
Telomere Maintenance and DNA Repair: A Bidirectional Relationship in Cancer Biology and Therapy

 , , , , , , and
, , , , , , and 
Simple Summary
Abstract
1. Introduction
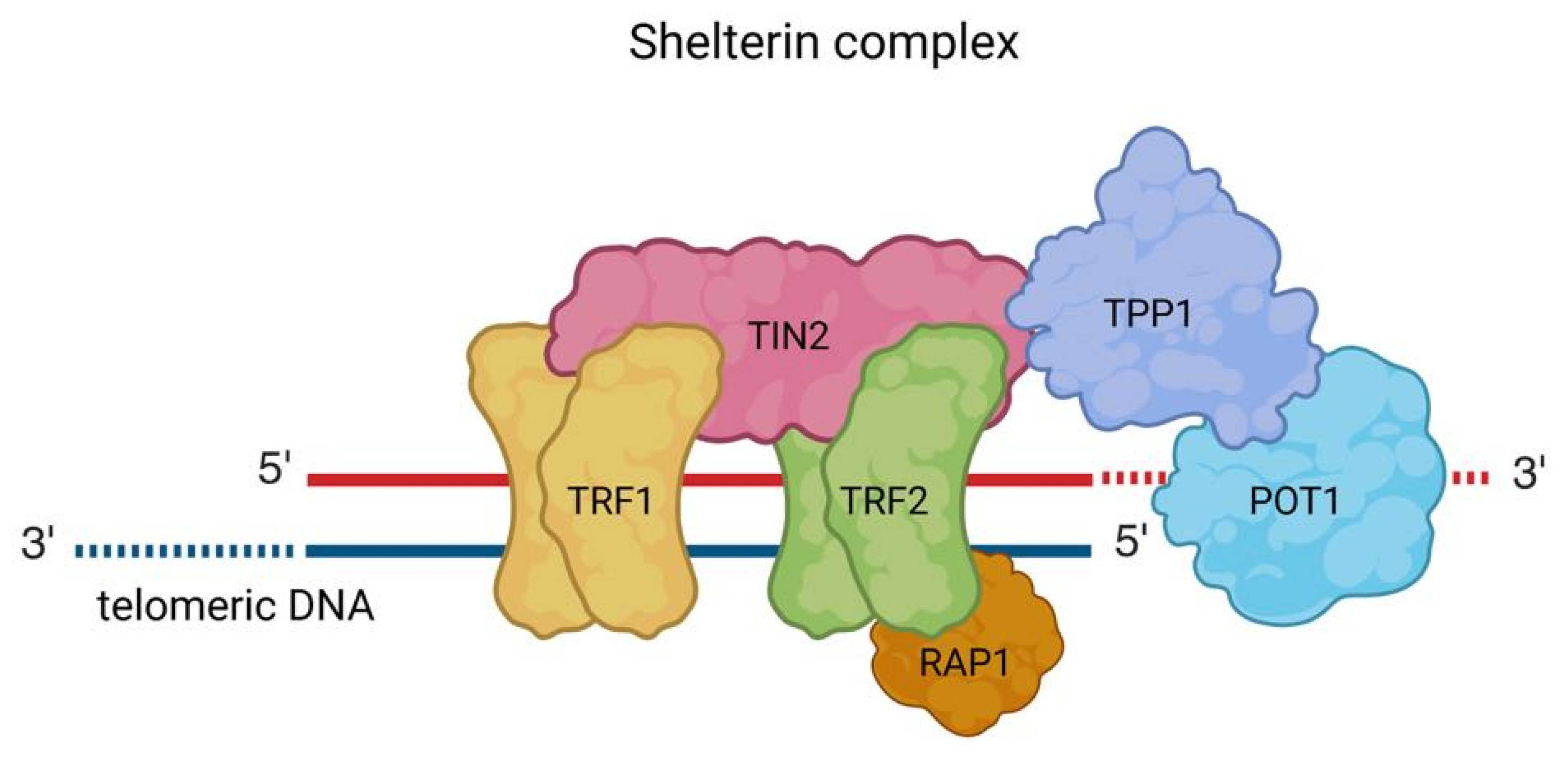
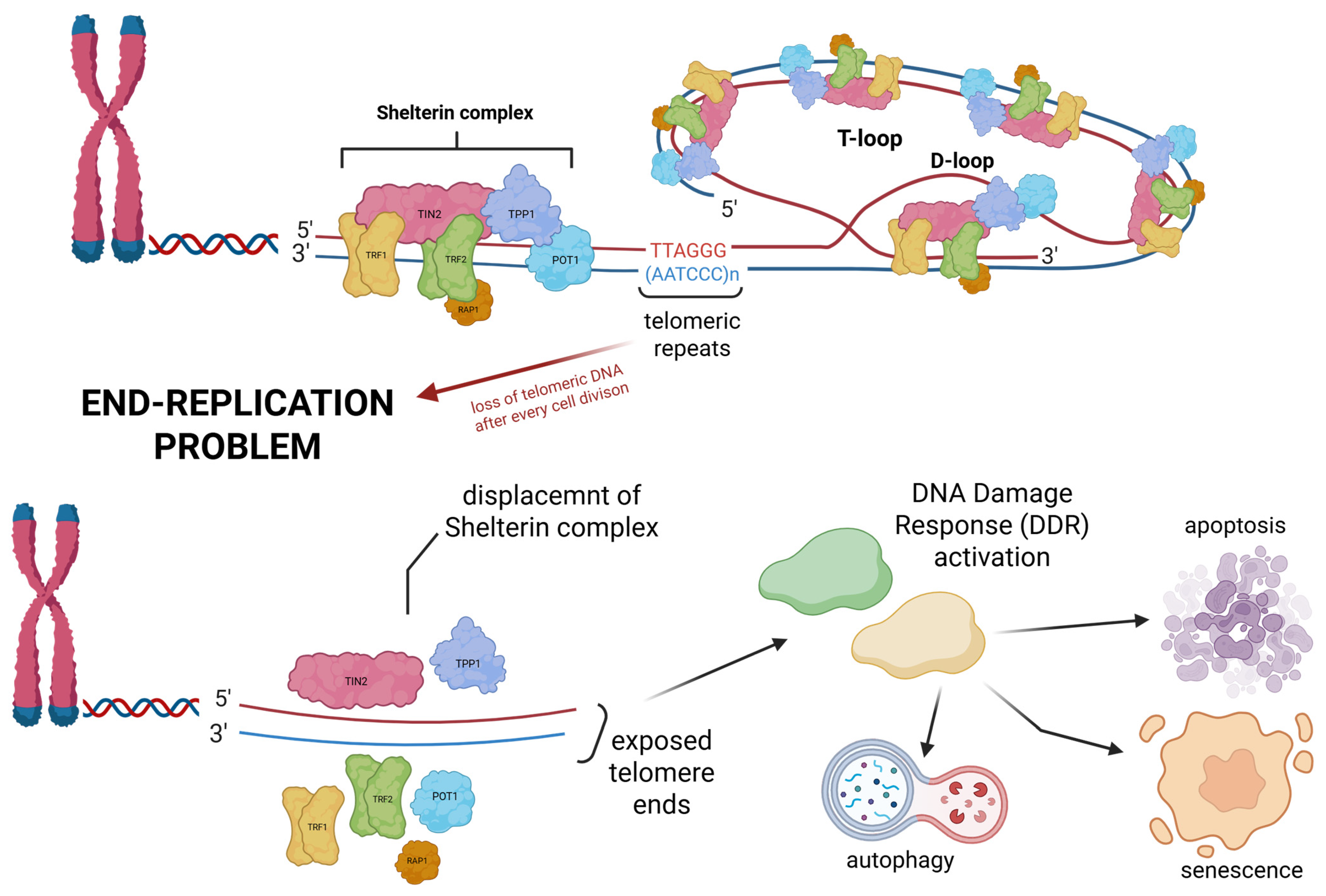
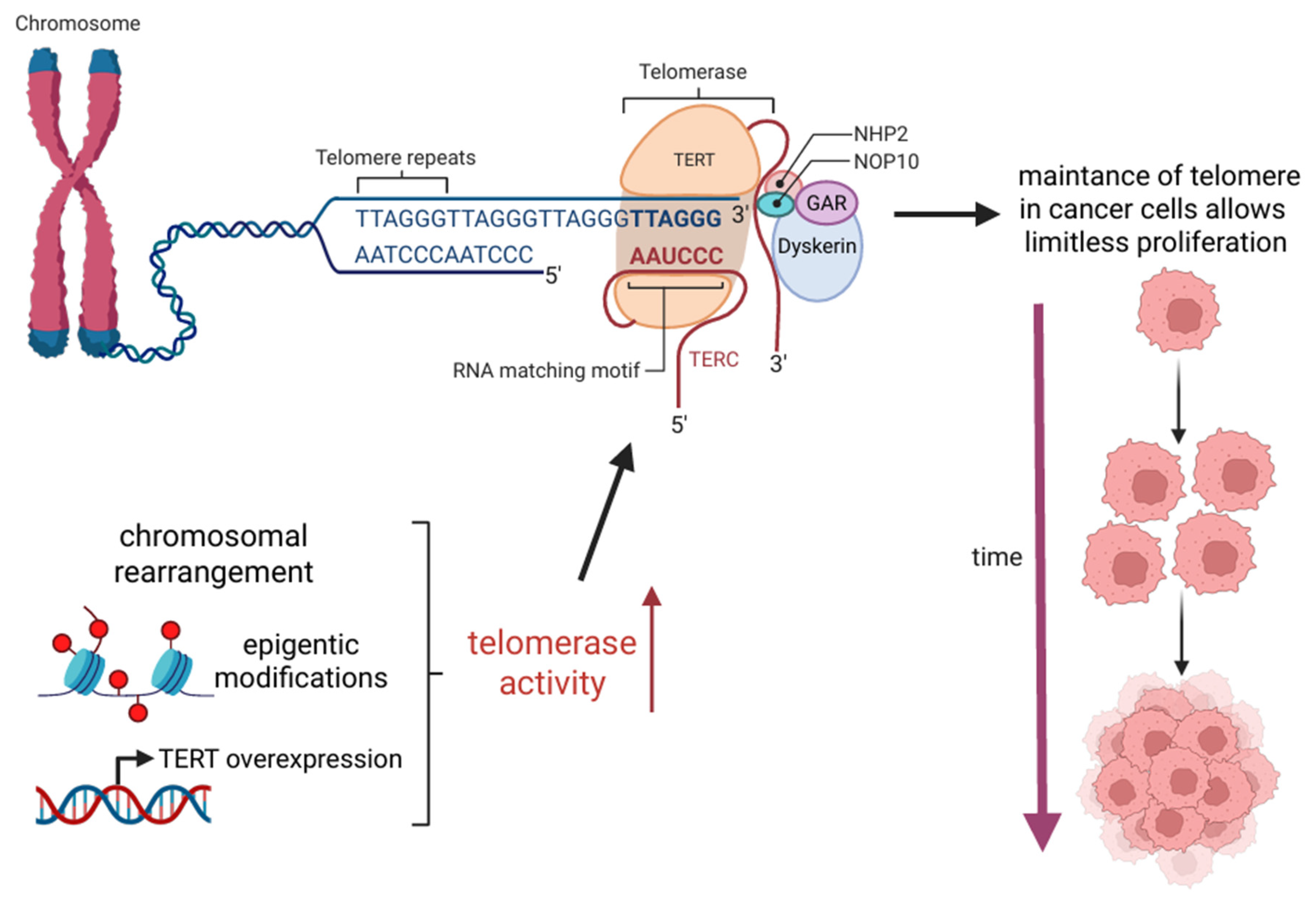
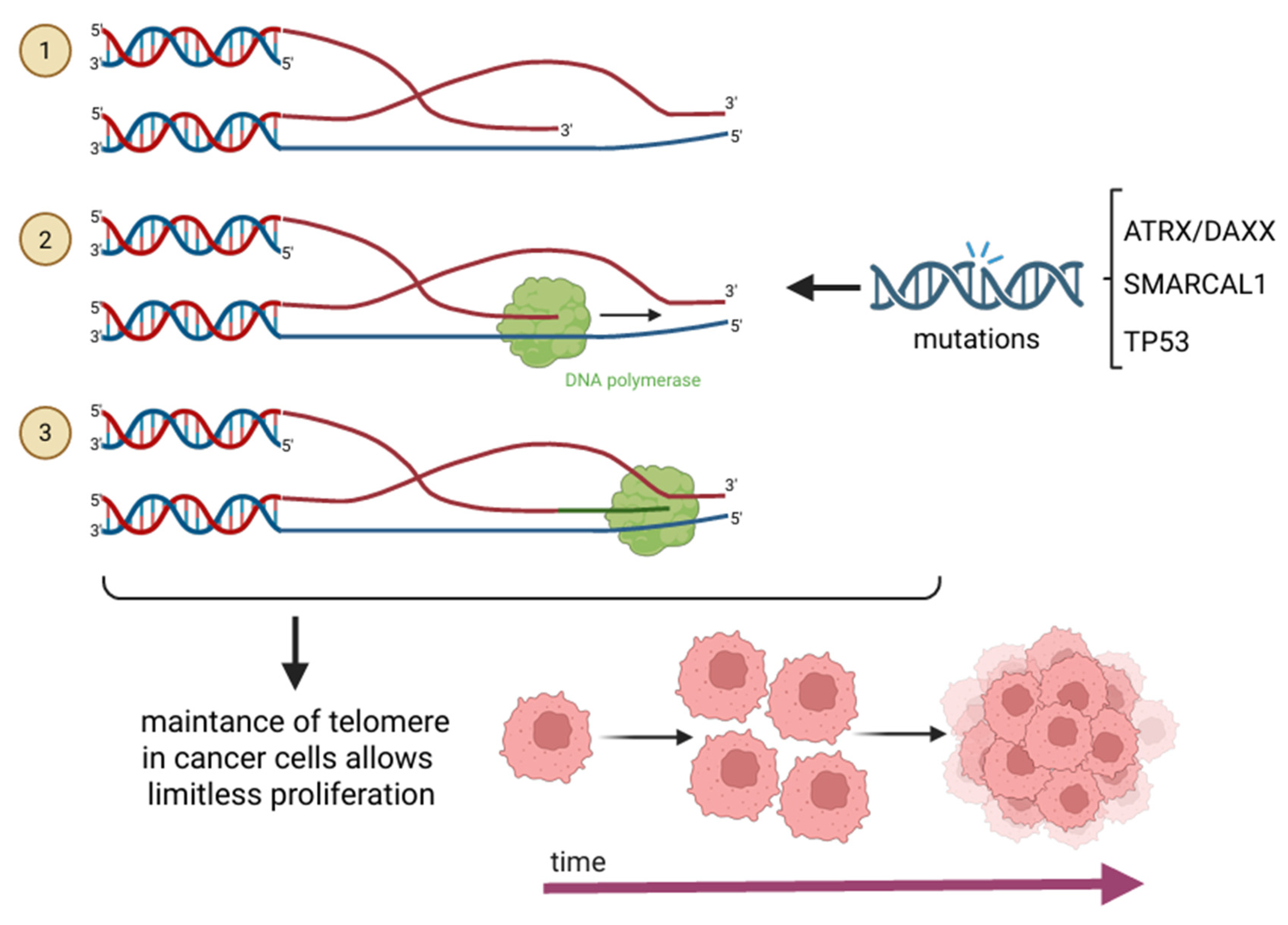
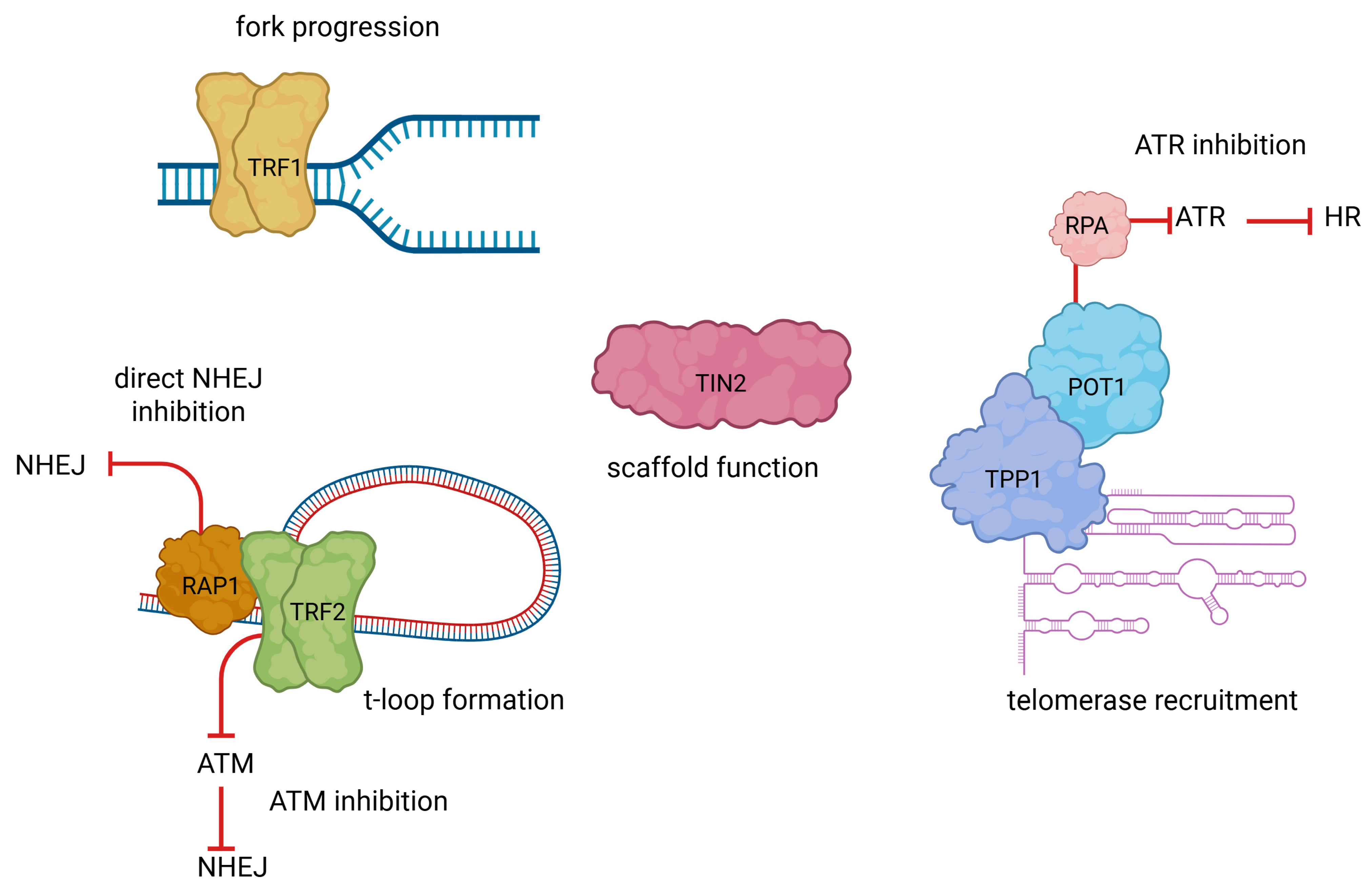
2. Mechanisms of Telomere Maintenance in Cancer
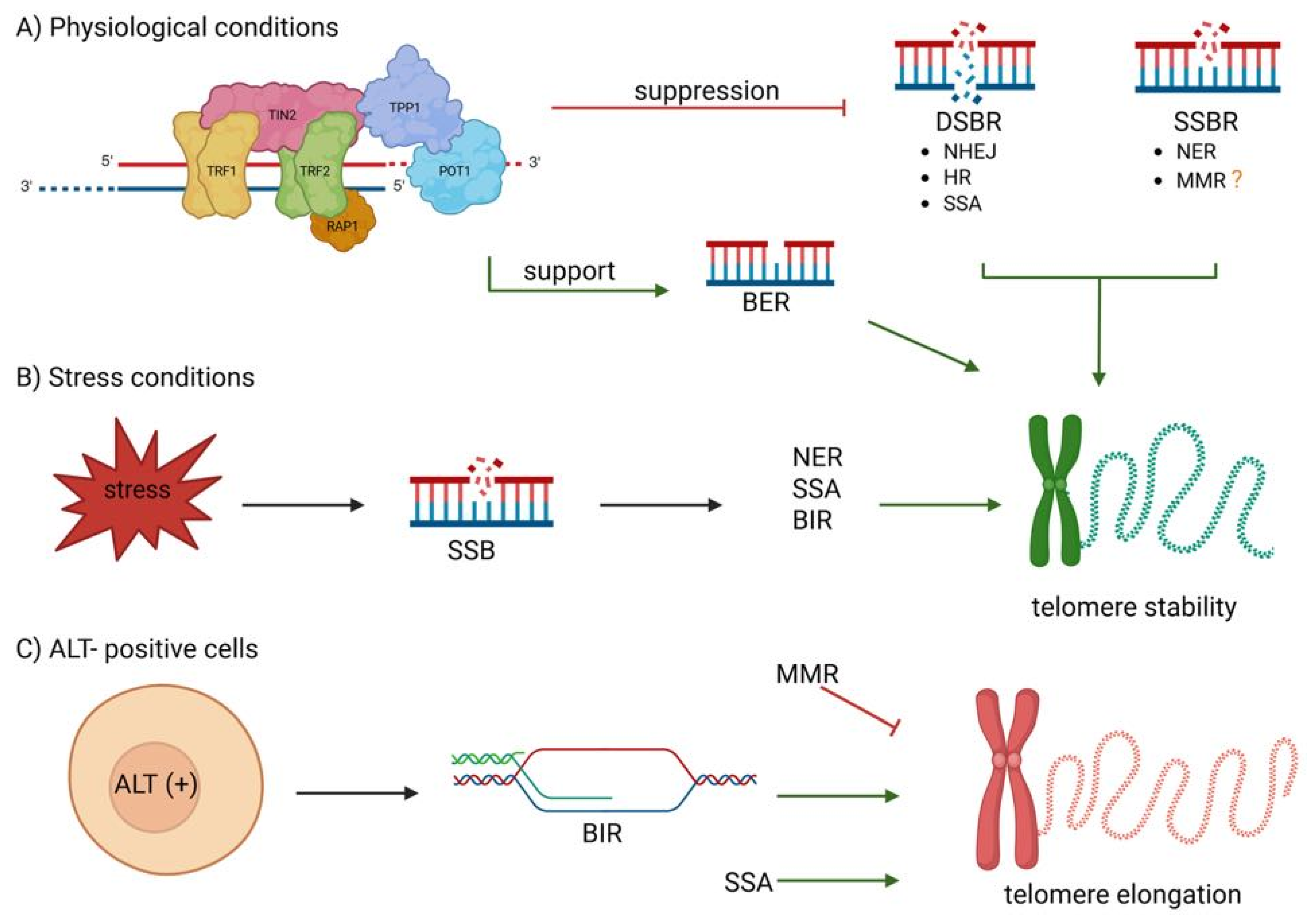
3. Telomere Maintenance and DNA Repair: A Bidirectional Relationship in Cancer Biology and Therapy
4. Therapeutic Targeting of Telomerase and ALT Pathways
4.1. Oligonucleotide-Based Inhibitors
4.2. Small Molecule Inhibitors
4.3. Immunotherapy
4.4. Gene Therapy
4.5. Naturally Occurring Compounds
4.6. Targeting G4-Quadruplexes
4.7. Inhibition of the ATR Kinase
4.8. RAD51 Inhibitors
4.9. CHK1 Inhibitors
5. Challenges in Targeting Telomere Dynamics for Cancer Therapy
5.1. Diagnostic and Prognostic Applications
5.2. Prognosis, Drug Response, and Therapeutic Targets
5.3. Challenges in Telomere-Based Therapeutics
6. Summary and Future Perspectives
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADR | Adverse Drug Reactions |
| ALT | Alternative Lengthening of Telomeres |
| ATM | Ataxia–Telangiectasia Mutated |
| ATR | Ataxia–Telangiectasia And Rad3-Related Kinases: |
| ATRX | Alpha Thalassemia/Mental Retardation Syndrome X-Linked |
| BER | Base Excision Repair |
| BIR | Break-Induced Replication |
| BLM | Bloom Syndrome Protein |
| BRCA | Adverse Drug Reactions |
| CAF | Cancer-Associated Fibroblasts |
| CST complex | Ctc1–Stn1–Ten1 Complex |
| DAXX | Death Domain-Associated Protein |
| DDR | Dna Damage Response |
| DNA-PK | Dna-Dependent Protein Kinase |
| DR | Direct Repair |
| DSB | Double-Strand Break |
| EBV | Epstein-Barr Virus |
| ecDNA | Extrachromosomal Dna |
| EEC | Endometrioid Endometrial Cancer |
| EGCG | Epigallocatechin Gallate |
| FVYL | Conserved Hydrophobic Pocket (Phenylalanine/Valine/Tyrosine/Leucine) |
| GnRHR | Gonadotropin-Releasing Hormone Receptor |
| HGSOC | High-Grade Serous Ovarian Cancer |
| HL | Hodgkin Lymphoma |
| HR | Homologous Recombination |
| hTERT | Human Telomerase Reverse Transcriptase |
| hTR | Human Telomerase |
| MMEJ | Microhomology-Mediated End-Joining |
| MMR | Mismatch Repair |
| MRN | Mre11–Rad50–Nbs1 Protein Complex |
| NER | Nucleotide Excision Repair |
| NHEJ | Non-Homologous End Joining |
| NSCLC | Non-Small Cell Lung Cancer |
| OvC | Ovarian Cancer |
| POT1 | Protection of Telomeres 1 |
| RAP1 | Repressor/Activator Protein 1 |
| RP2D | Recommended Phase 2 Dose |
| RPA | Replication Protein A |
| SSA | Single-Strand Annealing |
| TERC | Human Telomerase Rna |
| TERT | Telomerase Reverse Transcriptase |
| TIL | Tumor-Infiltrating Lymphocytes |
| TIN2 | Trf-Interacting Nuclear Protein 2 |
| TL | Telomere Length |
| TPP1 | Tripeptidyl-Peptidase 1 |
| TRF1 | Telomeric Repeat-Binding Factor 1 |
| TRF2 | Telomeric Repeat-Binding Factor 2 |
References
- Schellnegger, M.; Hofmann, E.; Carnieletto, M.; Kamolz, L.-P. Unlocking longevity: The role of telomeres and its targeting interventions. Front. Aging 2024, 5, 1339317. [Google Scholar]
- Reddel, R. Telomere Maintenance Mechanisms in Cancer: Clinical Implications. Curr. Pharm. Des. 2014, 20, 6361–6374. [Google Scholar] [PubMed]
- Ebata, H.; Loo, T.M.; Takahashi, A. Telomere Maintenance and the cGAS-STING Pathway in Cancer. Cells 2022, 11, 1958. [Google Scholar] [PubMed]
- de Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes. Dev. 2005, 19, 2100–2110. [Google Scholar]
- Bernal, A.; Tusell, L. Telomeres: Implications for Cancer Development. Int. J. Mol. Sci. 2018, 19, 294. [Google Scholar]
- Turner, K.; Vasu, V.; Griffin, D. Telomere Biology and Human Phenotype. Cells 2019, 8, 73. [Google Scholar] [CrossRef]
- Schmutz, I.; de Lange, T. Shelterin. Curr. Biol. 2016, 26, R397–R399. [Google Scholar]
- Victorelli, S.; Passos, J.F. Telomeres and Cell Senescence—Size Matters Not. eBioMedicine 2017, 21, 14–20. [Google Scholar]
- De Vitis, M.; Berardinelli, F.; Sgura, A. Telomere Length Maintenance in Cancer: At the Crossroad between Telomerase and Alternative Lengthening of Telomeres (ALT). Int. J. Mol. Sci. 2018, 19, 606. [Google Scholar]
- Waitkus, M.S.; Erman, E.N.; Reitman, Z.J.; Ashley, D.M. Mechanisms of telomere maintenance and associated therapeutic vulnerabilities in malignant gliomas. Neuro Oncol. 2024, 26, 1012–1024. [Google Scholar]
- George, S.L.; Parmar, V.; Lorenzi, F.; Marshall, L.V.; Jamin, Y.; Poon, E.; Angelini, P.; Chesler, L. Novel therapeutic strategies targeting telomere maintenance mechanisms in high-risk neuroblastoma. J. Exp. Clin. Cancer Res. 2020, 39, 78. [Google Scholar] [PubMed]
- Baird, D.M. Telomere Dynamics in Human Health and Disease. Cold Spring Harb. Perspect. Biol. 2024, 17, a041701. [Google Scholar]
- Baird, D.M.; Rowson, J.; Wynford-Thomas, D.; Kipling, D. Extensive allelic variation and ultrashort telomeres in senescent human cells. Nat. Genet. 2003, 33, 203–207. [Google Scholar] [PubMed]
- Schmidt, T.T.; Tyer, C.; Rughani, P.; Haggblom, C.; Jones, J.R.; Dai, X.; Frazer, K.A.; Gage, F.H.; Juul, S.; Hickey, S.; et al. High resolution long-read telomere sequencing reveals dynamic mechanisms in aging and cancer. Nat. Commun. 2024, 15, 5149. [Google Scholar] [PubMed]
- Trybek, T.; Kowalik, A.; Góźdź, S.; Kowalska, A. Telomeres and telomerase in oncogenesis (Review). Oncol. Lett. 2020, 20, 1015–1027. [Google Scholar]
- Ahmad, F.B.; Anderson, R.N. The Leading Causes of Death in the US for 2020. JAMA 2021, 325, 1829. [Google Scholar]
- Ahmad, F.B.; Cisewski, J.A.; Anderson, R.N. Mortality in the United States—Provisional Data, 2023. MMWR Morb. Mortal. Wkly. Rep. 2024, 73, 677–681. [Google Scholar]
- Cancer. Available online: https://www.who.int/health-topics/cancer#tab=tab_1 (accessed on 11 March 2025).
- Mai, S.; Garini, Y. The significance of telomeric aggregates in the interphase nuclei of tumor cells. J. Cell. Biochem. 2006, 97, 904–915. [Google Scholar]
- Xu, L.; Blackburn, E.H. Human Cancer Cells Harbor T-Stumps, a Distinct Class of Extremely Short Telomeres. Mol. Cell 2007, 28, 315–327. [Google Scholar]
- Knecht, H.; Johnson, N.; Bienz, M.N.; Brousset, P.; Memeo, L.; Shifrin, Y.; Alikhah, A.; Louis, S.F.; Mai, S. Analysis by TeloView® Technology Predicts the Response of Hodgkin’s Lymphoma to First-Line ABVD Therapy. Cancers 2024, 16, 2816. [Google Scholar]
- Vermolen, B.J.; Garini, Y.; Mai, S.; Mougey, V.; Fest, T.; Chuang, T.C.; Chuang, A.Y.; Wark, L.; Young, I.T. Characterizing the three-dimensional organization of telomeres. Cytom. Part A 2005, 67A, 144–150. [Google Scholar]
- Chuang, T.C.Y.; Moshir, S.; Garini, Y.; Chuang, A.Y.; Young, I.T.; Vermolen, B.; van den Doel, R.; Mougey, V.; Perrin, M.; Braun, M.; et al. The three-dimensional organization of telomeres in the nucleus of mammalian cells. BMC Biol. 2004, 2, 12. [Google Scholar]
- Louis, S.F.; Vermolen, B.J.; Garini, Y.; Young, I.T.; Guffei, A.; Lichtensztejn, Z.; Kuttler, F.; Chuang, T.C.; Moshir, S.; Mougey, V.; et al. c-Myc induces chromosomal rearrangements through telomere and chromosome remodeling in the interphase nucleus. Proc. Natl. Acad. Sci. USA 2005, 102, 9613–9618. [Google Scholar] [PubMed]
- McClintock, B. The stability of broken ends of chromosomes in Zea mays. Genetics 1941, 26, 234–282. [Google Scholar]
- Zinder, J.C.; Olinares, P.D.B.; Svetlov, V.; Bush, M.W.; Nudler, E.; Chait, B.T.; Walz, T.; de Lange, T. Shelterin is a dimeric complex with extensive structural heterogeneity. Proc. Natl. Acad. Sci. USA 2022, 119, e2201662119. [Google Scholar]
- Lim, C.J.; Cech, T.R. Shaping human telomeres: From shelterin and CST complexes to telomeric chromatin organization. Nat Rev. Mol. Cell Biol. 2021, 22, 283–298, Erratum in Nat. Rev. Mol. Cell Biol. 2021, 22, 299. https://doi.org/10.1038/s41580-021-00353-x. [Google Scholar] [CrossRef]
- Lajoie, V.; Lemieux, B.; Sawan, B.; Lichtensztejn, D.; Lichtensztejn, Z.; Wellinger, R.; Mai, S.; Knecht, H. LMP1 mediates multinuclearity through downregulation of shelterin proteins and formation of telomeric aggregates. Blood 2015, 125, 2101–2110. [Google Scholar]
- Gao, J.; Pickett, H.A. Targeting telomeres: Advances in telomere maintenance mechanism-specific cancer therapies. Nat. Rev. Cancer 2022, 22, 515–532. [Google Scholar]
- O’Sullivan, R.J.; Greenberg, R.A. Mechanisms of Alternative Lengthening of Telomeres. Cold Spring Harb. Perspect. Biol. 2025, 17, a041690. [Google Scholar]
- Liu, T.; Li, S.; Xia, C.; Xu, D. TERT promoter mutations and methylation for telomerase activation in urothelial carcinomas: New mechanistic insights and clinical significance. Front. Immunol. 2023, 13, 1071390. [Google Scholar]
- Li, S.; Hu, G.; Chen, Y.; Sang, Y.; Tang, Q.; Liu, R. TERT upstream promoter methylation regulates TERT expression and acts as a therapeutic target in TERT promoter mutation-negative thyroid cancer. Cancer Cell Int. 2024, 24, 271. [Google Scholar]
- Li, S.; Xue, J.; Jiang, K.; Chen, Y.; Zhu, L.; Liu, R. TERT promoter methylation is associated with high expression of TERT and poor prognosis in papillary thyroid cancer. Front. Oncol. 2024, 14, 1325345. [Google Scholar]
- Dratwa, M.; Wysoczańska, B.; Łacina, P.; Kubik, T.; Bogunia-Kubik, K. TERT—Regulation and Roles in Cancer Formation. Front. Immunol. 2020, 11, 589929. [Google Scholar]
- Cesare, A.J.; Reddel, R.R. Alternative lengthening of telomeres: Models, mechanisms and implications. Nat. Rev. Genet. 2010, 11, 319–330. [Google Scholar]
- Barthel, F.P.; Wei, W.; Tang, M.; Martinez-Ledesma, E.; Hu, X.; Amin, S.B.; Akdemir, K.C.; Seth, S.; Song, X.; Wang, Q.; et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat. Genet. 2017, 49, 349–357. [Google Scholar] [PubMed]
- Lovejoy, C.A.; Takai, K.; Huh, M.S.; Picketts, D.J.; de Lange, T. ATRX affects the repair of telomeric DSBs by promoting cohesion and a DAXX-dependent activity. PLoS Biol. 2020, 18, e3000594. [Google Scholar]
- Koschmann, C.; Calinescu, A.-A.; Nunez, F.J.; Mackay, A.; Fazal-Salom, J.; Thomas, D.; Mendez, F.; Kamran, N.; Dzaman, M.; Mulpuri, L.; et al. ATRX loss promotes tumor growth and impairs nonhomologous end joining DNA repair in glioma. Sci. Transl. Med. 2016, 8, 328ra28. [Google Scholar]
- Wang, Y.; Yang, J.; Wild, A.T.; Wu, W.H.; Shah, R.; Danussi, C.; Riggins, G.J.; Kannan, K.; Sulman, E.P.; Chan, T.A.; et al. G-quadruplex DNA drives genomic instability and represents a targetable molecular abnormality in ATRX-deficient malignant glioma. Nat. Commun. 2019, 10, 943. [Google Scholar]
- Liu, H.; Xu, C.; Diplas, B.H.; Brown, A.; Strickland, L.M.; Yao, H.; Ling, J.; McLendon, R.E.; Keir, S.T.; Ashley, D.M.; et al. Cancer-associated SMARCAL1 loss-of-function mutations promote alternative lengthening of telomeres and tumorigenesis in telomerase-negative glioblastoma cells. Neuro Oncol. 2023, 25, 1563–1575. [Google Scholar]
- Ida, C.M.; Jenkins, R.B. SMARCAL1: Expanding the spectrum of genes associated with alternative lengthening of telomeres. Neuro Oncol. 2023, 25, 1576–1577. [Google Scholar]
- Mangerel, J.; Price, A.; Castelo-Branco, P.; Brzezinski, J.; Buczkowicz, P.; Rakopoulos, P.; Merino, D.; Baskin, B.; Wasserman, J.; Mistry, M.; et al. Alternative lengthening of telomeres is enriched in, and impacts survival of TP53 mutant pediatric malignant brain tumors. Acta Neuropathol. 2014, 128, 853–862. [Google Scholar] [PubMed]
- Borodovsky, A.; Meeker, A.K.; Kirkness, E.F.; Zhao, Q.; Eberhart, C.G.; Gallia, G.L.; Riggins, G.J. A model of a patient-derived IDH1 mutant anaplastic astrocytoma with alternative lengthening of telomeres. J. Neuro-Oncol. 2015, 121, 479–487. [Google Scholar]
- de Lange, T. Shelterin-Mediated Telomere Protection. Annu. Rev. Genet. 2018, 52, 223–247. [Google Scholar] [PubMed]
- Maestroni, L.; Matmati, S.; Coulon, S. Solving the Telomere Replication Problem. Genes 2017, 8, 55. [Google Scholar] [CrossRef]
- Ghilain, C.; Gilson, E.; Giraud-Panis, M.-J. Multifunctionality of the Telomere-Capping Shelterin Complex Explained by Variations in Its Protein Composition. Cells 2021, 10, 1753. [Google Scholar] [CrossRef]
- Hu, H.; Yan, H.L.; Nguyen, T.H.D. Structural biology of shelterin and telomeric chromatin: The pieces and an unfinished puzzle. Biochem. Soc. Trans. 2024, 52, 1551–1564. [Google Scholar]
- Mir, S.M.; Samavarchi Tehrani, S.; Goodarzi, G.; Jamalpoor, Z.; Asadi, J.; Khelghati, N.; Qujeq, D.; Maniati, M. Shelterin Complex at Telomeres: Implications in Ageing. Clin. Interv. Aging 2020, 15, 827–839. [Google Scholar]
- Okamoto, K.; Bartocci, C.; Ouzounov, I.; Diedrich, J.K.; Yates, I.I.I.J.R.; Denchi, E.L. A two-step mechanism for TRF2-mediated chromosome-end protection. Nature 2013, 494, 502–505. [Google Scholar]
- Imran, S.A.M.; Yazid, M.D.; Cui, W.; Lokanathan, Y. The Intra- and Extra-Telomeric Role of TRF2 in the DNA Damage Response. Int. J. Mol. Sci. 2021, 22, 9900. [Google Scholar]
- Pinzaru, A.M.; Kareh, M.; Lamm, N.; Lazzerini-Denchi, E.; Cesare, A.J.; Sfeir, A. Replication stress conferred by POT1 dysfunction promotes telomere relocalization to the nuclear pore. Genes. Dev. 2020, 34, 1619–1636. [Google Scholar]
- Harman, A.; Bryan, T.M. Telomere maintenance and the DNA damage response: A paradoxical alliance. Front. Cell Dev. Biol. 2024, 12, 1472906. [Google Scholar]
- Aksenova, A.Y.; Mirkin, S.M. At the Beginning of the End and in the Middle of the Beginning: Structure and Maintenance of Telomeric DNA Repeats and Interstitial Telomeric Sequences. Genes 2019, 10, 118. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Liu, M.; Zhang, Y.; Zhou, H.; Wang, Y.; Xu, Z.-X. Telomere-related DNA damage response pathways in cancer therapy: Prospective targets. Front. Pharmacol. 2024, 15, 1379166. [Google Scholar]
- Cicconi, A.; Chang, S. Shelterin and the replisome: At the intersection of telomere repair and replication. Curr. Opin. Genet. Dev. 2020, 60, 77–84. [Google Scholar]
- Tire, B.; Talibova, G.; Ozturk, S. The crosstalk between telomeres and DNA repair mechanisms: An overview to mammalian somatic cells, germ cells, and preimplantation embryos. J. Assist. Reprod. Genet. 2024, 41, 277–291. [Google Scholar]
- Fouquerel, E.; Parikh, D.; Opresko, P. DNA damage processing at telomeres: The ends justify the means. DNA Repair 2016, 44, 159–168. [Google Scholar]
- Ruis, P.; Boulton, S.J. The end protection problem—An unexpected twist in the tail. Genes. Dev. 2021, 35, 1–21. [Google Scholar]
- Lototska, L.; Yue, J.; Li, J.; Giraud-Panis, M.J.; Songyang, Z.; Royle, N.J.; Liti, G.; Ye, J.; Gilson, E.; Mendez-Bermudez, A. Human RAP1 specifically protects telomeres of senescent cells from DNA damage. EMBO Rep. 2020, 21, e49076. [Google Scholar]
- Liang, F.; Rai, R.; Sodeinde, T.; Chang, S. TRF2–RAP1 represses RAD51-dependent homology-directed telomere repair by promoting BLM-mediated D-loop unwinding and inhibiting BLM–DNA2-dependent 5′-end resection. Nucleic Acids Res. 2024, 52, 9695–9709. [Google Scholar]
- Gu, P.; Jia, S.; Takasugi, T.; Smith, E.; Nandakumar, J.; Hendrickson, E.; Chang, S. CTC1-STN1 coordinates G- and C-strand synthesis to regulate telomere length. Aging Cell 2018, 17, e12783. [Google Scholar]
- Sui, J.; Zhang, S.; Chen, B.P.C. DNA–dependent protein kinase in telomere maintenance and protection. Cell Mol. Biol. Lett. 2020, 25, 2. [Google Scholar]
- Camfield, S.; Chakraborty, S.; Dwivedi, S.K.D.; Pramanik, P.K.; Mukherjee, P.; Bhattacharya, R. Secrets of DNA-PKcs beyond DNA repair. npj Precis. Oncol. 2024, 8, 154. [Google Scholar] [PubMed]
- Glousker, G.; Briod, A.; Quadroni, M.; Lingner, J. Human shelterin protein POT1 prevents severe telomere instability induced by homology-directed DNA repair. EMBO J. 2020, 39, e104500. [Google Scholar] [PubMed]
- Claussin, C.; Chang, M. The many facets of homologous recombination at telomeres. Microb. Cell 2015, 2, 308–321. [Google Scholar]
- Badie, S.; Escandell, J.M.; Bouwman, P.; Carlos, A.R.; Thanasoula, M.; Gallardo, M.M.; Suram, A.; Jaco, I.; Benitez, J.; Herbig, U. BRCA2 acts as a RAD51 loader to facilitate telomere replication and capping. Nat. Struct. Mol. Biol. 2010, 17, 1461–1469. [Google Scholar]
- Dilley, R.L.; Greenberg, R.A. ALTernative Telomere Maintenance and Cancer. Trends Cancer 2015, 1, 145–156. [Google Scholar]
- Tan, J.; Duan, M.; Yadav, T.; Phoon, L.; Wang, X.; Zhang, J.M.; Zou, L.; Lan, L. An R-loop-initiated CSB–RAD52–POLD3 pathway suppresses ROS-induced telomeric DNA breaks. Nucleic Acids Res. 2020, 48, 1285–1300. [Google Scholar]
- Rosso, I.; Jones-Weinert, C.; Rossiello, F.; Cabrini, M.; Brambillasca, S.; Munoz-Sagredo, L.; Lavagnino, Z.; Martini, E.; Tedone, E.; Garre’, M.; et al. Alternative lengthening of telomeres (ALT) cells viability is dependent on C-rich telomeric RNAs. Nat. Commun. 2023, 14, 7086. [Google Scholar]
- Miller, A.S.; Balakrishnan, L.; Buncher, N.A.; Opresko, P.L.; Bambara, R.A. Telomere proteins POT1, TRF1 and TRF2 augment long-patch base excision repair in vitro. Cell Cycle 2012, 11, 998–1007. [Google Scholar]
- Jia, P.; Her, C.; Chai, W. DNA excision repair at telomeres. DNA Repair 2015, 36, 137–145. [Google Scholar]
- Yang, Z.; Sharma, K.; de Lange, T. TRF1 uses a noncanonical function of TFIIH to promote telomere replication. Genes Dev. 2022, 36, 956–969. [Google Scholar] [PubMed]
- Gregg, S.Q.; Robinson, A.R.; Niedernhofer, L.J. Physiological consequences of defects in ERCC1–XPF DNA repair endonuclease. DNA Repair 2011, 10, 781–791. [Google Scholar] [PubMed]
- Krokan, H.E.; Bjoras, M. Base Excision Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar]
- Fouquerel, E.; Barnes, R.P.; Uttam, S.; Watkins, S.C.; Bruchez, M.P.; Opresko, P.L. Targeted and Persistent 8-Oxoguanine Base Damage at Telomeres Promotes Telomere Loss and Crisis. Mol. Cell 2019, 75, 117–130.e6. [Google Scholar]
- Barroso-González, J.; García-Expósito, L.; Galaviz, P.; Lynskey, M.L.; Allen, J.A.M.; Hoang, S.; Watkins, S.C.; Pickett, H.A.; O’Sullivan, R.J. Anti-recombination function of MutSα restricts telomere extension by ALT-associated homology-directed repair. Cell Rep. 2021, 37, 110088. [Google Scholar]
- Liu, C.-C.; Capart, M.M.M.; Lin, J.-J. Mismatch repair enzymes regulate telomere recombination in Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 2024, 707, 149768. [Google Scholar]
- Pal, D.; Sharma, U.; Singh, S.K.; Kakkar, N.; Prasad, R. Over-Expression of Telomere Binding Factors (TRF1 & TRF2) in Renal Cell Carcinoma and Their Inhibition by Using SiRNA Induce Apoptosis, Reduce Cell Proliferation and Migration Invitro. PLoS ONE 2015, 10, e0115651. [Google Scholar]
- Chen, W.; Wang, Y.; Li, F.; Lin, W.; Liang, Y.; Ma, Z. Expression of Telomere Repeat Binding Factor 1 and TRF2 in Prostate Cancer and Correlation with Clinical Parameters. Biomed. Res. Int. 2017, 2017, 9764752. [Google Scholar]
- Hu, H.; Zhang, Y.; Zou, M.; Yang, S.; Liang, X.-Q. Expression of TRF1, TRF2, TIN2, TERT, KU70, and BRCA1 proteins is associated with telomere shortening and may contribute to multistage carcinogenesis of gastric cancer. J. Cancer Res. Clin. Oncol. 2010, 136, 1407–1414. [Google Scholar]
- Shen, E.; Xiu, J.; Lopez, G.Y.; Bentley, R.; Jalali, A.; Heimberger, A.B.; Bainbridge, M.N.; Bondy, M.L.; Walsh, K.M. POT1 mutation spectrum in tumour types commonly diagnosed among POT1-associated hereditary cancer syndrome families. J. Med. Genet. 2020, 57, 664–670. [Google Scholar]
- Jäger, K.; Walter, M. Therapeutic Targeting of Telomerase. Genes 2016, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-M.; Zou, L. Alternative lengthening of telomeres: From molecular mechanisms to therapeutic outlooks. Cell Biosci. 2020, 10, 30. [Google Scholar]
- Lennox, A.L.; Huang, F.; Behrs, M.K.; González-Sales, M.; Bhise, N.; Wan, Y.; Sun, L.; Berry, T.; Feller, F.; Morcos, P.N. Imetelstat, a novel, first-in-class telomerase inhibitor: Mechanism of action, clinical, and translational science. Clin. Transl. Sci. 2024, 17, e70076. [Google Scholar] [PubMed]
- Schrank, Z.; Khan, N.; Osude, C.; Singh, S.; Miller, R.J.; Merrick, C.; Mabel, A.; Kuckovic, A.; Puri, N. Oligonucleotides Targeting Telomeres and Telomerase in Cancer. Molecules 2018, 23, 2267. [Google Scholar] [CrossRef]
- Ozawa, T.; Gryaznov, S.M.; Hu, L.J.; Pongracz, K.; Santos, R.A.; Bollen, A.W.; Lamborn, K.R.; Deen, D.F. Antitumor effects of specific telomerase inhibitor GRN163 in human glioblastoma xenografts. Neuro Oncol. 2004, 6, 218–226. [Google Scholar]
- Hashizume, R.; Ozawa, T.; Gryaznov, S.M.; Bollen, A.W.; Lamborn, K.R.; Frey, W.H., 2nd; Deen, D.F. New therapeutic approach for brain tumors: Intranasal delivery of telomerase inhibitor GRN163. Neuro-Oncol. 2008, 10, 112–120. [Google Scholar]
- Thompson, P.A.; Drissi, R.; Muscal, J.A.; Panditharatna, E.; Fouladi, M.; Ingle, A.M.; Ahern, C.H.; Reid, J.M.; Lin, T.; Weigel, B.J.; et al. A Phase I Trial of Imetelstat in Children with Refractory or Recurrent Solid Tumors: A Children’s Oncology Group Phase I Consortium Study (ADVL1112). Clin. Cancer Res. 2013, 19, 6578–6584. [Google Scholar]
- Salloum, R.; Hummel, T.R.; Kumar, S.S.; Dorris, K.; Li, S.; Lin, T.; Daryani, V.M.; Stewart, C.F.; Miles, L.; Poussaint, T.Y.; et al. A molecular biology and phase II study of imetelstat (GRN163L) in children with recurrent or refractory central nervous system malignancies: A pediatric brain tumor consortium study. J. Neuro-Oncol. 2016, 129, 443–451. [Google Scholar]
- Chiappori, A.A.; Kolevska, T.; Spigel, D.R.; Hager, S.; Rarick, M.; Gadgeel, S.; Blais, N.; Von Pawel, J.; Hart, L.; Reck, M.; et al. A randomized phase II study of the telomerase inhibitor imetelstat as maintenance therapy for advanced non-small-cell lung cancer. Ann. Oncol. 2015, 26, 354–362. [Google Scholar]
- Siteni, S.; Grichuk, A.; Shay, J.W. Telomerase in Cancer Therapeutics. Cold Spring Harb. Perspect. Biol. 2024, 16, a041703. [Google Scholar]
- Zhang, Y.; Yang, X.; Zhou, H.; Yao, G.; Zhou, L.; Qian, C. BIBR1532 inhibits proliferation and enhances apoptosis in multiple myeloma cells by reducing telomerase activity. PeerJ 2023, 11, e16404. [Google Scholar] [PubMed]
- Lavanya, C.; Venkataswamy, M.M.; Sibin, M.K.; Srinivas Bharath, M.M.; Chetan, G.K. Down regulation of human telomerase reverse transcriptase (hTERT) expression by BIBR1532 in human glioblastoma LN18 cells. Cytotechnology 2018, 70, 1143–1154. [Google Scholar] [PubMed]
- Sameni, S.; Viswanathan, R.; Ng, G.Y.-Q.; Martinez-Lopez, W.; Hande, M.P. Telomerase Inhibition by MST-312 Sensitizes Breast Cancer Cells to the Anti-cancer Properties of Plumbagin. Genome Integr. 2023, 14, 20230002. [Google Scholar]
- Bahmei, A.; Karimi, F.; Mahini, S.M.; Irandoost, H.; Tandel, P.; Niknam, H.; Tamaddon, G. Targeting telomerase with MST-312 leads to downregulation of CCND1, MDM2, MYC, and HSP90AA1 and induce apoptosis in Jurkat cell line. Med. Oncol. 2024, 41, 267. [Google Scholar]
- Kim, J.W.; Park, M.; Kim, S.; Lim, S.C.; Kim, H.S.; Kang, K.W. Anti-metastatic effect of GV1001 on prostate cancer cells; roles of GnRHR-mediated Gαs-cAMP pathway and AR-YAP1 axis. Cell Biosci. 2021, 11, 191. [Google Scholar]
- Kim, J.H.; Cho, Y.-R.; Ahn, E.-K.; Kim, S.; Han, S.; Kim, S.J.; Bae, G.U.; Oh, J.S.; Seo, D.W. A novel telomerase-derived peptide GV1001-mediated inhibition of angiogenesis: Regulation of VEGF/VEGFR-2 signaling pathways. Transl. Oncol. 2022, 26, 101546. [Google Scholar]
- Kim, G.E.; Jung, A.R.; Kim, M.Y.; Lee, J.B.; Im, J.H.; Lee, K.W.; Park, Y.H.; Lee, J.Y. GV1001 Induces Apoptosis by Reducing Angiogenesis in Renal Cell Carcinoma Cells Both In Vitro and In Vivo. Urology 2018, 113, 129–137. [Google Scholar]
- Jo, J.H.; Kim, Y.-T.; Choi, H.S.; Kim, H.G.; Lee, H.S.; Choi, Y.W.; Kim, D.U.; Lee, K.H.; Kim, E.J.; Han, J.H.; et al. Efficacy of GV1001 with gemcitabine/capecitabine in previously untreated patients with advanced pancreatic ductal adenocarcinoma having high serum eotaxin levels (KG4/2015): An open-label, randomised, Phase 3 trial. Br. J. Cancer 2024, 130, 43–52. [Google Scholar]
- Chen, W.; Kim, S.; Kim, S.Y.; Beheshtian, C.; Kim, N.; Shin, K.H.; Kim, R.H.; Kim, S.; Park, N.H. GV1001, hTERT Peptide Fragment, Prevents Doxorubicin-Induced Endothelial-to-Mesenchymal Transition in Human Endothelial Cells and Atherosclerosis in Mice. Cells 2025, 14, 98. [Google Scholar]
- Pateras, I.S.; Kotsakis, A.; Avgeris, M.; Baliou, E.; Kouroupakis, P.; Patsea, E.; Georgoulias, V.; Menez-Jamet, J.; Kinet, J.P.; Kosmatopoulos, K. Clinical Activity of an hTERT-Specific Cancer Vaccine (Vx-001) in ‘Immune Desert’ NSCLC. Cancers 2021, 13, 1658. [Google Scholar]
- Kotsakis, A.; Papadimitraki, E.; Vetsika, E.K.; Aggouraki, D.; Dermitzaki, E.K.; Hatzidaki, D.; Kentepozidis, N.; Mavroudis, D.; Georgoulias, V. A phase II trial evaluating the clinical and immunologic response of HLA-A2+ non-small cell lung cancer patients vaccinated with an hTERT cryptic peptide. Lung Cancer 2014, 86, 59–66. [Google Scholar] [PubMed]
- Gridelli, C.; Ciuleanu, T.; Domine, M.; Szczesna, A.; Bover, I.; Cobo, M.; Kentepozidis, N.; Zarogoulidis, K.; Kalofonos, C.; Kazarnowisz, A.; et al. Clinical activity of a htert (vx-001) cancer vaccine as post-chemotherapy maintenance immunotherapy in patients with stage IV non-small cell lung cancer: Final results of a randomised phase 2 clinical trial. Br. J. Cancer 2020, 122, 1461–1466. [Google Scholar]
- Han, S.R.; Lee, C.H.; Im, J.Y.; Kim, J.H.; Kim, J.H.; Kim, S.J.; Cho, Y.W.; Kim, E.; Kim, Y.; Ryu, J.H.; et al. Targeted suicide gene therapy for liver cancer based on ribozyme-mediated RNA replacement through post-transcriptional regulation. Mol. Ther. Nucleic Acids 2021, 23, 154–168. [Google Scholar] [PubMed]
- Shirakawa, Y.; Tazawa, H.; Tanabe, S.; Kanaya, N.; Noma, K.; Koujima, T.; Kashima, H.; Kato, T.; Kuroda, S.; Kikuchi, S.; et al. Phase I dose-escalation study of endoscopic intratumoral injection of OBP-301 (Telomelysin) with radiotherapy in oesophageal cancer patients unfit for standard treatments. Eur. J. Cancer 2021, 153, 98–108. [Google Scholar] [PubMed]
- Heo, J.; Liang, J.-D.; Kim, C.W.; Woo, H.Y.; Shih, I.L.; Su, T.H.; Lin, Z.Z.; Yoo, S.Y.; Chang, S.; Urata, Y.; et al. Safety and dose escalation of the targeted oncolytic adenovirus OBP-301 for refractory advanced liver cancer: Phase I clinical trial. Mol. Ther. 2023, 31, 2077–2088. [Google Scholar]
- Rowaiye, A.B.; Mendes, Y.J.T.; Olofinsae, S.A.; Oche, J.B.; Oluwakemi, H.O.; Okpalefe, O.A.; Ogidigo, J.O. Camptothecin shows better promise than Curcumin in the inhibition of the Human Telomerase: A computational study. Heliyon 2021, 7, e07742. [Google Scholar]
- Xiao, Z.; Zhang, A.; Lin, J.; Zheng, Z.; Shim, X.; Di, W.; Qi, W.; Zhu, Y.; Zhou, G.; Fang, Y. Telomerase: A Target for Therapeutic Effects of Curcumin and a Curcumin Derivative in Aβ1-42 Insult In Vitro. PLoS ONE 2014, 9, e101251. [Google Scholar]
- Mirzazadeh, A.; Kheirollahi, M.; Farashahi, E.; Sadeghian-Nodoushan, F.; Sheikhha, M.; Aflatoonian, B. Assessment Effects of Resveratrol on Human Telomerase Reverse Transcriptase Messenger Ribonucleic Acid Transcript in Human Glioblastoma. Adv. Biomed. Res. 2017, 6, 73. [Google Scholar]
- Samad, M.A.; Saiman, M.Z.; Abdul Majid, N.; Karsani, S.A.; Yaacob, J.S. Berberine Inhibits Telomerase Activity and Induces Cell Cycle Arrest and Telomere Erosion in Colorectal Cancer Cell Line, HCT 116. Molecules 2021, 26, 376. [Google Scholar] [CrossRef]
- Sinicrope, F.A.; Viggiano, T.R.; Buttar, N.S.; Song, L.M.W.K.; Schroeder, K.W.; Kraichely, R.E.; Larson, M.V.; Sedlack, R.E.; Kisiel, J.B.; Gostout, C.J.; et al. Randomized Phase II Trial of Polyphenon E versus Placebo in Patients at High Risk of Recurrent Colonic Neoplasia. Cancer Prev. Res. 2021, 14, 573–580. [Google Scholar]
- Figueiredo, J.; Mergny, J.-L.; Cruz, C. G-quadruplex ligands in cancer therapy: Progress, challenges, and clinical perspectives. Life Sci. 2024, 340, 122481. [Google Scholar] [PubMed]
- Kosiol, N.; Juranek, S.; Brossart, P.; Heine, A.; Paeschke, K. G-quadruplexes: A promising target for cancer therapy. Mol. Cancer 2021, 20, 40. [Google Scholar] [PubMed]
- Harkness, R.W.; Mittermaier, A.K. G-quadruplex dynamics. Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 1544–1554. [Google Scholar] [PubMed]
- Nakanishi, C.; Seimiya, H. G-quadruplex in cancer biology and drug discovery. Biochem. Biophys. Res. Commun. 2020, 531, 45–50. [Google Scholar]
- Lv, L.; Zhang, L. Characterization of G-Quadruplexes in Enterovirus A71 Genome and Their Interaction with G-Quadruplex Ligands. Microbiol. Spectr. 2022, 10, 45–50. [Google Scholar]
- Linder, J.; Garner, T.P.; Williams, H.E.L.; Searle, M.S.; Moody, C.J. Telomestatin: Formal Total Synthesis and Cation-Mediated Interaction of Its seco-Derivatives with G-Quadruplexes. J. Am. Chem. Soc. 2011, 133, 1044–1051. [Google Scholar]
- Islam, M.K.; Jackson, P.J.; Rahman, K.M.; Thurston, D.E. Recent Advances in Targeting the Telomeric G-Quadruplex DNA Sequence with Small Molecules as a Strategy for Anticancer Therapies. Future Med. Chem. 2016, 8, 1259–1290. [Google Scholar]
- Leonetti, C.; Scarsella, M.; Riggio, G.; Rizzo, A.; Salvati, E.; D’Incalci, M.; Staszewsky, L.; Frapolli, R.; Stevens, M.F.; Stoppacciaro, A.; et al. G-Quadruplex Ligand RHPS4 Potentiates the Antitumor Activity of Camptothecins in Preclinical Models of Solid Tumors. Clin. Cancer Res. 2008, 14, 7284–7291. [Google Scholar]
- Goncalves, T.; Zoumpoulidou, G.; Alvarez-Mendoza, C.; Mancusi, C.; Collopy, L.C.; Strauss, S.J.; Mittnacht, S.; Tomita, K. Selective Elimination of Osteosarcoma Cell Lines with Short Telomeres by Ataxia Telangiectasia and Rad3-Related Inhibitors. ACS Pharmacol. Transl. Sci. 2020, 3, 1253–1264. [Google Scholar]
- Flynn, R.L.; Cox, K.E.; Jeitany, M.; Wakimoto, H.; Bryll, A.R.; Ganem, N.J.; Bersani, F.; Pineda, J.R.; Suvà, M.L.; Benes, C.H.; et al. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 2015, 347, 273–277. [Google Scholar]
- Deeg, K.I.; Chung, I.; Bauer, C.; Rippe, K. Cancer Cells with Alternative Lengthening of Telomeres do not Display a General Hypersensitivity to ATR Inhibition. Front. Oncol. 2016, 6, 186. [Google Scholar]
- Kim, S.T.; Smith, S.A.; Mortimer, P.; Loembé, A.B.; Cho, H.; Kim, K.M.; Smith, C.; Willis, S.; Irurzun-Arana, I.; Berges, A.; et al. Phase I Study of Ceralasertib (AZD6738), a Novel DNA Damage Repair Agent, in Combination with Weekly Paclitaxel in Refractory Cancer. Clin. Cancer Res. 2021, 27, 4700–4709. [Google Scholar] [PubMed]
- Dillon, M.T.; Guevara, J.; Mohammed, K.; Patin, E.C.; Smith, S.A.; Dean, E.; Jones, G.N.; Willis, S.E.; Petrone, M.; Silva, C.; et al. Durable responses to ATR inhibition with ceralasertib in tumors with genomic defects and high inflammation. J. Clin. Investig. 2024, 134, e175369. [Google Scholar] [PubMed]
- Kwon, M.; Kim, G.; Kim, R.; Kim, K.T.; Kim, S.T.; Smith, S.; Mortimer, P.G.S.; Hong, J.Y.; Loembé, A.B.; Irurzun-Arana, I.; et al. Phase II study of ceralasertib (AZD6738) in combination with durvalumab in patients with advanced gastric cancer. J. Immunother. Cancer 2022, 10, e005041. [Google Scholar]
- Kim, R.; Kwon, M.; An, M.; Kim, S.T.; Smith, S.A.; Loembé, A.B.; Mortimer, P.G.S.; Armenia, J.; Lukashchuk, N.; Shah, N.; et al. Phase II study of ceralasertib (AZD6738) in combination with durvalumab in patients with advanced/metastatic melanoma who have failed prior anti-PD-1 therapy. Ann. Oncol. 2022, 33, 193–203. [Google Scholar]
- American Association for Cancer Research. Preliminary Activity Seen with RAD51 Inhibitor. Cancer Discov. 2021, 11, OF1. [Google Scholar]
- Chen, Q.; Cai, D.; Li, M.; Wu, X. The homologous recombination protein RAD51 is a promising therapeutic target for cervical carcinoma. Oncol. Rep. 2017, 38, 767–774. [Google Scholar]
- King, H.O.; Brend, T.; Payne, H.L.; Wright, A.; Ward, T.A.; Patel, K.; Egnuni, T.; Stead, L.F.; Patel, A.; Wurdak, H.; et al. RAD51 Is a Selective DNA Repair Target to Radiosensitize Glioma Stem Cells. Stem Cell Rep. 2017, 8, 125–139. [Google Scholar]
- Jiang, K.; Deng, M.; Du, W.; Liu, T.; Li, J.; Zhou, Y. Functions and inhibitors of CHK1 in cancer therapy. Med. Drug Discov. 2024, 22, 100185. [Google Scholar]
- Do, K.T.; Kochupurakkal, B.; Kelland, S.; de Jonge, A.; Hedglin, J.; Powers, A.; Quinn, N.; Gannon, C.; Vuong, L.; Parmar, K.; et al. Phase 1 Combination Study of the CHK1 Inhibitor Prexasertib and the PARP Inhibitor Olaparib in High-grade Serous Ovarian Cancer and Other Solid Tumors. Clin. Cancer Res. 2021, 27, 4710–4716. [Google Scholar]
- Maron, S.B.; Sharma, M.R.; Spira, A.I.; Rivera, I.R.; Chawla, S.; Philipovskiy, A.; Johnson, M.; Falchook, G.; Wainberg, Z.; Shitara, K.; et al. Preclinical and phase 1/2 data of the CHK1 inhibitor BBI-355 in development for esophageal and gastric cancers (EGC) with EGFR or FGFR2 amplifications. J. Clin. Oncol. 2025, 43, 4. [Google Scholar]
- Farhoudi, L.; Maryam Hosseinikhah, S.; Vahdat-Lasemi, F.; Sukhorukov, V.N.; Kesharwani, P.; Sahebkar, A. Polymeric micelles paving the Way: Recent breakthroughs in camptothecin delivery for enhanced chemotherapy. Int. J. Pharm. 2024, 659, 124292. [Google Scholar] [PubMed]
- Saghatelyan, T.; Tananyan, A.; Janoyan, N.; Tadevosyan, A.; Petrosyan, H.; Hovhannisyan, A.; Hayrapetyan, L.; Arustamyan, M.; Arnhold, J.; Rotmann, A.R.; et al. Efficacy and safety of curcumin in combination with paclitaxel in patients with advanced, metastatic breast cancer: A comparative, randomized, double-blind, placebo-controlled clinical trial. Phytomedicine 2020, 70, 153218. [Google Scholar] [PubMed]
- Chen, Y.-X.; Gao, Q.-Y.; Zou, T.-H.; Wang, B.M.; Liu, S.D.; Sheng, J.Q.; Ren, J.L.; Zou, X.P.; Liu, Z.J.; Song, Y.Y.; et al. Berberine versus placebo for the prevention of recurrence of colorectal adenoma: A multicentre, double-blinded, randomised controlled study. Lancet Gastroenterol. Hepatol. 2020, 5, 267–275. [Google Scholar]
- Nemunaitis, J.; Tong, A.W.; Nemunaitis, M.; Senzer, N.; Phadke, A.P.; Bedell, C.; Adams, N.; Zhang, Y.A.; Maples, P.B.; Chen, S.; et al. A Phase I Study of Telomerase-specific Replication Competent Oncolytic Adenovirus (Telomelysin) for Various Solid Tumors. Mol. Ther. 2010, 18, 429–434. [Google Scholar]
- Liu, X.-G.; Li, M.; Mai, S.-J.; Cai, R.-J. Telomere length-related signature as a novel biomarker of prognosis and immune response in non-small cell lung cancer. Eur. Rev. Med. Pharmacol. Sci. 2022, 26, 1304–1319. [Google Scholar]
- Benati, M.; Montagnana, M.; Danese, E.; Mazzon, M.; Paviati, E.; Garzon, S.; Laganà, A.S.; Casarin, J.; Giudici, S.; Raffaelli, R.; et al. Aberrant Telomere Length in Circulating Cell-Free DNA as Possible Blood Biomarker with High Diagnostic Performance in Endometrial Cancer. Pathol. Oncol. Res. 2020, 26, 2281–2289. [Google Scholar]
- Peker Eyüboğlu, İ.; Koca, S.; Çelik, B.; Amuran, G.G.; Uğurlu, M.Ü.; Alan, Ö.; Telli, A.T.; Yumuk, P.F.; Akkiprik, M. Neoadjuvant Chemotherapy Shortens the cfDNA Telomere Length in Breast Cancer Patients. Int. J. Breast Cancer 2024, 2024, 6117394. [Google Scholar]
- Tomasova, K.; Seborova, K.; Kroupa, M.; Horak, J.; Kavec, M.; Vodickova, L.; Rob, L.; Hruda, M.; Mrhalova, M.; Bartakova, A.; et al. Telomere length as a predictor of therapy response and survival in patients diagnosed with ovarian carcinoma. Heliyon 2024, 10, e33525. [Google Scholar]
- Matsuda, Y.; Ye, J.; Yamakawa, K.; Mukai, Y.; Azuma, K.; Wu, L.; Masutomi, K.; Yamashita, T.; Daigo, Y.; Miyagi, Y.; et al. Association of longer telomere length in cancer cells and cancer-associated fibroblasts with worse prognosis. JNCI J. Natl. Cancer Inst. 2023, 115, 208–218. [Google Scholar]
- Trachu, N.; Reungwetwattana, T.; Meanwatthana, J.; Sukasem, C.; Majam, T.; Saengsiwaritt, W.; Jittikoon, J.; Udomsinprasert, W. Leukocytes telomere length as a biomarker of adverse drug reactions induced by Osimertinib in advanced non-small cell lung cancer. Sci. Rep. 2024, 14, 26543. [Google Scholar]
- Macha, S.J.; Koneru, B.; Burrow, T.A.; Zhu, C.; Savitski, D.; Rahman, R.L.; Ronaghan, C.A.; Nance, J.; McCoy, K.; Eslinger, C.; et al. Alternative Lengthening of Telomeres in Cancer Confers a Vulnerability to Reactivation of p53 Function. Cancer Res. 2022, 82, 3345–3358. [Google Scholar] [PubMed]
- Burrow, T.A.; Koneru, B.; Macha, S.J.; Sun, W.; Barr, F.G.; Triche, T.J.; Reynolds, C.P. Prevalence of alternative lengthening of telomeres in pediatric sarcomas determined by the telomeric DNA C-circle assay. Front. Oncol. 2024, 14, 1399442. [Google Scholar]
- Armando, R.; Cabrera, M.; Vilarullo, R.; Sun, W.; Barr, F.G.; Triche, T.J.; Reynolds, C.P. In vitro characterization and rational analog design of a novel inhibitor of telomerase assembly in MDA MB 231 breast cancer cell line. Oncol. Rep. 2022, 48, 188. [Google Scholar]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar]
- Recagni, M.; Bidzinska, J.; Zaffaroni, N.; Folini, M. The Role of Alternative Lengthening of Telomeres Mechanism in Cancer: Translational and Therapeutic Implications. Cancer 2020, 12, 949. [Google Scholar]
- Chen, Y.-Y.; Dagg, R.; Zhang, Y.; Lee, J.H.Y.; Lu, R.; La Rotta, M.N.; Sampl, S.; Korkut-Demirbaş, M.; Holzmann, K.; Lau, L.M.S.; et al. The C-Circle Biomarker Is Secreted by Alternative-Lengthening-of-Telomeres Positive Cancer Cells inside Exosomes and Provides a Blood-Based Diagnostic for ALT Activity. Cancer 2021, 13, 5369. [Google Scholar]
- Baylie, T.; Jemal, M.; Baye, G.; Getinet, M.; Amare, G.A.; Adugna, A.; Abebaw, D.; Hibstu, Z.; Tegegne, B.A.; Gugsa, E.; et al. The role of telomere and telomerase in cancer and novel therapeutic target: Narrative review. Front. Oncol. 2025, 15, 1542930. [Google Scholar]
- Wang, K.; Wang, R.-L.; Liu, J.-J.; Zhou, J.; Li, X.; Hu, W.W.; Jiang, W.J.; Hao, N.B. The prognostic significance of hTERT overexpression in cancers. Medicine 2018, 97, e11794. [Google Scholar]
- Raza, S.; Rajak, S.; Srivastava, J.; Tewari, A.; Gupta, P.; Chakravarti, B.; Ghosh, S.; Chaturvedi, C.P.; Sinha, R.A. ULK1 inhibition attenuates telomerase activity in hepatic cells. Biochim. Biophys. Acta Mol. Cell Res. 2022, 1869, 119355. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapeutic Strategy | Compound | Therapeutic Action | Key Clinical Outcomes | Reference |
|---|---|---|---|---|
| Telomerase-related strategies | ||||
| Oligonucleotide-based inhibitors | Imetelstat (GRN163L) | Inhibits hTR, induces telomere shortening and apoptosis | Imetelstat might be beneficial in the therapy of relapsed/refractory myelofibrosis | [84,85,88] |
| GRN163 | Similar to GRN163L, less effective due to reduced uptake | [86,87] | ||
| Small molecule inhibitors | BIBR1532 | Binds FVYL motif in hTERT; blocks telomerase, induces senescence | There are no current clinical trials | [91,92] |
| MST-312 | Telomere shortening; G2/M arrest and DDR activation | There are no current clinical trials | [94,95] | |
| Natural telomerase inhibitors | EGCG | Telomerase inhibition; apoptosis induction | Phase 2 trial; EGCG was well tolerated and slightly reduced the recurrence of colonic neoplasia (29% compared to 35% in placebo) | [111] |
| Camptothecin | Telomerase inhibition; DNA damage response | Although camptothecin itself was limited due to toxicity, a Phase 2 trial evaluating its derivative, irinotecan, in combination with bevacizumab demonstrated promising activity in patients with recurrent glioblastoma | [108,133] | |
| Curcumin | Telomerase inhibition; anti-oncogenic signalling | Phase 2 combining curcumin with paclitaxel in metastatic breast cancer patients showed a higher objective response rate compared to paclitaxel with placebo | [107,134] | |
| Resveratrol | Induces apoptosis and telomerase inhibition | Promising preclinical trials; definitive clinical efficacy results are pending | [110] | |
| Berberine | Telomerase inhibition; miRNA modulation | Phase 3 trial; berberine was well tolerated and reduced the recurrence rate of colorectal adenomas compared to placebo (36% vs. 47%) | [109,135] | |
| Immunotherapeutic telomerase vaccines | GV1001 | Anti-proliferative via GnRHR signalling; reduces angiogenesis | Phase 3 trial; combination therapy of GV1001 with chemotherapy in pancreatic cancer improved median overall survival and prolonged time to disease progression compared to chemotherapy alone | [97,98,99] |
| Vx-001 | Triggers hTERT-specific T-cell response | Phase 2 trial; median overall survival in patients with non-small cell lung cancer was increased | [101,103] | |
| Telomerase-targeted gene therapies | Suicide gene therapy | Cytotoxic gene proteins are selectively delivered to tumor cells. These genes are commonly regulated by hTERT promoter, allowing for targeted expression specifically in telomerase-active cancer cells. | There are no current clinical trials | [104] |
| Telomelysin | Oncolytic viruses are engineered to selectively replicate within telomerase-positive tumor cells and destroy cancer cells | Phase 1 clinical trials showed that Telomelysin was well-tolerated and significantly improved the results | [105,106,136] | |
| Inhibition of ALT | ||||
| G4 quadruplex stabilizing agents | Telomestatin | Stabilizes telomeric G4 structures; inhibits telomerase | Preclinical trials; promising results in vitro but not advanced to clinical trials due to challenges in synthesis and stability | [117] |
| RHPS4 | Induces telomere stability via shelterin disruption | There are no current clinical trials | [113,118] | |
| ATR inhibitors | VE-821 | Induce replication stress and telomeric DNA damage in ALT cells | There are no current clinical trials | [120,121] |
| Ceralasertib (AZD-6738) | Ceralasertib has demonstrated safety and well-tolerance along with anti-tumor response in a few clinical studies | |||
| RAD51 inhibitors | CYT-0851 | Blockage of HR process that ALT cells rely on to elongate telomeres | Phase 1 study showed responses and well-tolerance with manageable side effects, with improved | [127] |
| ri-1 | Showed promising results in preclinical studies but there are no current clinical trials | [128,129] | ||
| BO2 | ||||
| CHK1 inhibitors | Prexasertib | Impairing HR, thereby inducing DNA damage and replication stress within the cancer cells | Phase 1 clinical trials showed Prexasertib, in combination with Olaparib, shows therapeutic promise in patients with HGSOC who carry BRCA mutations and are PARP inhibitor-resistant | [131] |
| BBI-355 | Prevents cancer cells from properly managing and repairing DNA damage and replication stress | Preliminary clinical trial data suggest that the treatment is well tolerated | [132] | |
| Application Area | Cancer Type | Biomarker/Target | Key Findings | Method | Clinical Relevance | Telomere Connection (Targeted Molecule/Pathway) | Reference |
|---|---|---|---|---|---|---|---|
| Prognostic model | NSCLC | 18 telomere-related genes | Distinguishes high/low risk; predicts survival and PD-L1 therapy response | Gene expression profiling | Helps personalize immunotherapy | Telomere-related gene expression | [137] |
| Diagnostic biomarker | EEC | cfDNA relative TL | High sensitivity/specificity for diagnosis | Blood-based cfDNA measurement | Non-invasive early detection | Relative TL in cfDNA | [138] |
| Treatment monitoring | Breast cancer | cfDNA TL | Decreases post-chemotherapy in responders | Liquid biopsy | Tracks treatment response | cfDNA TL fluctuation | [139] |
| Predictive biomarker | Ovarian cancer | PBL TL, tumor TL | Shorter PBL TL = better chemosensitivity | qPCR + gene/methylation analysis | Predicts therapy response | Peripheral blood and tumor TL | [140] |
| Prognostic marker | Adenocarcinoma | TL in cancer cells and CAFs | Longer TL = worse prognosis | Tissue-based TL analysis | Correlates with survival | TL in cancer cells and fibroblasts | [141] |
| Adverse drug reaction predictor | NSCLC | Blood leukocyte TL | Shorter TL = higher risk of ADRs to Osimertinib | Leukocyte TL measurement | Patient safety stratification | TL as an indicator of drug tolerance | [142] |
| Therapeutic strategy | Various | hTERT | Imetelstat effective; manageable toxicity | Oligonucleotide therapy | Treats hematologic and solid tumors | Direct hTERT inhibition | [84] |
| Therapeutic target | ALT-positive tumors | p53 via APR-246 | Effective in combination with irinotecan | Drug combination therapy | Promising for ALT+ cancers | ALT pathway and p53 restoration | [143] |
| Diagnostic tool | ALT tumors | C-circles in exosomes | ALT-specific and stable in blood | Liquid biopsy | Non-invasive monitoring of ALT activity | ALT-specific telomeric DNA circles (C-circles) | [144] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rembiałkowska, N.; Sędzik, M.; Kisielewska, M.; Łuniewska, W.; Sebastianka, K.; Molik, K.; Skinderowicz, K.; Kuźnicki, J.; Tunikowska, J.; Kulbacka, J. Telomere Maintenance and DNA Repair: A Bidirectional Relationship in Cancer Biology and Therapy. Cancers 2025, 17, 2284. https://doi.org/10.3390/cancers17142284
Rembiałkowska N, Sędzik M, Kisielewska M, Łuniewska W, Sebastianka K, Molik K, Skinderowicz K, Kuźnicki J, Tunikowska J, Kulbacka J. Telomere Maintenance and DNA Repair: A Bidirectional Relationship in Cancer Biology and Therapy. Cancers. 2025; 17(14):2284. https://doi.org/10.3390/cancers17142284
Chicago/Turabian StyleRembiałkowska, Nina, Mikołaj Sędzik, Monika Kisielewska, Wiktoria Łuniewska, Kamil Sebastianka, Klaudia Molik, Katarzyna Skinderowicz, Jacek Kuźnicki, Joanna Tunikowska, and Julita Kulbacka. 2025. "Telomere Maintenance and DNA Repair: A Bidirectional Relationship in Cancer Biology and Therapy" Cancers 17, no. 14: 2284. https://doi.org/10.3390/cancers17142284
APA StyleRembiałkowska, N., Sędzik, M., Kisielewska, M., Łuniewska, W., Sebastianka, K., Molik, K., Skinderowicz, K., Kuźnicki, J., Tunikowska, J., & Kulbacka, J. (2025). Telomere Maintenance and DNA Repair: A Bidirectional Relationship in Cancer Biology and Therapy. Cancers, 17(14), 2284. https://doi.org/10.3390/cancers17142284