
The Role of ENHO in Pancreatic Adenocarcinoma: A Bioinformatics Approach †
 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Patient Population
2.2. Differential Gene and miRNA Expression
2.3. Survival Analysis
2.4. Mutation Analysis
2.5. Drug Sensitivity
2.6. Immune Infiltration
2.7. Protein–Protein Interactions and Co-Expressed Genes
2.8. Gene Set Enrichment Analysis and Transcription Factor Activity
2.9. mRNA-miRNA Interactions
3. Results
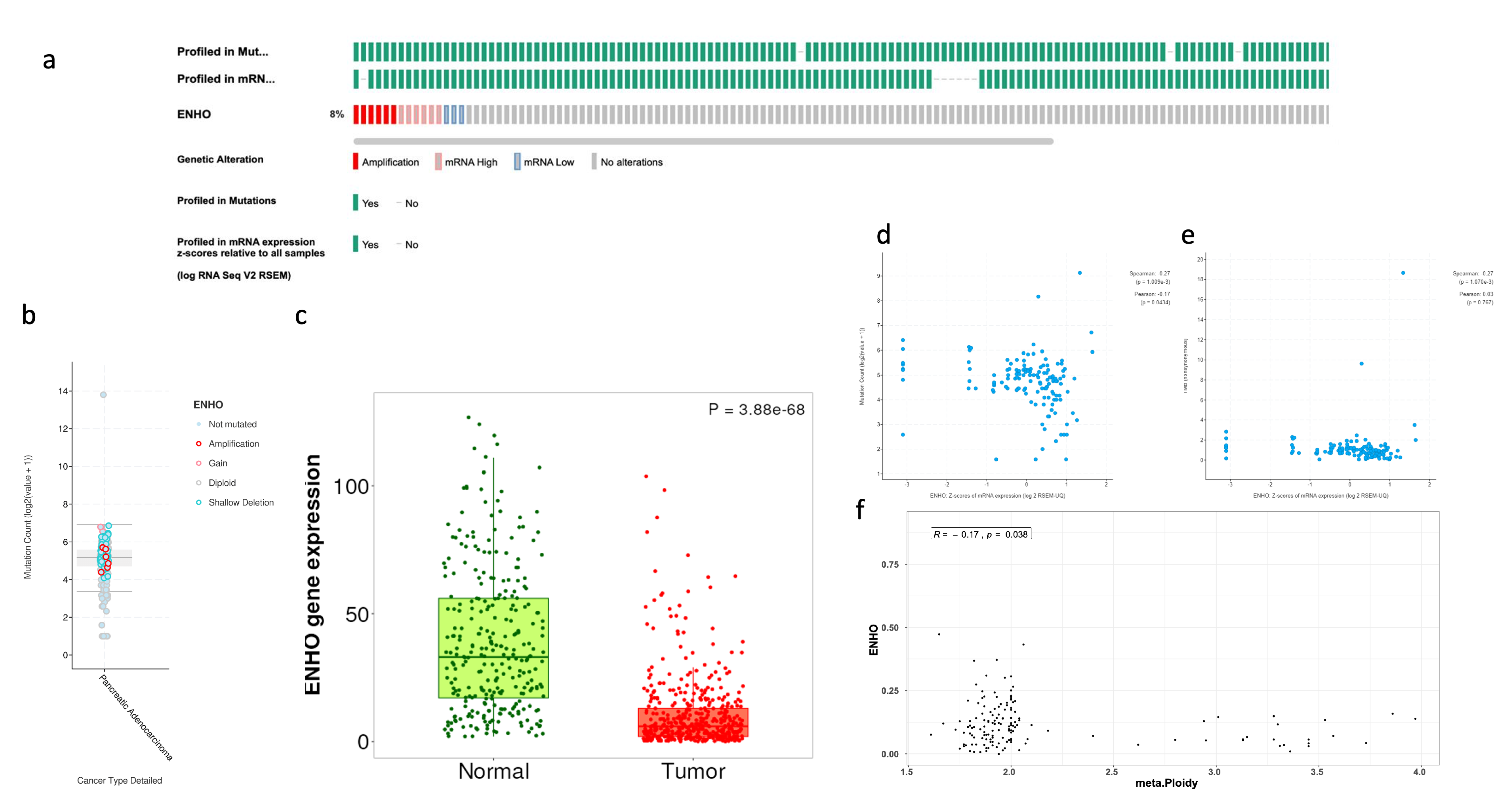
3.1. Comprehensive Analysis of ENHO Gene Alterations, Expression, and Clinical Correlations
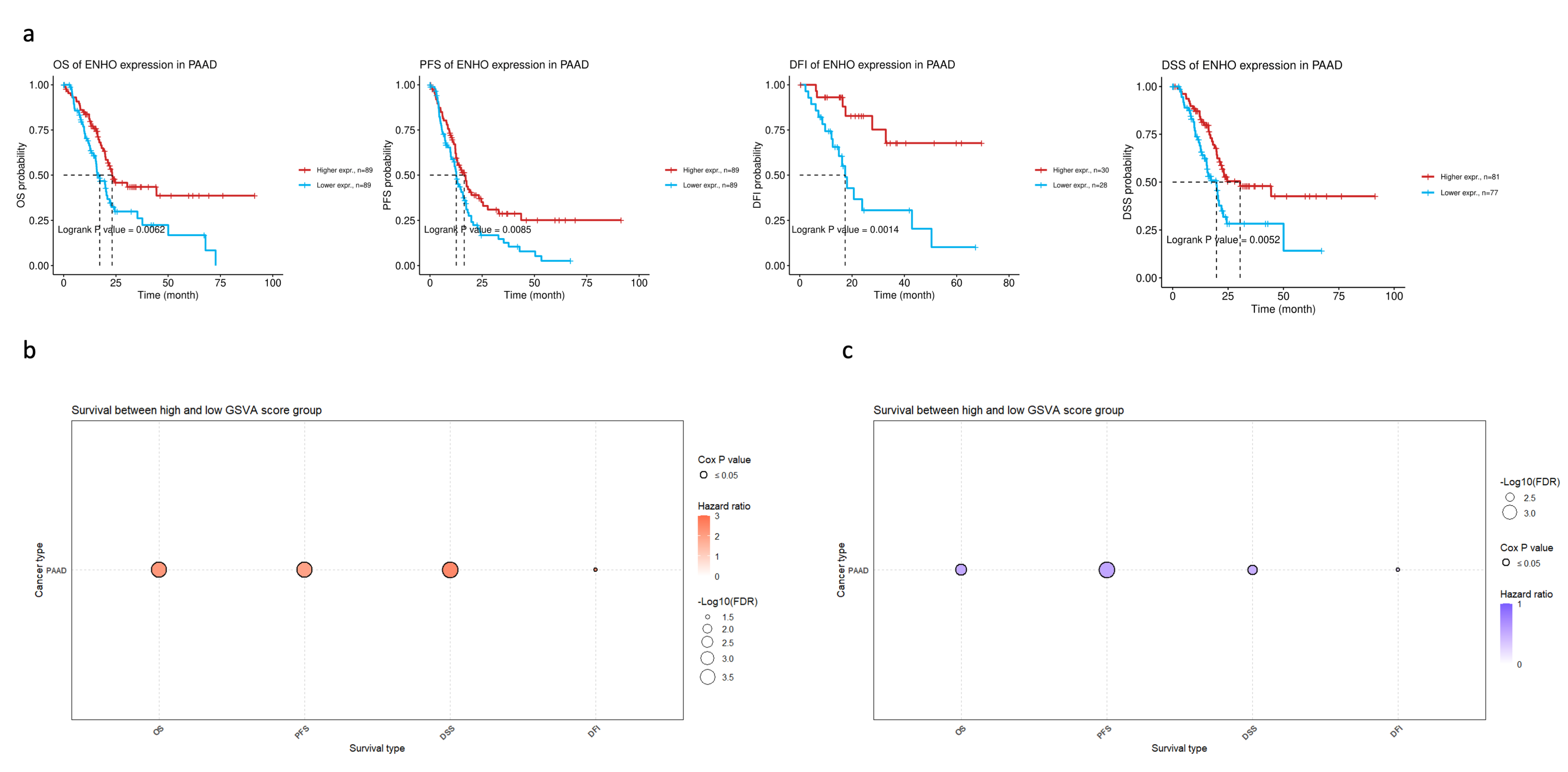
3.2. ENHO Expression in Different Survival Outcomes
3.3. ENHO Expression and Drug Sensitivity
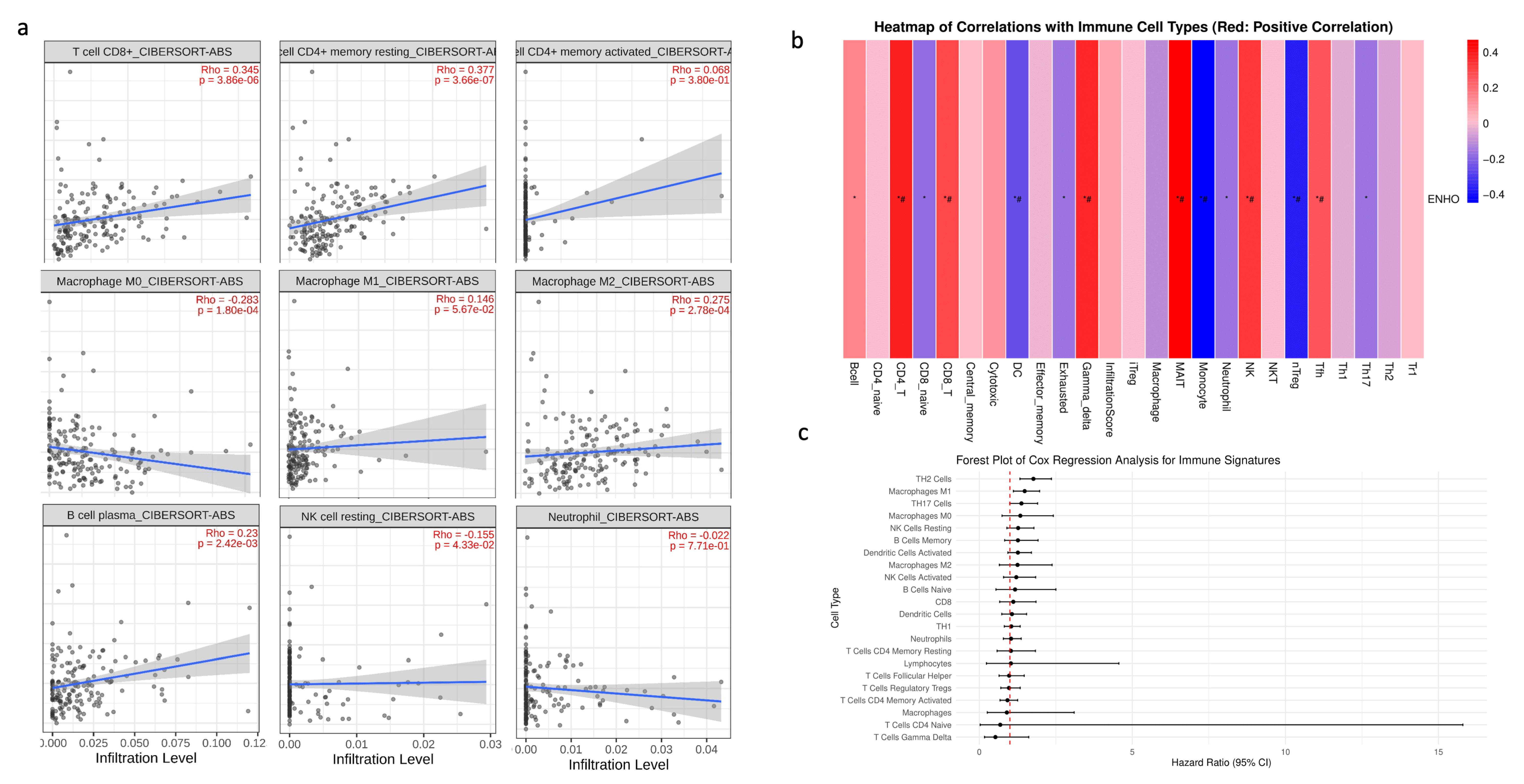
3.4. Immune Cell Infiltration
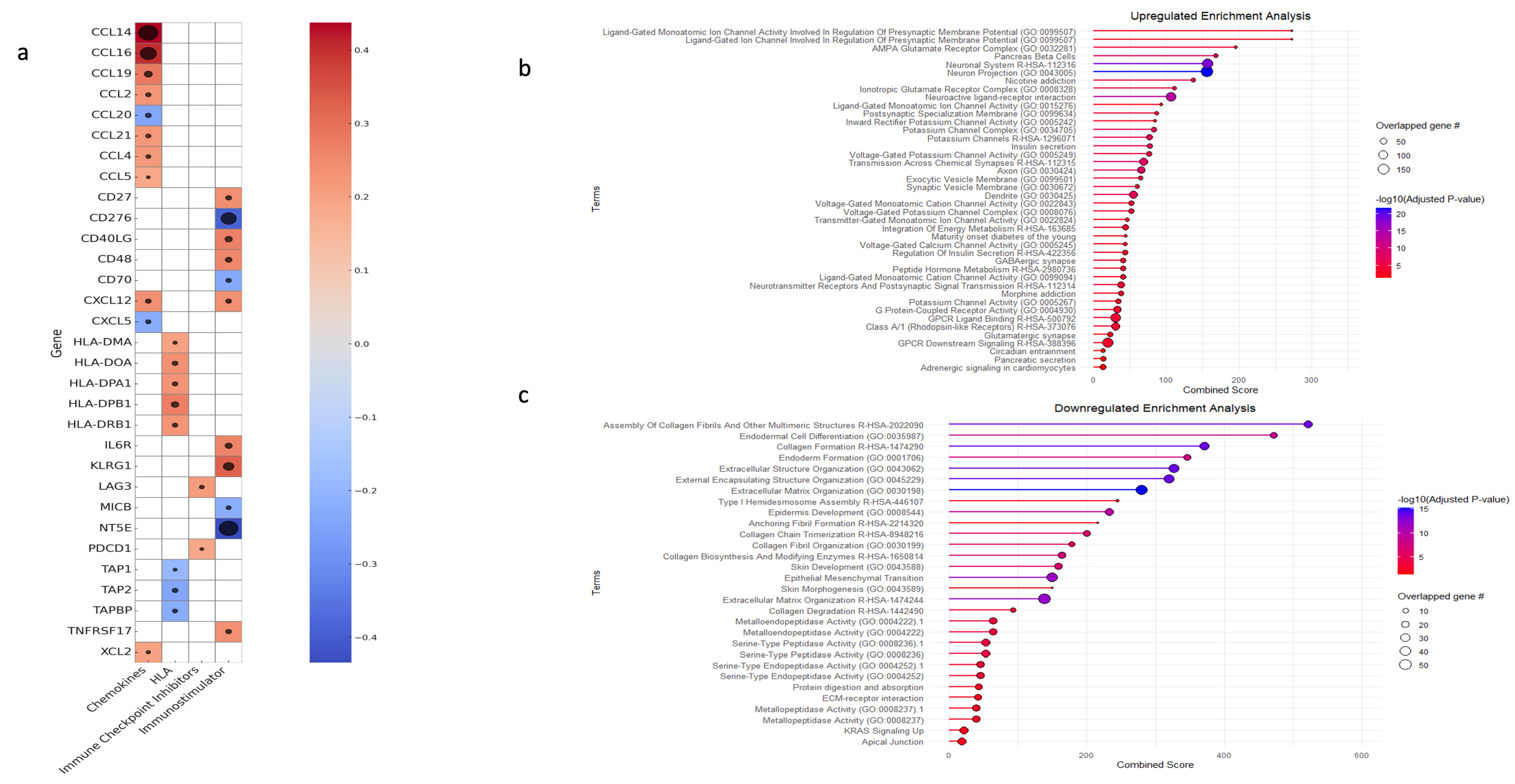
3.5. Gene Set Enrichment and Variation Analysis
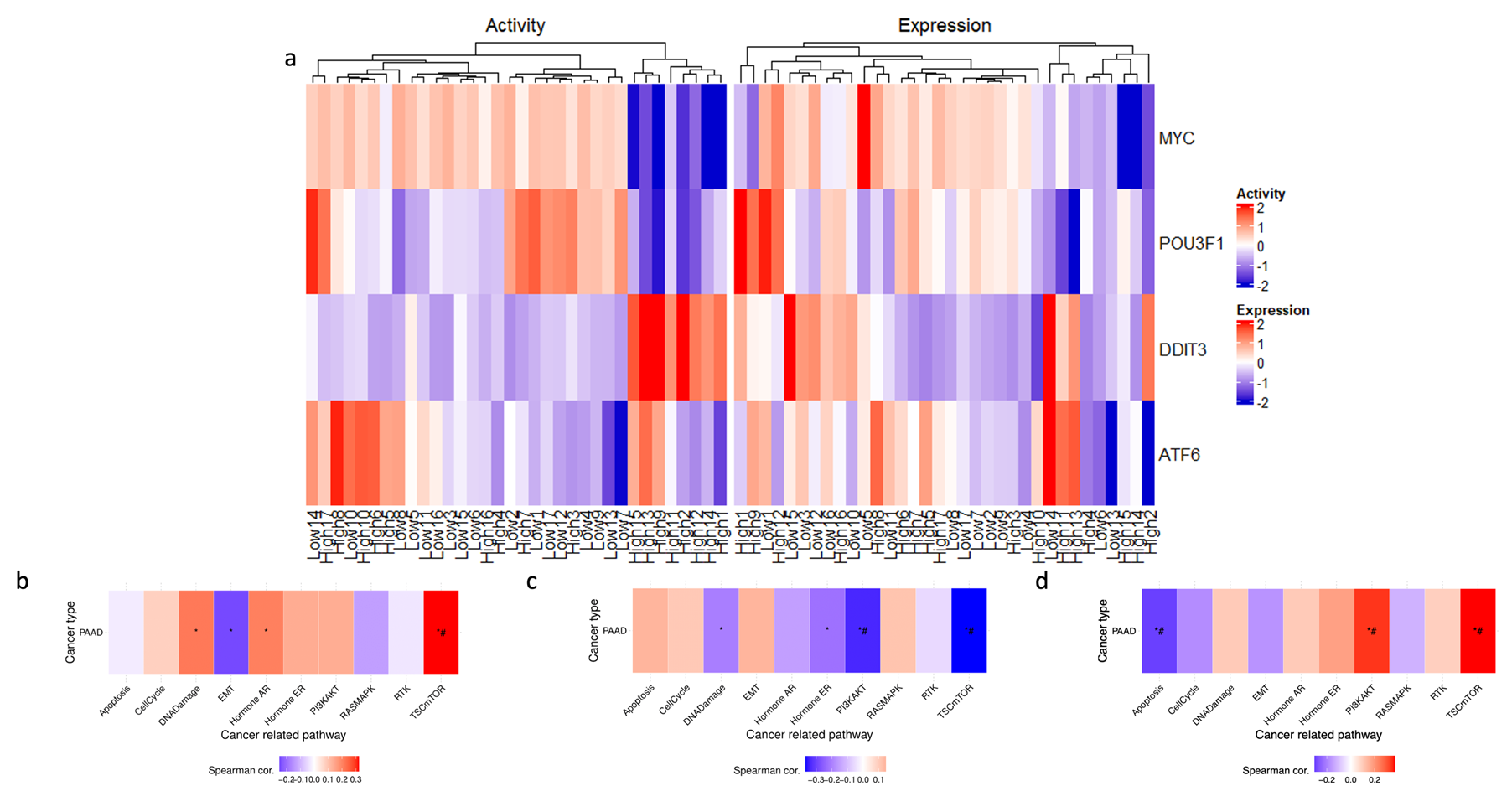
3.6. Transcription Factor Activity
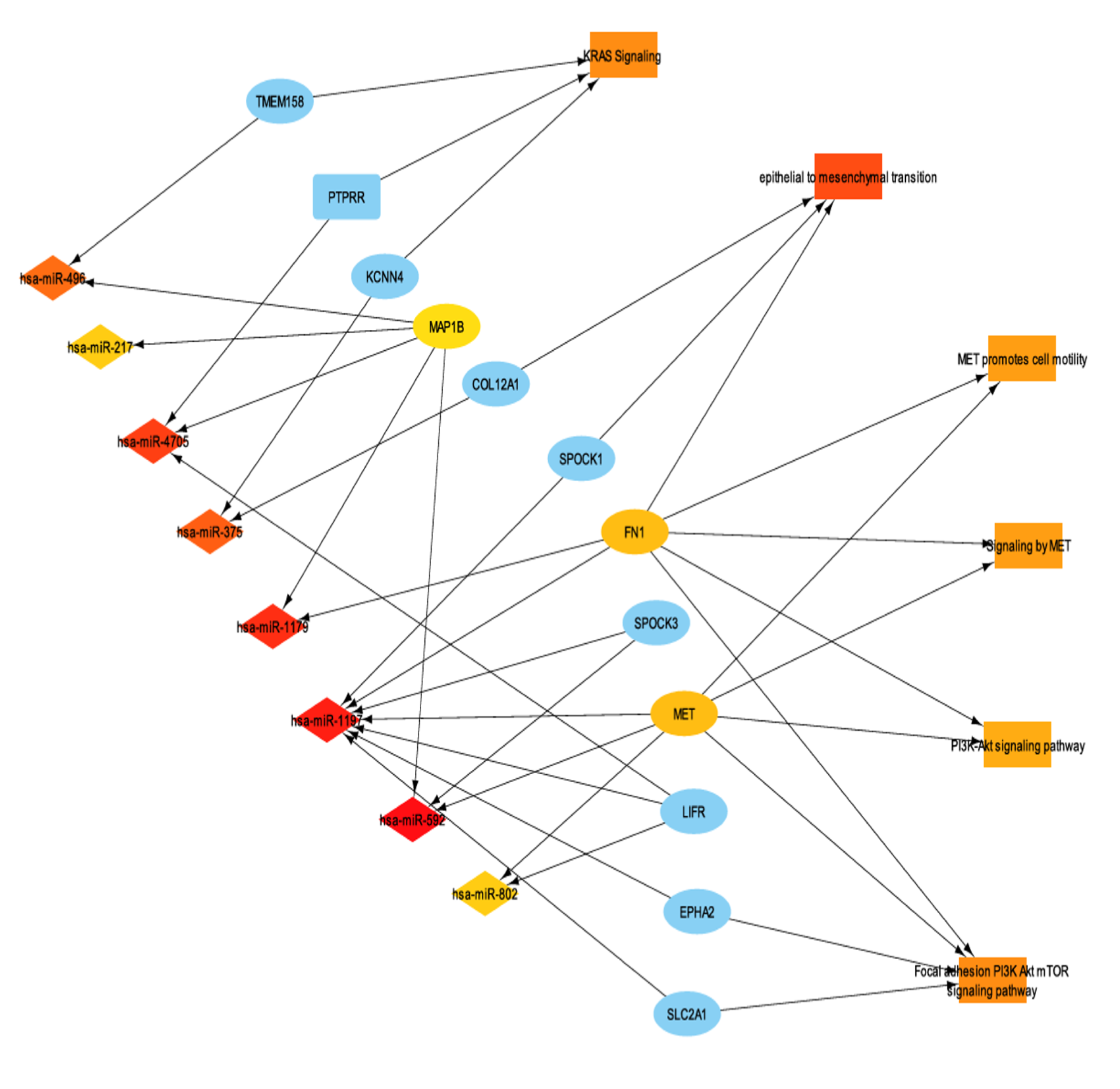
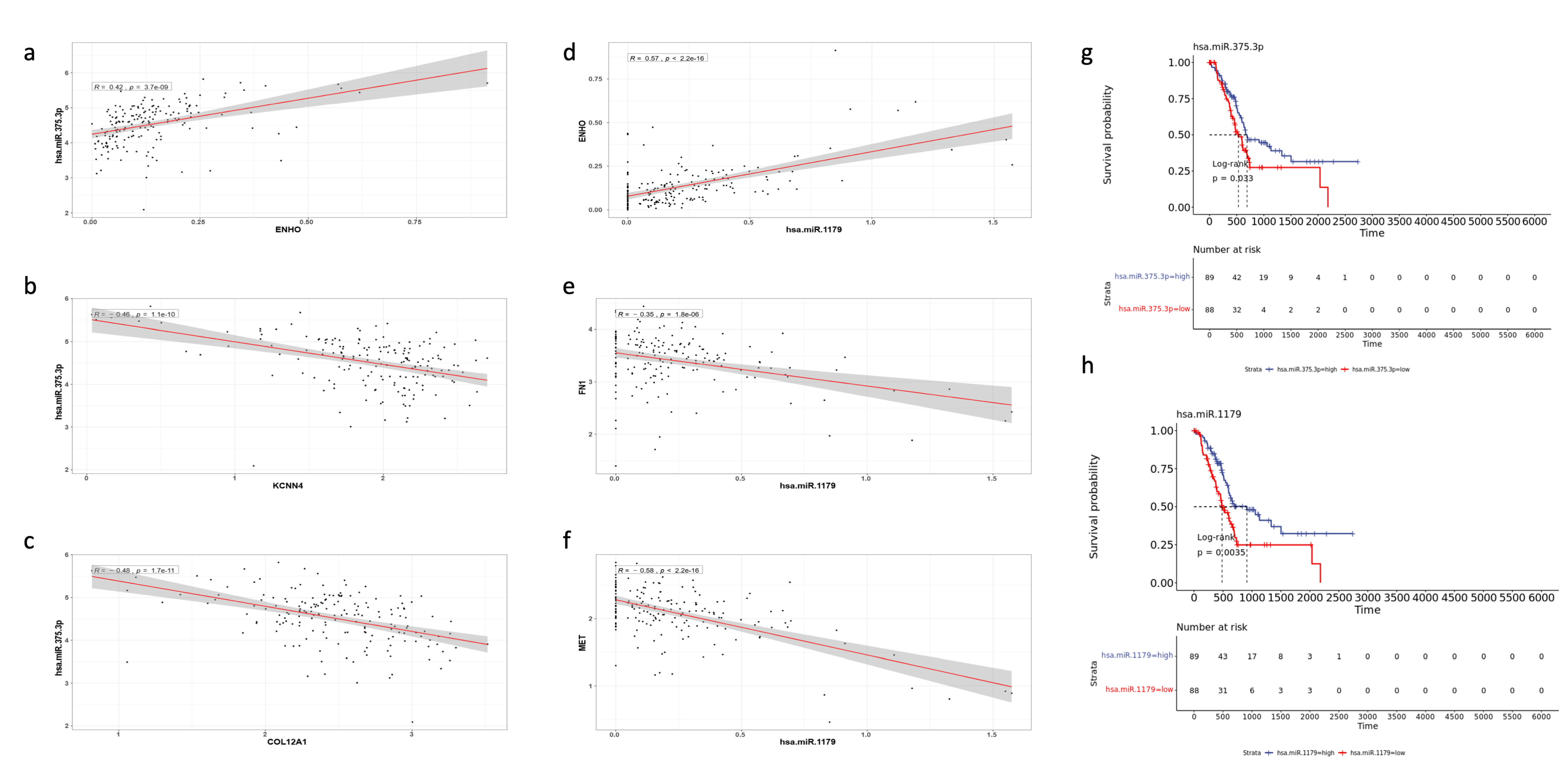
3.7. miRNA Differential Expression and miRNA-mRNA Interaction Networks
4. Discussion
4.1. ENHO Dysregulation in Pancreatic Cancer
4.2. ENHO and Tumor Microenvironment (TME)
4.3. Drug Sensitivity and Clinical Implications
4.4. Consistencies and Discrepancies with Prior Research
4.5. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Yang, G.; Yang, J.; Ren, B.; Wang, H.; Chen, G.; Zhao, F.; You, L.; Wang, W.; Zhao, Y. Metabolism of pancreatic cancer: Paving the way to better anticancer strategies. Mol. Cancer 2020, 19, 50. [Google Scholar] [CrossRef]
- Siegel, R.L.; Kratzer, T.B.; Giaquinto, A.N.; Sung, H.; Jemal, A. Cancer statistics, 2025. CA A Cancer J. Clin. 2025, 75, 10–45. [Google Scholar] [CrossRef]
- Ali, I.I.; D’Souza, C.; Singh, J.; Adeghate, E. Adropin’s Role in Energy Homeostasis and Metabolic Disorders. Int. J. Mol. Sci. 2022, 23, 8318. [Google Scholar] [CrossRef]
- Lovren, F.; Pan, Y.; Quan, A.; Singh, K.K.; Shukla, P.C.; Gupta, M.; Al-Omran, M.; Teoh, H.; Verma, S. Adropin is a novel regulator of endothelial function. Circulation 2010, 122, S185–S192. [Google Scholar] [CrossRef]
- Jasaszwili, M.; Billert, M.; Strowski, M.Z.; Nowak, K.W.; Skrzypski, M. Adropin as A Fat-Burning Hormone with Multiple Functions-Review of a Decade of Research. Molecules 2020, 25, 549. [Google Scholar] [CrossRef]
- Jia, L.; Liao, L.; Jiang, Y.; Hu, X.; Lu, G.; Xiao, W.; Gong, W.; Jia, X.; Xiaoqin, J. Low-dose adropin stimulates inflammasome activation of macrophage via mitochondrial ROS involved in colorectal cancer progression. BMC Cancer 2023, 23, 1042. [Google Scholar] [CrossRef]
- Hu, J.; Wu, Q.; Ding, Q.; Wu, W.; Li, Q.; Zheng, Z. High Level of Adropin Promotes the Progression of Pancreatic Ductal Adenocarcinoma. Curr. Cancer Drug Targets 2024, 24, 629–641. [Google Scholar] [CrossRef]
- Bartha, Á.; Győrffy, B. TNMplot.com: A Web Tool for the Comparison of Gene Expression in Normal, Tumor and Metastatic Tissues. Int. J. Mol. Sci. 2021, 22, 2622. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. Available online: https://pubmed.ncbi.nlm.nih.gov/25516281/ (accessed on 17 September 2024). [CrossRef]
- Therneau, T.M.; Grambsch, P.M. Modeling Survival Data: Extending the Cox Model; Springer: New York, NY, USA, 2000. [Google Scholar]
- Kassambara, A.; Kosinski, M.; Biecek, P. Survminer: Drawing Survival Curves Using “ggplot2” [Internet]. 2021. Available online: https://github.com/kassambara/survminer (accessed on 30 April 2024).
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–14. [Google Scholar] [CrossRef] [PubMed]
- GSCA: Gene Set Cancer Analysis [Internet]. Available online: https://guolab.wchscu.cn/GSCA/#/ (accessed on 14 December 2024).
- TCGExplorer [Internet]. Available online: https://tcgex.iyte.edu.tr/ (accessed on 14 December 2024).
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.E.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal 2013, 6, pl1. Available online: https://pubmed.ncbi.nlm.nih.gov/23550210/ (accessed on 23 August 2024). [CrossRef] [PubMed]
- de Bruijn, I.; Kundra, R.; Mastrogiacomo, B.; Tran, T.N.; Sikina, L.; Mazor, T.; Li, X.; Ochoa, A.; Zhao, G.; Lai, B.; et al. Analysis and Visualization of Longitudinal Genomic and Clinical Data from the AACR Project GENIE Biopharma Collaborative in cBioPortal. Cancer Res. 2023, 83, 3861–3867. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. Available online: https://pubmed.ncbi.nlm.nih.gov/22588877/ (accessed on 23 August 2024). [CrossRef]
- Miao, Y.R.; Zhang, Q.; Lei, Q.; Luo, M.; Xie, G.Y.; Wang, H.; Guo, A.Y. ImmuCellAI: A Unique Method for Comprehensive T-Cell Subsets Abundance Prediction and its Application in Cancer Immunotherapy. Adv. Sci. 2020, 7, 1902880. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-Protein Interaction Networks, Integrated Over the Tree of Life. Nucleic Acids Res. 2015, 43, D447–D452. Available online: https://pubmed.ncbi.nlm.nih.gov/25352553/ (accessed on 23 August 2024). [CrossRef]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. Available online: https://pubmed.ncbi.nlm.nih.gov/33780170/ (accessed on 23 August 2024). [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A Comprehensive Gene Set Enrichment Analysis Web Server 2016 Update. Nucleic Acids Res. 2016, 44, W90–W97. Available online: https://pubmed.ncbi.nlm.nih.gov/27141961/ (accessed on 23 August 2024). [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’Ayan, A. Enrichr: Interactive and Collaborative HTML5 Gene List Enrichment Analysis Tool. BMC Bioinform. 2013, 14, 128. Available online: https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-14-128 (accessed on 23 August 2024). [CrossRef]
- Su, K.; Katebi, A.; Kohar, V.; Clauss, B.; Gordin, D.; Qin, Z.S.; Karuturi, R.K.M.; Li, S.; Lu, M. NetAct: A Computational Platform to Construct Core Transcription Factor Regulatory Networks Using Gene Activity. Genome Biol. 2022, 23, 270. Available online: https://genomebiology.biomedcentral.com/articles/10.1186/s13059-022-02835-3 (accessed on 14 December 2024). [CrossRef]
- LinkedOmics Suite [Internet]. Available online: https://www.linkedomics.org/#/ (accessed on 14 December 2024).
- Manojlovic, N.; Savic, G.; Manojlovic, S. Neoadjuvant treatment of pancreatic ductal adenocarcinoma: Whom, when and how. World J. Gastrointest. Surg. 2024, 16, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Younis, O.M.; Yasin, J.A.; Saeed, A.; Qubbaj, F.B.; Al-Sharif, Z.K.; Saeed, A. ENHO’s Protective Role in Pancreatic Adenocarcinoma. In Proceedings of the ESMO Gastrointestinal Cancers Congress 2024, Munich, Germany, 26–29 June 2024. [Google Scholar] [CrossRef]
- Labbé, K.; LeBon, L.; King, B.; Vu, N.; Stoops, E.H.; Ly, N.; Lefebvre, A.E.Y.T.; Seitzer, P.; Krishnan, S.; Heo, J.-M.; et al. Specific activation of the integrated stress response uncovers regulation of central carbon metabolism and lipid droplet biogenesis. Nat. Commun. 2024, 15, 8301. [Google Scholar] [CrossRef] [PubMed]
- Flieswasser, T.; Eynde, A.V.D.; Van Audenaerde, J.; De Waele, J.; Lardon, F.; Riether, C.; de Haard, H.; Smits, E.; Pauwels, P.; Jacobs, J. The CD70-CD27 axis in oncology: The new kids on the block. J. Exp. Clin. Cancer Res. 2022, 41, 12. [Google Scholar] [CrossRef]
- Getu, A.A.; Tigabu, A.; Zhou, M.; Lu, J.; Fodstad, Ø.; Tan, M. New frontiers in immune checkpoint B7-H3 (CD276) research and drug development. Mol. Cancer 2023, 22, 43. [Google Scholar] [CrossRef]
- Yang, M.; Tang, X.; Zhang, Z.; Gu, L.; Wei, H.; Zhao, S.; Zhong, K.; Mu, M.; Huang, C.; Jiang, C.; et al. Tandem CAR-T cells targeting CD70 and B7-H3 exhibit potent preclinical activity against multiple solid tumors. Theranostics 2020, 10, 7622–7634. [Google Scholar] [CrossRef]
- Kordaß, T.; Osen, W.; Eichmüller, S.B. Controlling the immune suppressor: Transcription factors and microRNAs regulating CD73/NT5E. Front. Immunol. 2018, 9, 813. [Google Scholar] [CrossRef]
- Li, Y.; Su, Z.; Wei, B.; Qin, M.; Liang, Z. Bioinformatics analysis identified MMP14 and COL12A1 as immune-related biomarkers associated with pancreatic adenocarcinoma prognosis. Math. Biosci. Eng. MBE 2021, 18, 5921–5942. [Google Scholar] [CrossRef]
- Xiao, H.N.; Zhao, Z.Y.; Li, J.P.; Li, A.Y. Comprehensive pan-cancer analysis: Essential role of ABCB family genes in cancer. Transl. Cancer Res. 2024, 13, 1642–1664. [Google Scholar] [CrossRef]
- Riedl, J.M.; Posch, F.; Horvath, L.; Gantschnigg, A.; Renneberg, F.; Schwarzenbacher, E.; Moik, F.; Barth, D.A.; Rossmann, C.H.; Stotz, M.; et al. Gemcitabine/nab-Paclitaxel versus FOLFIRINOX for palliative first-line treatment of advanced pancreatic cancer: A propensity score analysis. Eur. J. Cancer 2021, 151, 3–13. [Google Scholar] [CrossRef]
- Chen, J.; Liao, S.; Xiao, Z.; Pan, Q.; Wang, X.; Shen, K.; Wang, S.; Yang, L.; Guo, F.; Liu, H.-F.; et al. The development and improvement of immunodeficient mice and humanized immune system mouse models. Front. Immunol. 2022, 13, 1007579. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, Q.; Lin, X.; Chen, M.; Liu, Q. A review of Adropin as the medium of dialogue between energy regulation and immune regulation. Oxid. Med. Cell. Longev. 2020, 2020, 3947806. [Google Scholar] [CrossRef] [PubMed]
- Stelcer, E.; Milecka, P.; Komarowska, H.; Jopek, K.; Tyczewska, M.; Szyszka, M.; Lesniczak, M.; Suchorska, W.; Bekova, K.; Szczepaniak, B.; et al. Adropin Stimulates Proliferation and Inhibits Adrenocortical Steroidogenesis in the Human Adrenal Carcinoma (HAC15) Cell Line. Front. Endocrinol. 2020, 11, 561370. [Google Scholar] [CrossRef] [PubMed]
- Tuna, B.G.; Atalay, P.B.; Altunbek, M.; Kalkan, B.M.; Dogan, S. Effects of Chronic and Intermittent Calorie Restriction on Adropin Levels in Breast Cancer. Nutr. Cancer 2017, 69, 1003–1010. [Google Scholar] [CrossRef]
- Rao, A.; Herr, D.R. G protein-coupled receptor GPR19 regulates E-cadherin expression and invasion of breast cancer cells. Biochim. Biophys. acta. Mol. Cell Res. 2017, 1864, 1318–1327. [Google Scholar] [CrossRef]
- Roca, H.; Hernandez, J.; Weidner, S.; McEachin, R.C.; Fuller, D.; Sud, S.; Schumann, T.; Wilkinson, J.E.; Zaslavsky, A.; Li, H.; et al. Transcription factors OVOL1 and OVOL2 induce the mesenchymal to epithelial transition in human cancer. PLoS ONE 2013, 8, e76773. [Google Scholar] [CrossRef]
- Nergiz, S.; Altinkaya, S.O.; Kurt Ömürlü, İ.; Yuksel, H.; Küçük, M.; Demircan Sezer, S. Circulating adropin levels in patients with endometrium cancer. Gynecol. Endocrinol. Off. J. Int. Soc. Gynecol. Endocrinol. 2015, 31, 730–735. [Google Scholar] [CrossRef]
- Lin, C.; Hu, Z.; Yuan, G.; Su, H.; Zeng, Y.; Guo, Z.; Zhong, F.; Jiang, K.; He, S. MicroRNA-1179 inhibits the proliferation, migration, and invasion of human pancreatic cancer cells by targeting E2F5. Chem.-Biol. Interact. 2018, 291, 65–71. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Chang, X.; Liu, X.; Wang, H.; Yang, X.; Gu, Y. Glycolysis in the progression of pancreatic cancer. Am. J. Cancer Res. 2022, 12, 861–872. [Google Scholar]
- Zheng, D.; Deng, Y.; Deng, L.; He, Z.; Sun, X.; Gong, Y.; Shi, B.; Lu, D.; Yu, C. CDCA7 enhances STAT3 transcriptional activity to regulate aerobic glycolysis and promote pancreatic cancer progression and gemcitabine resistance. Cell Death Dis. 2025, 16, 68. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Younis, O.M.; Al-Sharif, Z.K.; Saeed, A.E.; Qubbaj, F.B.; Yasin, J.A.; Nour, T.; Alami Idrissi, Y.; Saeed, A. The Role of ENHO in Pancreatic Adenocarcinoma: A Bioinformatics Approach. Cancers 2025, 17, 2139. https://doi.org/10.3390/cancers17132139
Younis OM, Al-Sharif ZK, Saeed AE, Qubbaj FB, Yasin JA, Nour T, Alami Idrissi Y, Saeed A. The Role of ENHO in Pancreatic Adenocarcinoma: A Bioinformatics Approach. Cancers. 2025; 17(13):2139. https://doi.org/10.3390/cancers17132139
Chicago/Turabian StyleYounis, Osama M., Zeid K. Al-Sharif, Ahmad E. Saeed, Fares B. Qubbaj, Jehad A. Yasin, Tasnim Nour, Yassine Alami Idrissi, and Anwaar Saeed. 2025. "The Role of ENHO in Pancreatic Adenocarcinoma: A Bioinformatics Approach" Cancers 17, no. 13: 2139. https://doi.org/10.3390/cancers17132139
APA StyleYounis, O. M., Al-Sharif, Z. K., Saeed, A. E., Qubbaj, F. B., Yasin, J. A., Nour, T., Alami Idrissi, Y., & Saeed, A. (2025). The Role of ENHO in Pancreatic Adenocarcinoma: A Bioinformatics Approach. Cancers, 17(13), 2139. https://doi.org/10.3390/cancers17132139