

Establishment of Human Lung Cancer Organoids Using Small Biopsy and Surgical Tissues
 , ,
, ,  ,
, 
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Human Tissues
2.2. Establishment of PDOs
2.3. PBMC Isolation
2.4. IHC Staining of Paraffin-Embedded Organoids
2.5. Targeted Exome Sequencing and EGFR Mutation Analyses
2.6. Xenograft Establishment Using PDTOs
2.7. Culture of PDX Cells
2.8. High-Throughput Drug Screening Using PDTOs
2.9. GFP Lentivirus Transfection of PDTOs
2.10. Co-Culture of PDTOs with CAFs and Immune Cells
2.11. Statistical Analysis
2.12. Data Availability
3. Results
3.1. Clinical Characteristics of the Study Subjects
3.2. Establishment of PDOs and PDTOs
3.3. Factors Associated with Successful PDTOs Establishment
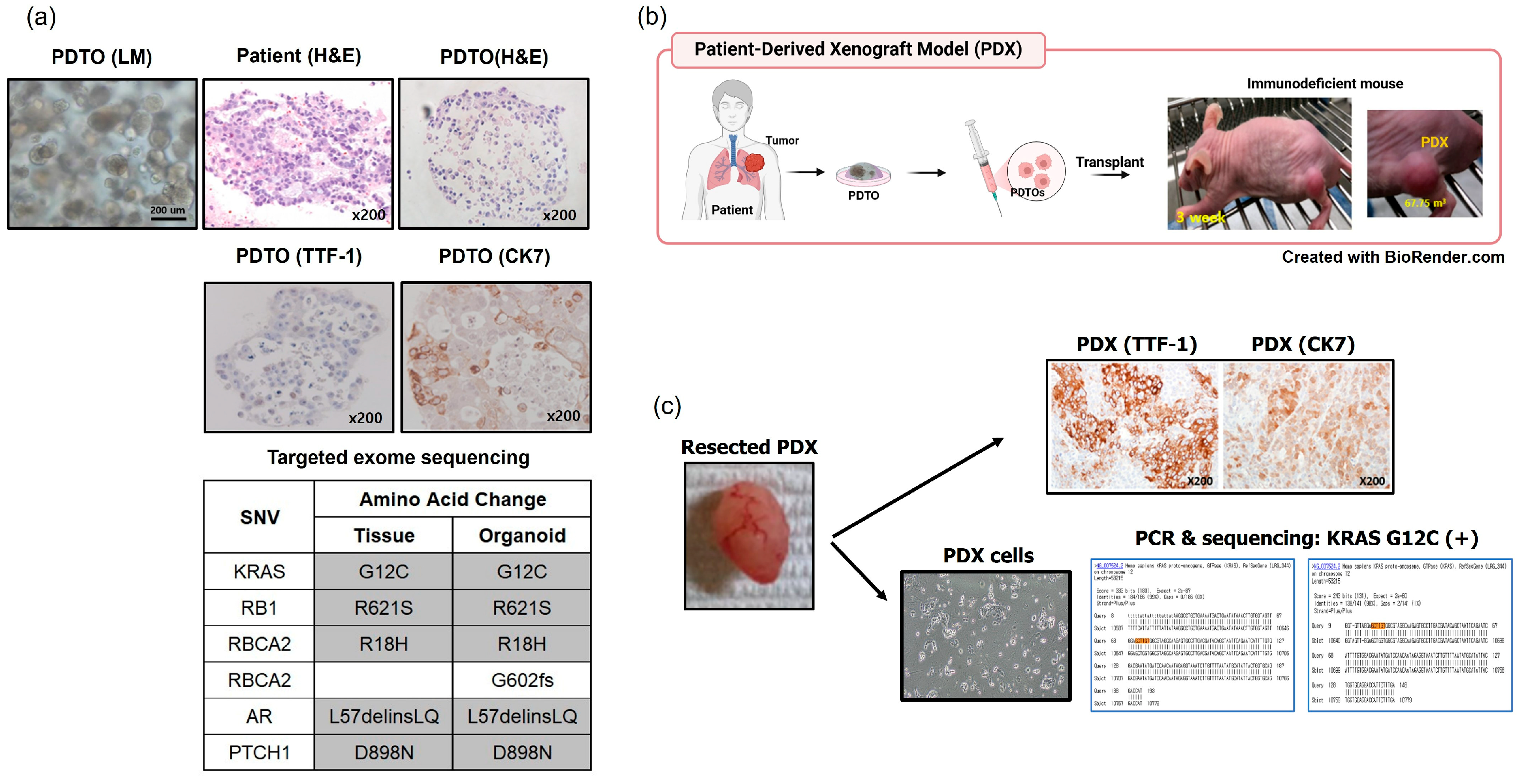
3.4. In Vivo Tumorigenesis Assays of PDTOs Using Patient-Derived Xenografts (PDXs)
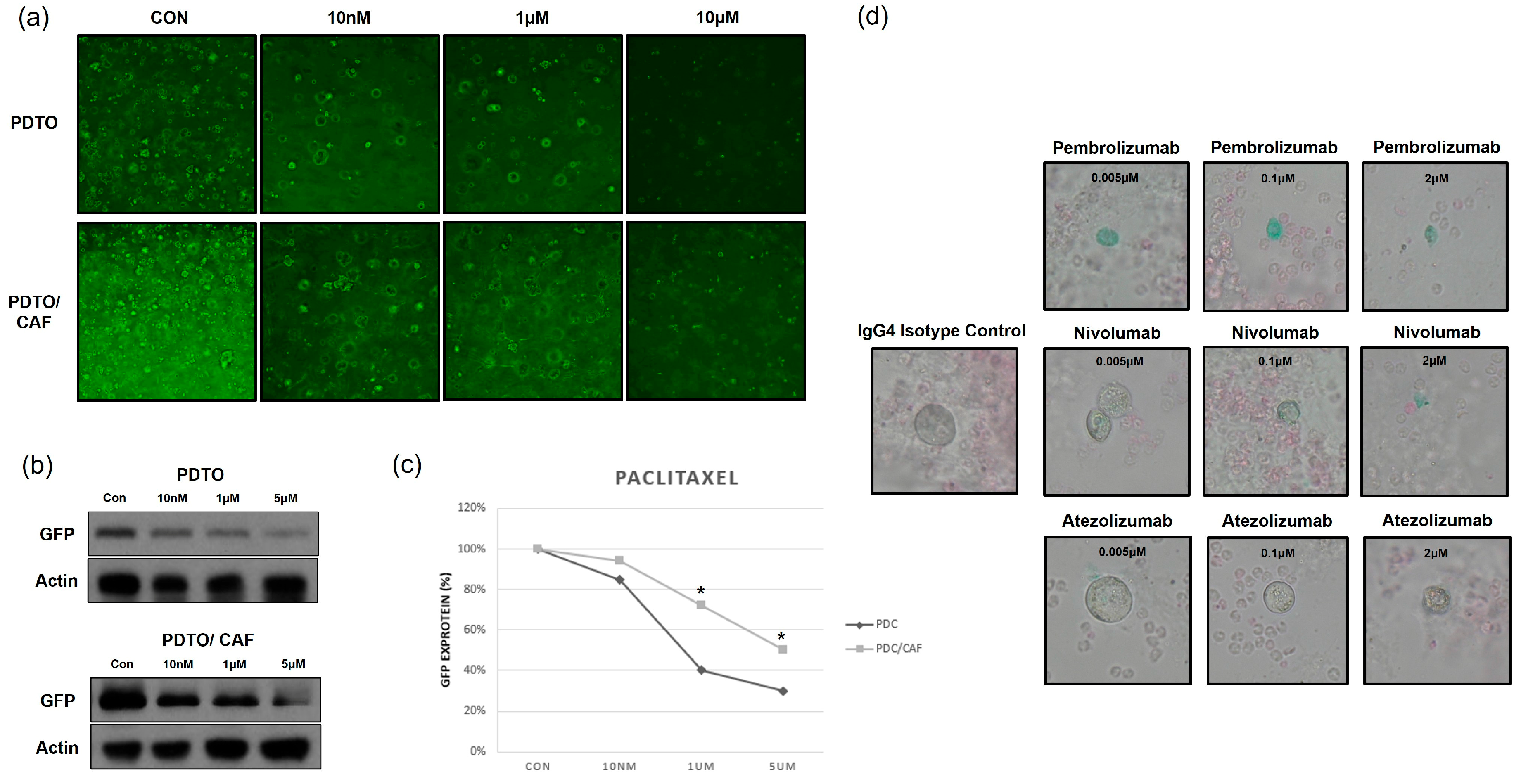
3.5. PDTO-Based High-Throughput Drug Screening
3.6. Co-Culture of PDTOs and Cancer-Associated Fibroblasts (CAFs) or Immune Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 1 July 2023).
- Sharma, R. Mapping of global, regional and national incidence, mortality and mortality-to-incidence ratio of lung cancer in 2020 and 2050. Int. J. Clin. Oncol. 2022, 27, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Rossi, R.; De Angelis, M.L.; Xhelili, E.; Sette, G.; Eramo, A.; De Maria, R.; Cesta Incani, U.; Francescangeli, F.; Zeuner, A. Lung Cancer Organoids: The Rough Path to Personalized Medicine. Cancers 2022, 14, 3703. [Google Scholar] [CrossRef]
- Bleijs, M.; van de Wetering, M.; Clevers, H.; Drost, J. Xenograft and organoid model systems in cancer research. Embo J. 2019, 38, e101654. [Google Scholar] [CrossRef] [PubMed]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef]
- Ramón, Y.C.S.; Sesé, M.; Capdevila, C.; Aasen, T.; De Mattos-Arruda, L.; Diaz-Cano, S.J.; Hernández-Losa, J.; Castellví, J. Clinical implications of intratumor heterogeneity: Challenges and opportunities. J. Mol. Med. 2020, 98, 161–177. [Google Scholar] [CrossRef]
- Wu, F.; Fan, J.; He, Y.; Xiong, A.; Yu, J.; Li, Y.; Zhang, Y.; Zhao, W.; Zhou, F.; Li, W.; et al. Single-cell profiling of tumor heterogeneity and the microenvironment in advanced non-small cell lung cancer. Nat. Commun. 2021, 12, 2540. [Google Scholar] [CrossRef]
- Hynds, R.E.; Frese, K.K.; Pearce, D.R.; Grönroos, E.; Dive, C.; Swanton, C. Progress towards non-small-cell lung cancer models that represent clinical evolutionary trajectories. Open Biol. 2021, 11, 200247. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhao, J.; Zhang, Y.; Li, K.; Li, T.; Chen, X.; Zhao, S.; Zhao, S.; Liu, K.; Dong, Z. Establishment of lung cancer patient-derived xenograft models and primary cell lines for lung cancer study. J. Transl. Med. 2018, 16, 138. [Google Scholar] [CrossRef]
- Idrisova, K.F.; Simon, H.U.; Gomzikova, M.O. Role of Patient-Derived Models of Cancer in Translational Oncology. Cancers 2022, 15, 139. [Google Scholar] [CrossRef]
- Barkauskas, C.E.; Chung, M.I.; Fioret, B.; Gao, X.; Katsura, H.; Hogan, B.L. Lung organoids: Current uses and future promise. Development 2017, 144, 986–997. [Google Scholar] [CrossRef]
- Lee, S.H.; Hu, W.; Matulay, J.T.; Silva, M.V.; Owczarek, T.B.; Kim, K.; Chua, C.W.; Barlow, L.J.; Kandoth, C.; Williams, A.B.; et al. Tumor Evolution and Drug Response in Patient-Derived Organoid Models of Bladder Cancer. Cell 2018, 173, 515–528.e17. [Google Scholar] [CrossRef]
- Clevers, H. Modeling Development and Disease with Organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, G.; Ponnusamy, M.P.; Batra, S.K. Concise Review: Current Status of Three-Dimensional Organoids as Preclinical Models. Stem Cells 2018, 36, 1329–1340. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Heo, I.; Clevers, H. Disease Modeling in Stem Cell-Derived 3D Organoid Systems. Trends Mol. Med. 2017, 23, 393–410. [Google Scholar] [CrossRef]
- Mittal, R.; Woo, F.W.; Castro, C.S.; Cohen, M.A.; Karanxha, J.; Mittal, J.; Chhibber, T.; Jhaveri, V.M. Organ-on-chip models: Implications in drug discovery and clinical applications. J. Cell Physiol. 2019, 234, 8352–8380. [Google Scholar] [CrossRef]
- Gupta, N.; Liu, J.R.; Patel, B.; Solomon, D.E.; Vaidya, B.; Gupta, V. Microfluidics-based 3D cell culture models: Utility in novel drug discovery and delivery research. Bioeng. Transl. Med. 2016, 1, 63–81. [Google Scholar] [CrossRef]
- Lee, H.; Hwang, M.; Jang, S.; Um, S.W. Immune Regulatory Function of Cancer- Associated Fibroblasts in Non-small Cell Lung Cancer. Tuberc. Respir. Dis. 2023, 86, 304–318. [Google Scholar] [CrossRef]
- Euhus, D.M.; Hudd, C.; LaRegina, M.C.; Johnson, F.E. Tumor measurement in the nude mouse. J. Surg. Oncol. 1986, 31, 229–234. [Google Scholar] [CrossRef]
- Seino, T.; Kawasaki, S.; Shimokawa, M.; Tamagawa, H.; Toshimitsu, K.; Fujii, M.; Ohta, Y.; Matano, M.; Nanki, K.; Kawasaki, K.; et al. Human Pancreatic Tumor Organoids Reveal Loss of Stem Cell Niche Factor Dependence during Disease Progression. Cell Stem Cell 2018, 22, 454–467.e6. [Google Scholar] [CrossRef]
- van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef]
- Yu, J.; Huang, W. The Progress and Clinical Application of Breast Cancer Organoids. Int. J. Stem Cells 2020, 13, 295–304. [Google Scholar] [CrossRef]
- Gao, D.; Vela, I.; Sboner, A.; Iaquinta, P.J.; Karthaus, W.R.; Gopalan, A.; Dowling, C.; Wanjala, J.N.; Undvall, E.A.; Arora, V.K.; et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 2014, 159, 176–187. [Google Scholar] [CrossRef]
- Chari, A.; Kaufman, J.L.; Laubach, J.; Sborov, D.W.; Reeves, B.; Rodriguez, C.; Silbermann, R.; Costa, L.J.; Anderson, L.D., Jr.; Nathwani, N.; et al. Daratumumab in transplant-eligible patients with newly diagnosed multiple myeloma: Final analysis of clinically relevant subgroups in GRIFFIN. Blood Cancer J. 2024, 14, 107. [Google Scholar] [CrossRef]
- Miao, X.; Wang, C.; Chai, C.; Tang, H.; Hu, J.; Zhao, Z.; Luo, W.; Zhang, H.; Zhu, K.; Zhou, W.; et al. Establishment of gastric cancer organoid and its application in individualized therapy. Oncol. Lett. 2022, 24, 447. [Google Scholar] [CrossRef]
- Lee, D.; Kim, Y.; Chung, C. Scientific Validation and Clinical Application of Lung Cancer Organoids. Cells 2021, 10, 3012. [Google Scholar] [CrossRef]
- Sachs, N.; Papaspyropoulos, A.; Zomer-van Ommen, D.D.; Heo, I.; Bottinger, L.; Klay, D.; Weeber, F.; Huelsz-Prince, G.; Iakobachvili, N.; Amatngalim, G.D.; et al. Long-term expanding human airway organoids for disease modeling. EMBO J. 2019, 38, e100300. [Google Scholar] [CrossRef]
- Kim, M.; Mun, H.; Sung, C.O.; Cho, E.J.; Jeon, H.J.; Chun, S.M.; Jung, D.J.; Shin, T.H.; Jeong, G.S.; Kim, D.K.; et al. Patient-derived lung cancer organoids as in vitro cancer models for therapeutic screening. Nat. Commun. 2019, 10, 3991. [Google Scholar] [CrossRef]
- Shi, R.; Radulovich, N.; Ng, C.; Liu, N.; Notsuda, H.; Cabanero, M.; Martins-Filho, S.N.; Raghavan, V.; Li, Q.; Mer, A.S.; et al. Organoid Cultures as Preclinical Models of Non-Small Cell Lung Cancer. Clin. Cancer Res. 2020, 26, 1162–1174. [Google Scholar] [CrossRef]
- Palma, G.; Khurshid, F.; Lu, K.; Woodward, B.; Husain, H. Selective KRAS G12C inhibitors in non-small cell lung cancer: Chemistry, concurrent pathway alterations, and clinical outcomes. NPJ Precis. Oncol. 2021, 5, 98. [Google Scholar] [CrossRef] [PubMed]
- Endo, H.; Okami, J.; Okuyama, H.; Kumagai, T.; Uchida, J.; Kondo, J.; Takehara, T.; Nishizawa, Y.; Imamura, F.; Higashiyama, M.; et al. Spheroid culture of primary lung cancer cells with neuregulin 1/HER3 pathway activation. J. Thorac. Oncol. 2013, 8, 131–139. [Google Scholar] [CrossRef]
- Dijkstra, K.K.; Monkhorst, K.; Schipper, L.J.; Hartemink, K.J.; Smit, E.F.; Kaing, S.; de Groot, R.; Wolkers, M.C.; Clevers, H.; Cuppen, E.; et al. Challenges in Establishing Pure Lung Cancer Organoids Limit Their Utility for Personalized Medicine. Cell Rep. 2020, 31, 107588. [Google Scholar] [CrossRef]
- Sachs, N.; de Ligt, J.; Kopper, O.; Gogola, E.; Bounova, G.; Weeber, F.; Balgobind, A.V.; Wind, K.; Gracanin, A.; Begthel, H.; et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell 2018, 172, 373–386.e10. [Google Scholar] [CrossRef] [PubMed]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernández-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef]
- Shinozaki, T.; Togasaki, K.; Hamamoto, J.; Mitsuishi, A.; Fukushima, T.; Sugihara, K.; Ebisudani, T.; Okada, M.; Saito, A.; Shigematsu, L.; et al. Basal-shift transformation leads to EGFR therapy-resistance in human lung adenocarcinoma. Nat. Commun. 2025, 16, 4369. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Lee, D.; Kim, Y.; Park, Y.; Lee, Y.J.; Lee, J.E.; Yeo, M.K.; Kang, M.W.; Chong, Y.; Han, S.J.; et al. Cryobiopsy: A Breakthrough Strategy for Clinical Utilization of Lung Cancer Organoids. Cells 2023, 12, 1854. [Google Scholar] [CrossRef] [PubMed]
- Vilgelm, A.E.; Bergdorf, K.; Wolf, M.; Bharti, V.; Shattuck-Brandt, R.; Blevins, A.; Jones, C.; Phifer, C.; Lee, M.; Lowe, C.; et al. Fine-Needle Aspiration-Based Patient-Derived Cancer Organoids. iScience 2020, 23, 101408. [Google Scholar] [CrossRef]
- Ma, H.C.; Zhu, Y.J.; Zhou, R.; Yu, Y.Y.; Xiao, Z.Z.; Zhang, H.B. Lung cancer organoids, a promising model still with long way to go. Crit. Rev. Oncol. Hematol. 2022, 171, 103610. [Google Scholar] [CrossRef]
- Hu, Y.; Sui, X.; Song, F.; Li, Y.; Li, K.; Chen, Z.; Yang, F.; Chen, X.; Zhang, Y.; Wang, X.; et al. Lung cancer organoids analyzed on microwell arrays predict drug responses of patients within a week. Nat. Commun. 2021, 12, 2581. [Google Scholar] [CrossRef]
- Kim, S.Y.; Kim, S.M.; Lim, S.; Lee, J.Y.; Choi, S.J.; Yang, S.D.; Yun, M.R.; Kim, C.G.; Gu, S.R.; Park, C.; et al. Modeling Clinical Responses to Targeted Therapies by Patient-Derived Organoids of Advanced Lung Adenocarcinoma. Clin. Cancer Res. 2021, 27, 4397–4409. [Google Scholar] [CrossRef]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988.e16. [Google Scholar] [CrossRef]
- Thangam, T.; Parthasarathy, K.; Supraja, K.; Haribalaji, V.; Sounderrajan, V.; Rao, S.S.; Jayaraj, S. Lung Organoids: Systematic Review of Recent Advancements and its Future Perspectives. Tissue Eng. Regen. Med. 2024, 21, 653–671. [Google Scholar] [CrossRef] [PubMed]
- Long, X.; Xiong, W.; Zeng, X.; Qi, L.; Cai, Y.; Mo, M.; Jiang, H.; Zhu, B.; Chen, Z.; Li, Y. Cancer-associated fibroblasts promote cisplatin resistance in bladder cancer cells by increasing IGF-1/ERbeta/Bcl-2 signalling. Cell Death Dis. 2019, 10, 375. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Song, E. Turning foes to friends: Targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 2019, 18, 99–115. [Google Scholar] [CrossRef]
- Forsythe, S.D.; Erali, R.A.; Sasikumar, S.; Laney, P.; Shelkey, E.; D’Agostino, R., Jr.; Miller, L.D.; Shen, P.; Levine, E.A.; Soker, S.; et al. Organoid Platform in Preclinical Investigation of Personalized Immunotherapy Efficacy in Appendiceal Cancer: Feasibility Study. Clin. Cancer Res. 2021, 27, 5141–5150. [Google Scholar] [CrossRef] [PubMed]
- Dijkstra, K.K.; Cattaneo, C.M.; Weeber, F.; Chalabi, M.; van de Haar, J.; Fanchi, L.F.; Slagter, M.; van der Velden, D.L.; Kaing, S.; Kelderman, S.; et al. Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 2018, 174, 1586–1598.e12. [Google Scholar] [CrossRef]
- Ringel, T.; Frey, N.; Ringnalda, F.; Janjuha, S.; Cherkaoui, S.; Butz, S.; Srivatsa, S.; Pirkl, M.; Russo, G.; Villiger, L.; et al. Genome-Scale CRISPR Screening in Human Intestinal Organoids Identifies Drivers of TGF-β Resistance. Cell Stem Cell 2020, 26, 431–440.e8. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Number (%) or Mean ± SD |
|---|---|
| Age, years | 64.0 ± 9.3 |
| Male sex | 128 (78.5) |
| Method used to acquire tissue specimens | |
| EBUS-TBNA of metastatic lymph nodes | 100 (61.3) |
| Surgery on the primary tumor | 52 (31.9) |
| Bronchoscopy or EBUS-TBNA of the primary tumor | 9 (5.5) |
| Thoracentesis of malignant pleural effusions | 2 (1.2) |
| Histology | |
| Adenocarcinoma | 97 (59.5) |
| Squamous cell carcinoma | 36 (22.1) |
| Other NSCLC | 4 (1.2) |
| SCLC | 26 (15.9) |
| Stage | |
| I | 24 (14.7) |
| II | 18 (11.0) |
| III | 49 (30.0) |
| IV | 72 (44.2) |
| Establishment Rate | N (%) |
|---|---|
| PDOs | 56 (34.4%) |
| PDTOs | 25 (15.3%) |
| All confirmed with IHC staining and sequencing | |
| PDBOs | 25 (15.3%) |
| IHC staining results not compatible with the tumor histology | 7 (4.3%) |
| Sequencing results not compatible with patients’ tumors | 18 (11.1%) |
| Indeterminate organoids | 6 (3.6%) |
| Passage Failure; it was not possible to obtain sufficient cells for | |
| IHC staining or sequencing |
| Characteristics | Number (%) | p-Value |
|---|---|---|
| Histology | 0.976 | |
| Adenocarcinoma (N = 97) | 16 (16.4) | |
| Squamous cell carcinoma (N = 36) | 5 (13.9) | |
| Other NSCLC (N = 4) | 0 (0) | |
| SCLC (N = 26) | 4 (15.4) | |
| Tissue acquisition methods | 0.060 | |
| EBUS-TBNA of metastatic lymph nodes (N = 100) | 10 (10.0) | |
| Bronchoscopy or EBUS-TBNA of primary tumor (N = 9) | 3 (33.3) | |
| Surgery on primary tumor (N = 52) | 12 (23.1) | |
| Thoracentesis of malignant pleural effusions (N = 2) | 0 (0) | |
| Genotypes | 0.046 | |
| EGFR mutation (L858R, exon 19 del, exon 20 insertion, T790M) (+) (N = 31) | 4 (12.9) | |
| Other genetic alterations (N = 61) | 21 (34.4) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hwang, M.; Choe, J.; Shin, Y.J.; Seo, B.-G.; Park, K.-M.; Shin, S.H.; Jhun, B.W.; Yoo, H.; Jeong, B.-H.; Jeon, K.; et al. Establishment of Human Lung Cancer Organoids Using Small Biopsy and Surgical Tissues. Cancers 2025, 17, 2291. https://doi.org/10.3390/cancers17142291
Hwang M, Choe J, Shin YJ, Seo B-G, Park K-M, Shin SH, Jhun BW, Yoo H, Jeong B-H, Jeon K, et al. Establishment of Human Lung Cancer Organoids Using Small Biopsy and Surgical Tissues. Cancers. 2025; 17(14):2291. https://doi.org/10.3390/cancers17142291
Chicago/Turabian StyleHwang, Mina, Junsu Choe, Yong Jae Shin, Bo-Gyeong Seo, Kyung-Mi Park, Sun Hye Shin, Byung Woo Jhun, Hongseok Yoo, Byeong-Ho Jeong, Kyeongman Jeon, and et al. 2025. "Establishment of Human Lung Cancer Organoids Using Small Biopsy and Surgical Tissues" Cancers 17, no. 14: 2291. https://doi.org/10.3390/cancers17142291
APA StyleHwang, M., Choe, J., Shin, Y. J., Seo, B.-G., Park, K.-M., Shin, S. H., Jhun, B. W., Yoo, H., Jeong, B.-H., Jeon, K., Lee, K., Lee, J., Jeon, Y. J., Cho, J. H., Park, S. Y., Kim, H. K., & Um, S.-W. (2025). Establishment of Human Lung Cancer Organoids Using Small Biopsy and Surgical Tissues. Cancers, 17(14), 2291. https://doi.org/10.3390/cancers17142291