [CrossRef]
- Eckstein, M.; Strissel, P.; Strick, R.; Weyerer, V.; Wirtz, R.; Pfannstiel, C.; Wullweber, A.; Lange, F.; Erben, P.; Stoehr, R.; et al. Cytotoxic T-cell-related gene expression signature predicts improved survival in muscle-invasive urothelial bladder cancer patients after radical cystectomy and adjuvant chemotherapy. J. Immunother. Cancer 2020, 8, e000162. [Google Scholar] [CrossRef]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef]
- Wiedemann, G.M.; Geary, C.D.; Lau, C.M.; Sun, J.C. Cutting edge: STAT1-mediated epigenetic control of Rsad2 promotes clonal expansion of antiviral NK cells. J. Immunol. 2020, 205, 21–25. [Google Scholar] [CrossRef]
- Arnold, N.; Girke, T.; Sureshchandra, S.; Nguyen, C.; Rais, M.; Messaoudi, I. Genomic and functional analysis of the host response to acute simian varicella infection in the lung. Sci. Rep. 2016, 6, srep34164. [Google Scholar] [CrossRef]
- Larsson, I.; Uhlén, M.; Zhang, C.; Mardinoglu, A. Genome-scale metabolic modeling of glioblastoma reveals promising targets for drug development. Front. Genet. 2020, 11, 381. [Google Scholar] [CrossRef]
- Ho, S.-R.; Lee, Y.-C.; Ittmann, M.M.; Lin, F.-T.; Chan, K.S.; Lin, W.-C. RNF144A deficiency promotes PD-L1 protein stabilization and carcinogen-induced bladder tumorigenesis. Cancer Lett. 2021, 520, 344–360. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-L.; Zhang, Y.; Li, D.-D.; Zhang, F.-L.; Liu, H.-Y.; Liao, X.-H.; Xie, H.-Y.; Lu, Q.; Zhang, L.; Hong, Q.; et al. RNF144A functions as a tumor suppressor in breast cancer through ubiquitin ligase activity-dependent regulation of stability and oncogenic functions of HSPA2. Cell Death Differ. 2019, 27, 1105–1118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Q.; Yang, Y.; Wang, Q. Association between succinate receptor SUCNR1 expression and immune infiltrates in ovarian cancer. Front. Mol. Biosci. 2020, 7, 150. [Google Scholar] [CrossRef]
- Najm, R.; Hachim, M.Y.; Kandasamy, R.K. Divulging a pleiotropic role of succinate receptor SUCNR1 in renal cell carcinoma microenvironment. Cancers 2022, 14, 6064. [Google Scholar] [CrossRef] [PubMed]
- Hodson, D.J.; Shaffer, A.L.; Xiao, W.; Wright, G.W.; Schmitz, R.; Phelan, J.D.; Yang, Y.; Webster, D.E.; Rui, L.; Kohlhammer, H.; et al. Regulation of normal B-cell differentiation and malignant B-cell survival by OCT2. Proc. Natl. Acad. Sci. USA 2016, 113, E2039–E2046. [Google Scholar] [CrossRef]
- Corcoran, L.; Emslie, D.; Kratina, T.; Shi, W.; Hirsch, S.; Taubenheim, N.; Chevrier, S. Oct2 and Obf1 as facilitators of B:T cell collaboration during a humoral immune response. Front. Immunol. 2014, 5, 108. [Google Scholar] [CrossRef]
- Rodriguez, A.B.; Peske, J.D.; Woods, A.N.; Leick, K.M.; Mauldin, I.S.; Meneveau, M.O.; Young, S.J.; Lindsay, R.S.; Melssen, M.M.; Cyranowski, S.; et al. Immune mechanisms orchestrate tertiary lymphoid structures in tumors via cancer-associated fibroblasts. Cell Rep. 2021, 36, 109422. [Google Scholar] [CrossRef]
- Hennequin, A.; Derangère, V.; Boidot, R.; Apetoh, L.; Vincent, J.; Orry, D.; Fraisse, J.; Causeret, S.; Martin, F.; Arnould, L.; et al. Tumor infiltration by Tbet+ effector T cells and CD20+ B cells is associated with survival in gastric cancer patients. OncoImmunology 2016, 5, e1054598. [Google Scholar] [CrossRef]
- Konieczny, P.; Stepniak-Konieczna, E.; Sobczak, K. MBNL proteins and their target RNAs, interaction and splicing regulation. Nucleic Acids Res. 2014, 42, 10873–10887. [Google Scholar] [CrossRef]
- Sznajder, Ł.J.; Scotti, M.M.; Shin, J.; Taylor, K.; Ivankovic, F.; Nutter, C.A.; Aslam, F.N.; Subramony, S.H.; Ranum, L.P.W.; Swanson, M.S. Loss of MBNL1 induces RNA misprocessing in the thymus and peripheral blood. Nat. Commun. 2020, 11, 2022. [Google Scholar] [CrossRef] [PubMed]
- Delfín, D.A.; DeAguero, J.L.; McKown, E.N. The extracellular matrix protein ABI3BP in cardiovascular health and disease. Front. Cardiovasc. Med. 2019, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Han, X.; Zhang, Z.; Qiao, H.; Tang, H. ABI3BP is a prognosis biomarker related with clinicopathological features and immunity infiltration of lung tumor. Front. Genet. 2023, 13, 1085785. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Drouet, V.; Majounie, E.; Deramecourt, V.; Jacoupy, M.; Nicolas, A.; Cormier-Dequaire, F.; Hassoun, S.M.; Pujol, C.; Ciura, S.; et al. Loss of VPS13C function in autosomal-recessive parkinsonism causes mitochondrial dysfunction and increases PINK1/Parkin-dependent mitophagy. Am. J. Hum. Genet. 2016, 98, 500–513. [Google Scholar] [CrossRef]
- Wang, X.; Xu, P.; Bentley-DeSousa, A.; Hancock-Cerutti, W.; Cai, S.; Johnson, B.T.; Tonelli, F.; Shao, L.; Talaia, G.; Alessi, D.R.; et al. The bridge-like lipid transport protein VPS13C/PARK23 mediates ER-lysosome contacts following lysosome damage. Nat. Cell Biol. 2025, 27, 776–789. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Association Between Tumor Gene Expression and JAV-Immuno Score | Cell Type | Immune Compartment | Immune Process | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Gene Description | Statistic Value | p-Value | q-Value | Endothelial Cell | Fibroblast | B Cell | Monocyte | Natural Killer Cell | Dendritic Cell | Cytotoxic T Cell | Lymph Node | Germinal Center | MHC Class I Antigen Presentation | MHC Class II Antigen Presentation | Response to Stress |
| POU2F2 | POU class 2 homeobox 2 | 0.8141921 | 2.75 × 10−135 | 4.96 × 10−134 | 0.09011 | 0.2449 | 0.2558 | 0.2563 | 0.228 | 0.2125 | 0.02845 | 0.09261 | 0.09931 | 0 | 0.04072 | 0.004806 |
| MBNL1 | Muscle blind-like splicing regulator 1 | 0.6073972 | 2.31 × 10−58 | 2.77 × 10−57 | 0.2185 | 0.222 | 0.2186 | 0.2325 | 0.07344 | 0.04999 | 0.02845 | 0 | 0 | 0.008677 | 0.008505 | 0.1479 |
| ABI3BP | ABI family member 3 binding protein | 0.5517624 | 2.09 × 10−46 | 1.88 × 10−45 | 0.212 | 0.2435 | 0.2236 | 0.1964 | 0.1889 | 0.1974 | 0.02832 | 0.07382 | 0 | 0 | 0 | 0 |
| VPS13C | Vacuolar protein sorting 13 homolog C | 0.4595461 | 6.43 × 10−31 | 4.63 × 10−30 | 0.07422 | 0.1806 | 0.05065 | 0.06594 | 0.06424 | 0.05733 | 0.02213 | 0 | 0 | 0 | 0 | 0 |
| SUCNR1 | Succinate receptor 1 | 0.4390713 | 4.48 × 10−28 | 2.69 × 10−27 | 0.2147 | 0.2118 | 0.1985 | 0.2108 | 0.2092 | 0.208 | 0.15 | 0.09239 | 0 | 0.003451 | 0.00712 | 0.0858 |
| RSAD2 | Radical S-adenosyl methionine domain containing 2 | 0.3267927 | 1.49 × 10−15 | 6.72 × 10−15 | 0.2266 | 0.2148 | 0.234 | 0.2137 | 0.2234 | 0.229 | 0.1652 | 0.09274 | 0.07864 | 0 | 0.06117 | 0.1613 |
| CMPK2 | Cytidine/uridine monophosphate kinase 2 | 0.2787244 | 1.47 × 10−11 | 5.89 × 10−11 | 0.2066 | 0.2194 | 0.2099 | 0.2243 | 0.1865 | 0.2063 | 0.1437 | 0.07898 | 0.06812 | 0 | 0.0499 | 0.08855 |
| PI4KA | Phosphatidylinositol 4-kinase alpha | 0.152742 | 0.000265 | 0.000954 | 0.2101 | 0.2087 | 0.2186 | 0.1967 | 0.197 | 0.04704 | 0.02591 | 0 | 0 | 0.01603 | 0 | 0 |
| RNF144A | Ring finger protein 144A | 0.1471635 | 0.000444 | 0.00145 | 0.09449 | 0.2008 | 0.1847 | 0.1624 | 0.06401 | 0.1495 | 0.08942 | 0.07085 | 0.01967 | 0 | 0 | 0 |
| CPEB1 | Cytoplasmic polyadenylation element binding protein 1 | 0.1458114 | 0.000502 | 0.0015 | 0.1894 | 0.2084 | 0.1893 | 0.1863 | 0.07279 | 0.05963 | 0 | 0 | 0 | 0 | 0 | 0.01358 |
| NPY4R | Neuropeptide Y receptor Y4 | 0.1201768 | 0.00419 | 0.00944 | 0.04041 | 0.1401 | 0.1273 | 0.1208 | 0.1318 | 0.03498 | 0 | 0 | 0 | 0.003384 | 0 | 0 |
| ZNF573 | Zinc finger protein 573 | 0.1156841 | 0.00586 | 0.0113 | 0.06559 | 0.1173 | 0.1437 | 0.1506 | 0.1212 | 0.09401 | 0.04908 | 0.02246 | 0 | 0.005744 | 0 | 0 |
| SNAP29 | Synaptosome-associated protein 29 | 0.1154207 | 0.00598 | 0.0113 | 0.2049 | 0.1919 | 0.06076 | 0.06829 | 0.06407 | 0.04174 | 0.02845 | 0 | 0 | 0.006851 | 0.005367 | 0.1535 |
| ZNF781 | Zinc finger protein 781 | 0.1124934 | 0.00739 | 0.0121 | 0.1024 | 0.0865 | 0.1235 | 0.08621 | 0.09041 | 0.04321 | 0.005367 | 0.04858 | 0 | 0.003986 | 0.002624 | 0.000162 |
| SLC38A7 | Solute carrier family 38 member 7 | 0.0978701 | 0.0199 | 0.0286 | 0.1976 | 0.05722 | 0.192 | 0.06142 | 0.05257 | 0.03522 | 0.02277 | 0 | 0 | 0 | 0 | 0.08463 |
| DEDD2 | Death effector domain containing 2 | 0.0966533 | 0.0215 | 0.0297 | 0.08156 | 0.1777 | 0.1756 | 0.1643 | 0.1515 | 0.146 | 0.1052 | 0.06802 | 0 | 0 | 0.02047 | 0.06925 |
| C2CD4B | C2 calcium-dependent domain containing 4B | 0.0826085 | 0.0495 | 0.066 | 0.2162 | 0.1775 | 0.154 | 0.1431 | 0.1367 | 0.05737 | 0.01532 | 0.0178 | 0 | 0 | 0 | 0 |
| TMEM14E | Transmembrane protein 14E, pseudogene | 0.0678954 | 0.107 | 0.137 | 0.03418 | 0.0232 | 0.0338 | 0.03111 | 0.04544 | 0.02271 | 0.004312 | 0 | 0 | 0 | 0 | 0 |
| MMP16 | Matrix metallopeptidase 16 | 0.0639516 | 0.129 | 0.16 | 0.2134 | 0.2523 | 0.2056 | 0.2038 | 0.2085 | 0.06526 | 0.007903 | 0.09234 | 0 | 0 | 0 | 0 |
| LZTR1 | Leucine zipper-like transcription regulator 1 | 0.0550683 | 0.191 | 0.222 | 0.2021 | 0.1965 | 0.06215 | 0.1714 | 0.2034 | 0.03179 | 0.01318 | 0.08179 | 0 | 0.007257 | 0.008901 | 0.006873 |
| RPS17 | Ribosomal protein S17 | 0.0165515 | 0.694 | 0.757 | 0.2079 | 0.183 | 0.2076 | 0.2269 | 0.2022 | 0.179 | 0.1641 | 0 | 0 | 0.03428 | 0.07347 | 0.1058 |
| CNBD1 | Cyclic nucleotide binding domain containing 1 | 0.0040582 | 0.923 | 0.923 | 0.1355 | 0.1114 | 0.1151 | 0.1164 | 0.1073 | 0.09581 | 0.01741 | 0.04782 | 0 | 0.003208 | 0.003264 | 0.00432 |
| C2CD4A | C2 calcium-dependent domain containing 4A | −0.004789 | 0.909 | 0.923 | 0.171 | 0.1587 | 0.1331 | 0.1213 | 0.1156 | 0.05684 | 0.004867 | 0.04788 | 0 | 0 | 0.003982 | 0.08443 |
| DCAF4L2 | DDB1 and CUL4-associated factor 4-like 2 | −0.008794 | 0.835 | 0.884 | 0.06324 | 0.03736 | 0.06659 | 0.09815 | 0.1282 | 0.05735 | 0 | 0.06016 | 0 | 0.00528 | 0.01013 | 0.00442 |
| ZNF526 | Zinc finger protein 526 | −0.048678 | 0.248 | 0.279 | 0.1291 | 0.1437 | 0.07571 | 0.06573 | 0.04032 | 0.03047 | 0.004803 | 0 | 0 | 0 | 0 | 0 |
| CNOT1 | CCR4-NOT transcription complex subunit 1 | −0.055119 | 0.19 | 0.222 | 0.0678 | 0.21 | 0.2023 | 0.2026 | 0.03454 | 0.1721 | 0.01716 | 0 | 0 | 0.01848 | 0.003042 | 0.09112 |
| ZFP30 | ZFP30 zinc finger protein | −0.098922 | 0.0186 | 0.0279 | 0.1219 | 0.1227 | 0.1167 | 0.1308 | 0.1257 | 0.07336 | 0.02253 | 0 | 0 | 0 | 0 | 0 |
| ZNF607 | Zinc finger protein 607 | −0.110554 | 0.00848 | 0.0133 | 0.04603 | 0.0471 | 0.06989 | 0.08244 | 0.1002 | 0.03778 | 0.0391 | 0.02276 | 0 | 0.004409 | 0.003307 | 0.01889 |
| GPRIN2 | G protein-regulated inducer of neurite outgrowth 2 | −0.112585 | 0.00734 | 0.0121 | 0.06067 | 0.1465 | 0.1516 | 0.03462 | 0.1517 | 0.04801 | 0.00213 | 0.05896 | 0.02013 | 0 | 0.01973 | 0.00166 |
| TFG | Trafficking from ER to golgi regulator | −0.114673 | 0.00631 | 0.0114 | 0.2094 | 0.1879 | 0.2205 | 0.2067 | 0.06115 | 0.02826 | 0.02845 | 0.08952 | 0 | 0.003158 | 0.00423 | 0.005551 |
| SERPIND1 | Serpin family D member 1 | −0.116083 | 0.00569 | 0.0113 | 0.2145 | 0.1994 | 0.189 | 0.1905 | 0.05945 | 0.1672 | 0 | 0.09033 | 0 | 0 | 0.004157 | 0.1498 |
| CRKL | CRK-like proto-oncogene, adaptor protein | −0.130329 | 0.00189 | 0.00454 | 0.2009 | 0.2036 | 0.2249 | 0.1969 | 0.2029 | 0.1667 | 0.0242 | 0.09401 | 0 | 0.02339 | 0.02136 | 0.02151 |
| AIFM3 | Apoptosis-inducing factor mitochondria-associated 3 | −0.138936 | 0.000919 | 0.00236 | 0.19 | 0.1729 | 0.05 | 0.06931 | 0.0611 | 0.05562 | 0.006126 | 0.08887 | 0 | 0.0107 | 0.004403 | 0.1262 |
| ANXA8L1 | Annexin A8-like 1 | −0.14415 | 0.000582 | 0.00161 | 0.1466 | 0.1922 | 0.1392 | 0.1212 | 0.1354 | 0.1276 | 0.02692 | 0.0607 | 0 | 0 | 0 | 0 |
| GOT2 | Glutamic-oxaloacetic transaminase 2 | −0.381291 | 5.01 × 10−21 | 2.58 × 10−20 | 0.2346 | 0.2296 | 0.2188 | 0.2172 | 0.07141 | 0.1993 | 0.1728 | 0.09115 | 0.07472 | 0 | 0.006362 | 0.09641 |
| Treatment | TMB | POU2F2 Marker | No. of Patients (N = 457) | No. of Events | OS, Median (95% CI), Months | HR (POU2F2 Marker Absent vs. Present) (95% CI) | p-Value |
|---|---|---|---|---|---|---|---|
| Avelumab plus BSC | ≤Median | Absent | 22 | 10 | 36.99 (18.17–NE) | 0.46 (0.240–0.894) | 0.0218 |
| Avelumab plus BSC | ≤Median | Present | 112 | 82 | 17.77 (13.34–22.34) | ||
| Avelumab plus BSC | >Median | Absent | 11 | 7 | 19.25 (17.81–NE) | 1.38 (0.623–3.048) | 0.4281 |
| Avelumab plus BSC | >Median | Present | 98 | 48 | 35.12 (26.05–NE) | ||
| BSC alone | ≤Median | Absent | 18 | 13 | 13.68 (8.8–NE) | 1.14 (0.622–2.071) | 0.6788 |
| BSC alone | ≤Median | Present | 89 | 60 | 16.07 (10.25–24.18) | ||
| BSC alone | >Median | Absent | 21 | 14 | 14.78 (11.5–NE) | 1.14 (0.635–2.044) | 0.6608 |
| BSC alone | >Median | Present | 86 | 58 | 17.81 (13.54–26.64) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Powles, T.; Sridhar, S.S.; Bellmunt, J.; Sternberg, C.N.; Grivas, P.; Hunter, E.; Salter, M.; Powell, R.; Dring, A.; Green, J.; et al. Blood-Epigenetic Biomarker Associations with Tumor Immunophenotype in Patients with Urothelial Carcinoma from JAVELIN Bladder 100. Cancers 2025, 17, 2332. https://doi.org/10.3390/cancers17142332
Powles T, Sridhar SS, Bellmunt J, Sternberg CN, Grivas P, Hunter E, Salter M, Powell R, Dring A, Green J, et al. Blood-Epigenetic Biomarker Associations with Tumor Immunophenotype in Patients with Urothelial Carcinoma from JAVELIN Bladder 100. Cancers. 2025; 17(14):2332. https://doi.org/10.3390/cancers17142332
Chicago/Turabian StylePowles, Thomas, Srikala S. Sridhar, Joaquim Bellmunt, Cora N. Sternberg, Petros Grivas, Ewan Hunter, Matthew Salter, Ryan Powell, Ann Dring, Jayne Green, and et al. 2025. "Blood-Epigenetic Biomarker Associations with Tumor Immunophenotype in Patients with Urothelial Carcinoma from JAVELIN Bladder 100" Cancers 17, no. 14: 2332. https://doi.org/10.3390/cancers17142332
APA StylePowles, T., Sridhar, S. S., Bellmunt, J., Sternberg, C. N., Grivas, P., Hunter, E., Salter, M., Powell, R., Dring, A., Green, J., Akoulitchev, A., Ronen, R., Dutkowski, J., Amezquita, R., Huang, C.-H., Fernandez, D., Nameki, R., Ching, K. A., Pu, J., ... Davis, C. B. (2025). Blood-Epigenetic Biomarker Associations with Tumor Immunophenotype in Patients with Urothelial Carcinoma from JAVELIN Bladder 100. Cancers, 17(14), 2332. https://doi.org/10.3390/cancers17142332