
Quantifying Genetic and Environmental Factors Accounting for Multistage Progression of Precancerous Lesions and Oral Cancer: Applications to Risk-Guided Prevention

 , , , and
, , , and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Source
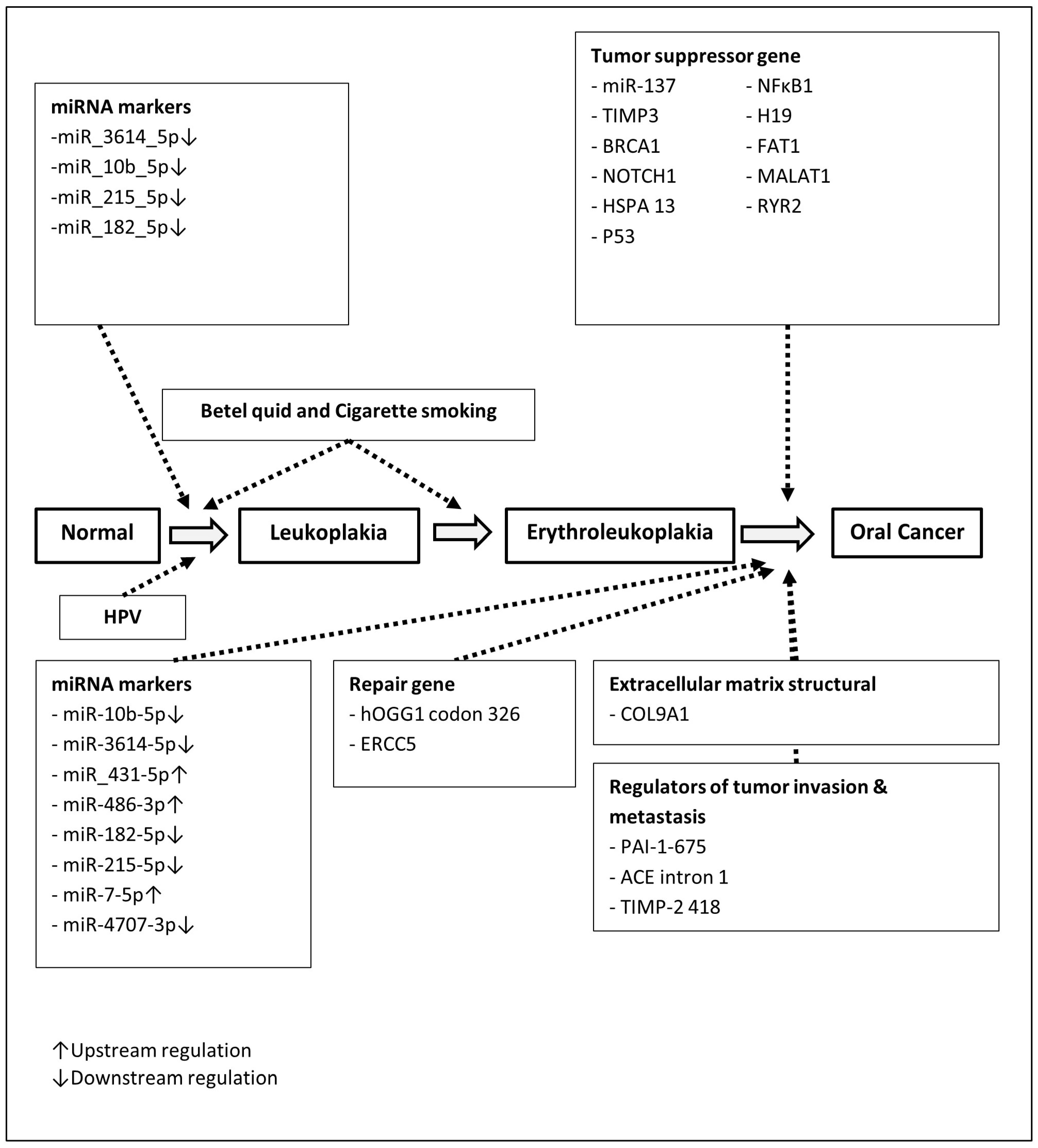
2.2. Multistate and Multifactorial Natural History Model of Precancerous and Cancer of Oral Cavity
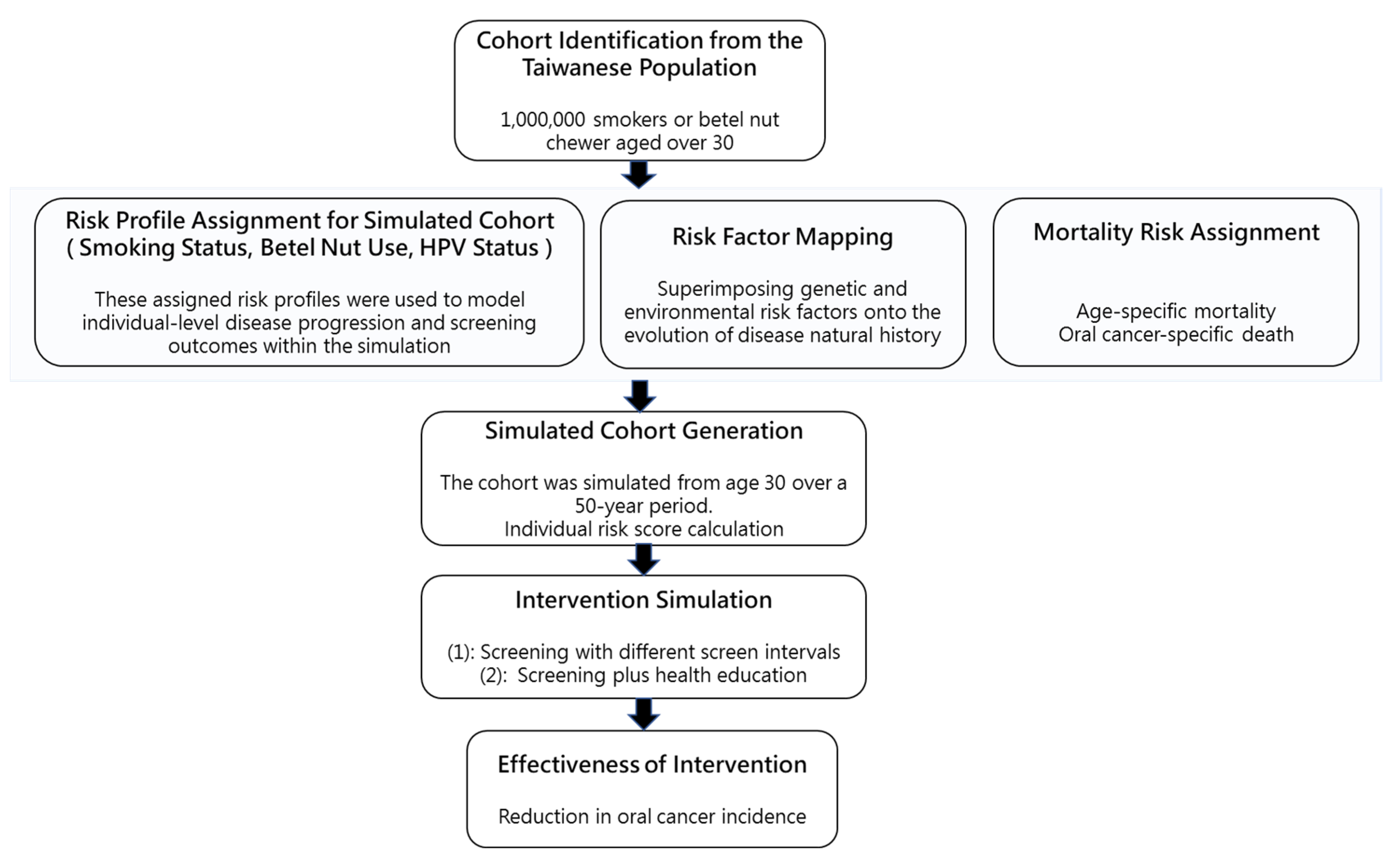
2.3. Computer Simulation
3. Results
3.1. Literature-Review-Derived Odds Ratios Associated with Environmental Factors and Genetic Susceptibility
3.2. Multistate Risk Score Developments
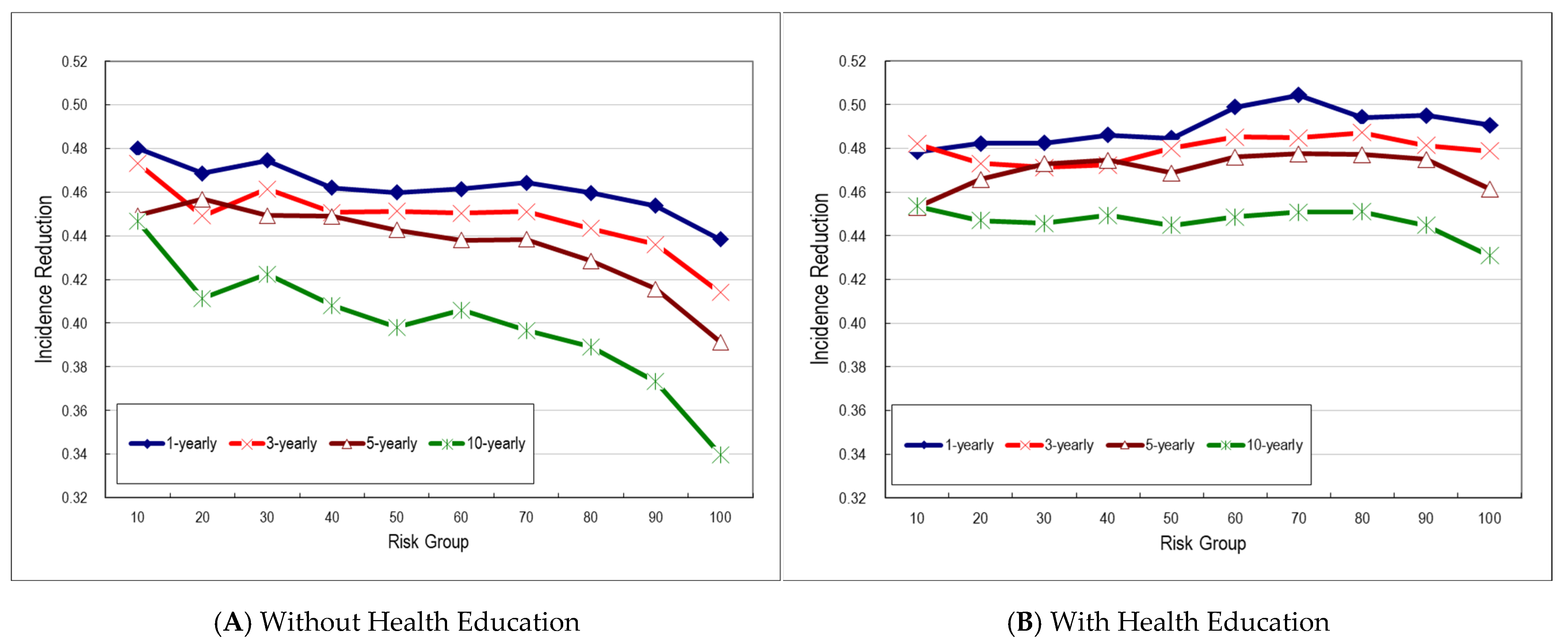
3.3. Decile Risk Spectrums of Multistate Transitions
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ram, H.; Sarkar, J.; Kumar, H.; Konwar, R.; Bhatt, M.L.; Mohammad, S. Oral cancer: Risk factors and molecular pathogenesis. J. Maxillofac. Oral. Surg. 2011, 10, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Chai, A.W.Y.; Lim, K.P.; Cheong, S.C. Translational genomics and recent advances in oral squamous cell carcinoma. Semin. Cancer Biol. 2020, 61, 71–83. [Google Scholar] [CrossRef]
- Tan, Y.; Wang, Z.; Xu, M.; Li, B.; Huang, Z.; Qin, S.; Nice, E.C.; Tang, J.; Huang, C. Oral squamous cell carcinomas: State of the field and emerging directions. Int. J. Oral. Sci. 2023, 15, 44. [Google Scholar] [CrossRef]
- Bouquot, J.E.; Whitaker, S.B. Oral leukoplakia--rationale for diagnosis and prognosis of its clinical subtypes or “phases”. Quintessence Int. 1994, 25, 133–140. [Google Scholar]
- Speight, P.M.; Khurram, S.A.; Kujan, O. Oral potentially malignant disorders: Risk of progression to malignancy. Oral. Surg. Oral. Med. Oral. Pathol. Oral. Radiol. 2018, 125, 612–627. [Google Scholar] [CrossRef] [PubMed]
- Chou, Y.E.; Hsieh, M.J.; Hsin, C.H.; Chiang, W.L.; Lai, Y.C.; Lee, Y.H.; Huang, S.C.; Yang, S.F.; Lin, C.W. CD44 gene polymorphisms and environmental factors on oral cancer susceptibility in Taiwan. PLoS ONE 2014, 9, e93692. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.H.; Chuang, C.Y.; Lin, C.W.; Su, S.C.; Chang, L.C.; Wu, S.W.; Liu, Y.F.; Yang, S.F. Effect of MACC1 Genetic Polymorphisms and Environmental Risk Factors in the Occurrence of Oral Squamous Cell Carcinoma. J. Pers. Med. 2021, 11, 490. [Google Scholar] [CrossRef]
- Chung, C.M.; Lee, C.H.; Chen, M.K.; Lee, K.W.; Lan, C.E.; Kwan, A.L.; Tsai, M.H.; Ko, Y.C. Combined Genetic Biomarkers and Betel Quid Chewing for Identifying High-Risk Group for Oral Cancer Occurrence. Cancer Prev. Res. 2017, 10, 355–362. [Google Scholar] [CrossRef]
- Shiu, M.N.; Chen, T.H. Impact of betel quid, tobacco and alcohol on three-stage disease natural history of oral leukoplakia and cancer: Implication for prevention of oral cancer. Eur. J. Cancer Prev. 2004, 13, 39–45. [Google Scholar] [CrossRef]
- Yen, A.M.; Chen, S.C.; Chang, S.H.; Chen, T.H. The effect of betel quid and cigarette on multistate progression of oral pre-malignancy. J. Oral. Pathol. Med. 2008, 37, 417–422. [Google Scholar] [CrossRef]
- Jithesh, P.V.; Risk, J.M.; Schache, A.G.; Dhanda, J.; Lane, B.; Liloglou, T.; Shaw, R.J. The epigenetic landscape of oral squamous cell carcinoma. Br. J. Cancer 2013, 108, 370–379. [Google Scholar] [CrossRef]
- Ali, J.; Sabiha, B.; Jan, H.U.; Haider, S.A.; Khan, A.A.; Ali, S.S. Genetic etiology of oral cancer. Oral. Oncol. 2017, 70, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Carenzo, A.; Serafini, M.S.; Roca, E.; Paderno, A.; Mattavelli, D.; Romani, C.; Saintigny, P.; Koljenović, S.; Licitra, L.; De Cecco, L.; et al. Gene Expression Clustering and Selected Head and Neck Cancer Gene Signatures Highlight Risk Probability Differences in Oral Premalignant Lesions. Cells 2020, 9, 1828. [Google Scholar] [CrossRef] [PubMed]
- Díez-Pérez, R.; Campo-Trapero, J.; Cano-Sánchez, J.; López-Durán, M.; Gonzalez-Moles, M.A.; Bascones-Ilundain, J.; Bascones-Martinez, A. Methylation in oral cancer and pre-cancerous lesions (Review). Oncol. Rep. 2011, 25, 1203–1209. [Google Scholar]
- Cervigne, N.K.; Reis, P.P.; Machado, J.; Sadikovic, B.; Bradley, G.; Galloni, N.N.; Pintilie, M.; Jurisica, I.; Perez-Ordonez, B.; Gilbert, R.; et al. Identification of a microRNA signature associated with progression of leukoplakia to oral carcinoma. Hum. Mol. Genet. 2009, 18, 4818–4829. [Google Scholar] [CrossRef] [PubMed]
- Deng, N.; Zhou, H.; Fan, H.; Yuan, Y. Single nucleotide polymorphisms and cancer susceptibility. Oncotarget 2017, 8, 110635–110649. [Google Scholar] [CrossRef]
- Brunotto, M.; Zarate, A.M.; Bono, A.; Barra, J.L.; Berra, S. Risk genes in head and neck cancer: A systematic review and meta-analysis of last 5 years. Oral. Oncol. 2014, 50, 178–188. [Google Scholar] [CrossRef]
- Sankaranarayanan, R.; Ramadas, K.; Thomas, G.; Muwonge, R.; Thara, S.; Mathew, B.; Rajan, B. Effect of screening on oral cancer mortality in Kerala, India: A cluster-randomised controlled trial. Lancet 2005, 365, 1927–1933. [Google Scholar] [CrossRef]
- Chuang, S.L.; Su, W.W.; Chen, S.L.; Yen, A.M.; Wang, C.P.; Fann, J.C.; Chiu, S.Y.; Lee, Y.C.; Chiu, H.M.; Chang, D.C.; et al. Population-based screening program for reducing oral cancer mortality in 2,334,299 Taiwanese cigarette smokers and/or betel quid chewers. Cancer 2017, 123, 1597–1609. [Google Scholar] [CrossRef]
- Ho, P.S.; Wang, W.C.; Huang, Y.T.; Yang, Y.H. Finding an oral potentially malignant disorder in screening program is related to early diagnosis of oral cavity cancer—Experience from real world evidence. Oral. Oncol. 2019, 89, 107–114. [Google Scholar] [CrossRef]
- Syrjänen, S.; Lodi, G.; von Bültzingslöwen, I.; Aliko, A.; Arduino, P.; Campisi, G.; Challacombe, S.; Ficarra, G.; Flaitz, C.; Zhou, H.M.; et al. Human papillomaviruses in oral carcinoma and oral potentially malignant disorders: A systematic review. Oral. Dis. 2011, 17 (Suppl. 1), 58–72. [Google Scholar] [CrossRef] [PubMed]
- Balakittnen, J.; Ekanayake Weeramange, C.; Wallace, D.F.; Duijf, P.H.G.; Cristino, A.S.; Hartel, G.; Barrero, R.A.; Taheri, T.; Kenny, L.; Vasani, S.; et al. A novel saliva-based miRNA profile to diagnose and predict oral cancer. Int. J. Oral. Sci. 2024, 16, 14. [Google Scholar] [CrossRef]
- Vairaktaris, E.; Yapijakis, C.; Serefoglou, Z.; Vylliotis, A.; Ries, J.; Nkenke, E.; Wiltfang, J.; Derka, S.; Vassiliou, S.; Springer, I.; et al. Plasminogen activator inhibitor-1 polymorphism is associated with increased risk for oral cancer. Oral. Oncol. 2006, 42, 888–892. [Google Scholar] [CrossRef]
- Tsou, Y.A.; Hua, C.H.; Tseng, H.C.; Hsu, C.F.; Tsai, C.W.; Sun, S.S.; Tsai, R.Y.; Tsai, M.H.; Bau, D.T. The joint effect of hOGG1 single nucleotide polymorphism and betel quid chewing on oral cancer in Taiwan. Anticancer. Res. 2010, 30, 4205–4208. [Google Scholar] [PubMed]
- Zavras, A.I.; Yoon, A.J.; Chen, M.K.; Lin, C.W.; Yang, S.F. Association between polymorphisms of DNA repair gene ERCC5 and oral squamous cell carcinoma. Oral. Surg. Oral. Med. Oral. Pathol. Oral. Radiol. 2012, 114, 624–629. [Google Scholar] [CrossRef]
- Langevin, S.M.; Stone, R.A.; Bunker, C.H.; Grandis, J.R.; Sobol, R.W.; Taioli, E. MicroRNA-137 promoter methylation in oral rinses from patients with squamous cell carcinoma of the head and neck is associated with gender and body mass index. Carcinogenesis 2010, 31, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Su, C.W.; Huang, Y.W.; Chen, M.K.; Su, S.C.; Yang, S.F.; Lin, C.W. Polymorphisms and Plasma Levels of Tissue Inhibitor of Metalloproteinase-3: Impact on Genetic Susceptibility and Clinical Outcome of Oral Cancer. Medicine 2015, 94, e2092. [Google Scholar] [CrossRef]
- Chen, F.; Liu, F.; Yan, L.; Lin, L.; Qiu, Y.; Wang, J.; Wu, J.; Bao, X.; Hu, Z.; Cai, L.; et al. A functional haplotype of NFKB1 influence susceptibility to oral cancer: A population-based and in vitro study. Cancer Med. 2018, 7, 2211–2218. [Google Scholar] [CrossRef]
- Yuan, Z.; Yu, Y.; Zhang, B.; Miao, L.; Wang, L.; Zhao, K.; Ji, Y.; Wang, R.; Ma, H.; Chen, N.; et al. Genetic variants in lncRNA H19 are associated with the risk of oral squamous cell carcinoma in a Chinese population. Oncotarget 2018, 9, 23915–23922. [Google Scholar] [CrossRef]
- Chung, C.M.; Hung, C.C.; Lee, C.H.; Lee, C.P.; Lee, K.W.; Chen, M.K.; Yeh, K.T.; Ko, Y.C. Variants in FAT1 and COL9A1 genes in male population with or without substance use to assess the risk factors for oral malignancy. PLoS ONE 2019, 14, e0210901. [Google Scholar] [CrossRef]
- Ding, Y.F.; Wen, Y.C.; Chuang, C.Y.; Lin, C.W.; Yang, Y.C.; Liu, Y.F.; Chang, W.M.; Chang, L.C.; Yang, S.F.; Chien, M.H. Combined Impacts of Genetic Variants of Long Non-Coding RNA MALAT1 and the Environmental Carcinogen on the Susceptibility to and Progression of Oral Squamous Cell Carcinoma. Front. Oncol. 2021, 11, 684941. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.H.; Hong, S.F.; Lo, Y.S.; Ho, H.Y.; Lin, C.C.; Chuang, Y.C.; Hsieh, M.J.; Chou, M.C. The Role of Ryanodine Receptor 2 Polymorphisms in Oral Squamous Cell Carcinoma Susceptibility and Clinicopathological Features. Int. J. Mol. Sci. 2024, 25, 10328. [Google Scholar] [CrossRef] [PubMed]
- Ramos-García, P.; González-Moles, M.; Warnakulasuriya, S. Significance of p53 overexpression in the prediction of the malignant transformation risk of oral potentially malignant disorders: A systematic review and meta-analysis. Oral. Oncol. 2022, 126, 105734. [Google Scholar] [CrossRef]
- Andersen, P.K. Repeated assessment of risk factors in survival analysis. Stat. Methods Med. Res. 1992, 1, 297–315. [Google Scholar] [CrossRef] [PubMed]
- Sankaranarayanan, R.; Ramadas, K.; Thara, S.; Muwonge, R.; Thomas, G.; Anju, G.; Mathew, B. Long term effect of visual screening on oral cancer incidence and mortality in a randomized trial in Kerala, India. Oral. Oncol. 2013, 49, 314–321. [Google Scholar] [CrossRef]
- Nielsen, K.J.; Jakobsen, K.K.; Jensen, J.S.; Grønhøj, C.; Von Buchwald, C. The Effect of Prophylactic HPV Vaccines on Oral and Oropharyngeal HPV Infection-A Systematic Review. Viruses 2021, 13, 1339. [Google Scholar] [CrossRef]
- Bouvard, V.; Nethan, S.T.; Singh, D.; Warnakulasuriya, S.; Mehrotra, R.; Chaturvedi, A.K.; Chen, T.H.; Ayo-Yusuf, O.A.; Gupta, P.C.; Kerr, A.R.; et al. IARC Perspective on Oral Cancer Prevention. N. Engl. J. Med. 2022, 387, 1999–2005. [Google Scholar] [CrossRef]
- Johnson, N.; Gupta, B.; Speicher, D.; Ray, C.; Al-hebshi, N.; Gupta, P. Etiology and Risk Factors. In Oral and Oropharyngeal Cancer: Causes, Prevention and Management of Malignancies of the Mouth and Oropharynx; Shah, J.P., Johnson, N.W., Eds.; CRC Press Taylor and Francis group: Boca Raton, FL, USA, 2018; pp. 19–93. [Google Scholar]
- Bagnardi, V.; Rota, M.; Botteri, E.; Tramacere, I.; Islami, F.; Fedirko, V.; Scotti, L.; Jenab, M.; Turati, F.; Pasquali, E.; et al. Alcohol consumption and site-specific cancer risk: A comprehensive dose-response meta-analysis. Br. J. Cancer 2015, 112, 580–593. [Google Scholar] [CrossRef]
- Moyer, V.A. Screening for oral cancer: U.S. Preventive Services Task Force recommendation statement. Ann. Intern. Med. 2014, 160, 55–60. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Variables | Coefficients | Rate Ratio | Reference |
|---|---|---|---|
| The effect of betel nut chewing on incidence of leukoplakia | |||
| Occasional use | 0 | 1.0 | [10] |
| 1–10 pieces per day | 0.936 | 2.55 (1.91–3.41) | |
| 11–20 pieces per day | 1.790 | 5.99 (4.25–8.45) | |
| 20+ pieces per day | 2.323 | 10.21 (7.53–13.84) | |
| The effect of smoking on incidence of leukoplakia | |||
| Occasional use | 0 | 1.0 | |
| 1–10 sticks per day | 0.732 | 2.08 (1.26–3.42) | |
| 11–20 sticks per day | 1.040 | 2.83 (1.74–4.59) | |
| 21–30 sticks per day | 1.278 | 3.59 (2.16–5.95) | |
| The effect of betel nut chewing on transition from leukoplakia to erythroleukoplakia | |||
| Occasional use | 0 | 1.0 | |
| 1–10 pieces per day | 0.513 | 1.67 (0.84–3.33) | |
| 11–20 pieces per day | 1.209 | 3.35 (1.65–6.83) | |
| 20+ pieces per day | 1.258 | 3.52 (1.80–6.86) | |
| The effect of smoking on transition from leukoplakia to erythroleukoplakia | |||
| Occasional use | 0 | 1.0 | |
| 1–10 sticks per day | 0 | 1.00 (0.42–2.39) | |
| 11–20 sticks per day | 0.199 | 1.22 (0.53–2.83) | |
| 21–30 sticks per day | 0.199 | 1.22 (0.51–2.93) | |
| Genes | OPMD N (%)/Mean (SE) | Healthy N (%)/Mean (SE) | OR | 95% CI | References |
|---|---|---|---|---|---|
| HPV | [21] | ||||
| Negative | 601 (63%) | 586 (87%) | |||
| Positive | 355 (37%) | 89 (13%) | 3.87 | 2.87–5.21 | |
| miR-3614-5p | −8.5 (3.9) | −9.2 (2.8) | 1.78 | 1.21–2.61 | [22] |
| miR-10b-5p | −8.5 (7.1) | −10.0 (2.8) | 1.83 | 1.19–2.81 | |
| miR-215-5p | −2.85 (1.6) | −3.0 (1.7) | 2.73 | 1.19–2.24 | |
| miR-182-5p | −4.5 (3.5) | −5.3 (2.8) | 1.51 | 1.02–2.24 |
| Genes | Cases N (%) | Control N (%) | OR | 95% CI | References |
|---|---|---|---|---|---|
| PAI-1–675 | [23] | ||||
| 5G/4G or 5G/5G | 59 (56.7) | 75 (70.8) | 1.00 | ||
| 4G/4G | 45 (43.3) | 31 (29.2) | 5.00 | 1.32–8.92 | |
| ACE intron 16 | |||||
| D/I or D/D | 130 (81.3) | 144 (94.1) | 1.00 | ||
| I/I | 30 (18.7) | 9 (5.9) | 9.16 | 1.14–73.50 | |
| TIMP-2–418 | |||||
| C/C and G/C | 66 (41.8) | 159 (94.6) | 1.00 | ||
| G/G | 92 (58.2) | 9 (5.4) | 26.33 | 12.39–55.95 | |
| hOGG1 codon 326 (rs1052133) | [24] | ||||
| CG/GG | 482 (77.7) | 516 (83.2) | 1.00 | ||
| CC | 138 (22.3) | 104 (16.8) | 1.42 | 1.08–1.89 | |
| ERCC5 (rs751402) | [25] | ||||
| CC | 98 (41.0) | 167 (49.7) | 1.00 | ||
| CT/TT | 141 (59.0) | 169 (50.3) | 1.42 | 1.02–1.99 | |
| miR-137 | [26] | ||||
| Unmethylated | 78 (78.8) | 96 (97.0) | 1.00 | ||
| Methylated | 21 (21.2) | 3 (3.0) | 4.80 | 1.23–18.82 | |
| TIMP3 (rs9862) | [27] | ||||
| CC | 192 (25.7) | 414 (34.5) | 1.00 | ||
| CT/TT | 555 (74.3) | 786 (65.5) | 1.52 | 1.24–1.86 | |
| BRCA1 (rs2070833) | [8] | ||||
| CC | 237 (53.0) | 374 (64.5) | 1.00 | ||
| CT/TT | 210 (47.0) | 206 (35.5) | 1.61 | 1.25–2.07 | |
| COL9A1 (rs550675) | |||||
| CC | 187 (41.8) | 314 (54.1) | 1.00 | ||
| CT/TT | 260 (58.2) | 266 (45.9) | 1.64 | 1.28–2.11 | |
| NOTCH1 (rs139994842) | |||||
| GG | 401 (89.7) | 560 (96.6) | 1.00 | ||
| AG/AA | 46 (10.3) | 20 (3.4) | 3.21 | 1.87–5.51 | |
| HSPA13 (rs2822641) | |||||
| GG | 386 (86.4) | 542 (93.4) | 1.00 | ||
| GT/TT | 61 (13.6) | 38 (6.6) | 2.25 | 1.47–3.45 | |
| NFκB1 (rs28362491) | [28] | ||||
| Ins/Ins + Ins/Del | 321 (76.2) | 393 (81.4) | 1.00 | ||
| Del/Del | 100 (23.8) | 90 (18.6) | 1.58 | 1.10–2.26 | |
| NFκB1 (rs72696119) | |||||
| CC + CG | 318 (75.53) | 388 (81.0) | 1.00 | ||
| GG | 103 (24.5) | 91 (19.0) | 1.62 | 1.14–2.32 | |
| H19 (rs217727) | [29] | ||||
| CC + TC | 380 (85.6) | 911 (92.6) | 1.00 | ||
| TT | 51 (11.5) | 73 (7.4) | 1.70 | 1.16–2.49 | |
| H19 (rs2839701) | |||||
| CC + CG | 393 (88.5) | 909 (92.4) | 1.00 | ||
| GG | 51 (11.5) | 75 (7.6) | 1.53 | 1.05–2.24 | |
| FAT1 (rs28647489) | [30] | ||||
| GA + AA | 285 (79.2) | 412 (84.8) | 1.00 | ||
| GG | 75 (20.8) | 74 (15.2) | 1.47 | 1.03–2.09 | |
| MALAT1 (rs3200401) | [31] | ||||
| CC | 948 (70.2) | 807 (67.3) | 1.00 | ||
| CT/TT | 402 (29.8) | 392 (32.7) | 0.78 | 0.63–0.96 | |
| RYR2 (rs12594) | [32] | ||||
| AA/AG | 509 (90.5) | 315 (94.9) | 1.00 | ||
| GG | 53 (9.5) | 17 (5.1) | 1.93 | 1.10–3.39 | |
| P53 | [33] | ||||
| Negative | 330 (54.2) | 421 (69.0) | 1.00 | ||
| Positive | 279 (45.8) | 189 (31.0) | 1.88 | 1.39–2.56 |
| Genes | Oral Cancer Mean (SE) | Healthy Mean (SE) | OR | 95% CI | References |
|---|---|---|---|---|---|
| miR-10b-5p | −8.2 (3.9) | −10.0 (2.8) | 3.91 | 2.02–7.57 | [22] |
| miR-3614-5p | −9.0 (3.5) | −9.2 (2.8) | 6.09 | 1.76–21.04 | |
| miR_431-5p | −5.8 (4.9) | −3.1 (2.8) | 0.40 | 0.21–0.76 | |
| miR-486-3p | −8.9 (4.9) | −6.0 (4.5) | 0.69 | 0.47–1.02 | |
| miR-182-5p | −5.0 (4.9) | −5.3 (2.8) | 1.70 | 0.69–4.20 | |
| miR-215-5p | −3.0 (2.6) | −3.0 (2.7) | 2.20 | 0.56–8.66 | |
| miR-7-5p | −3.8 (2.0) | −4.9 (3.9) | 1.64 | 0.69–3.90 | |
| miR-4707-3p | −7.0 (4.4) | −5.2 (4.4) | 0.79 | 0.39–1.57 |
| Characteristics | Risk Decile | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |
| Betel Nut Chewing | ||||||||||
| Occasional use | 36.9% | 28.9% | 26.4% | 24.8% | 23.6% | 22.3% | 21.2% | 20.1% | 19.0% | 17.0% |
| 1–10 pieces per day | 32.7% | 32.5% | 31.7% | 31.2% | 30.7% | 30.2% | 29.5% | 28.9% | 28.0% | 26.5% |
| 11–20 pieces per day | 23.2% | 28.8% | 30.9% | 32.1% | 33.1% | 34.1% | 35.2% | 36.3% | 37.3% | 39.1% |
| 20+ pieces per day | 7.2% | 9.9% | 11.0% | 11.9% | 12.6% | 13.3% | 14.1% | 14.6% | 15.8% | 17.4% |
| Smoking | ||||||||||
| No Smoking | 95.0% | 90.2% | 86.9% | 83.6% | 80.2% | 76.6% | 72.7% | 68.3% | 62.1% | 53.7% |
| 1–10 sticks per day | 4.9% | 9.0% | 11.1% | 12.9% | 14.6% | 16.2% | 17.3% | 18.0% | 19.1% | 17.6% |
| 11–20 sticks per day | 0.1% | 0.7% | 1.5% | 2.5% | 3.7% | 5.1% | 7.1% | 9.6% | 12.7% | 18.7% |
| 20+ sticks per day | 0.0% | 0.2% | 0.5% | 0.9% | 1.4% | 2.1% | 3.0% | 4.2% | 6.2% | 10.0% |
| HPV Infection | ||||||||||
| No | 95.1% | 91.2% | 88.9% | 86.7% | 84.8% | 82.8% | 80.7% | 78.0% | 74.8% | 67.5% |
| Yes | 4.9% | 8.8% | 11.1% | 13.3% | 15.2% | 17.2% | 19.3% | 22.0% | 25.2% | 32.5% |
| Characteristics | Risk Decile | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |
| Mean risk score | 3.4 | 4.8 | 5.6 | 6.3 | 6.9 | 7.5 | 8.2 | 8.9 | 9.9 | 11.8 |
| Lifetime risk (per 105) | 362 | 925 | 1576 | 2360 | 3348 | 4753 | 6698 | 9551 | 13239 | 24523 |
| Relative risk | 0.11 | 0.28 | 0.47 | 0.71 | 1.00 | 1.42 | 2.00 | 2.85 | 4.25 | 7.33 |
| MST of from eleukoplakia to oral cancer (year) | 45.5 | 18.7 | 10.8 | 6.9 | 4.5 | 3.1 | 2.0 | 1.3 | 0.7 | 0.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jumparway, D.; Su, C.-W.; Yen, A.M.-F.; Liu, Y.-T.; Chen, M.-K.; Liu, K.-J.; Sarakarn, P.; Chen, S.L.-S. Quantifying Genetic and Environmental Factors Accounting for Multistage Progression of Precancerous Lesions and Oral Cancer: Applications to Risk-Guided Prevention. Cancers 2025, 17, 2114. https://doi.org/10.3390/cancers17132114
Jumparway D, Su C-W, Yen AM-F, Liu Y-T, Chen M-K, Liu K-J, Sarakarn P, Chen SL-S. Quantifying Genetic and Environmental Factors Accounting for Multistage Progression of Precancerous Lesions and Oral Cancer: Applications to Risk-Guided Prevention. Cancers. 2025; 17(13):2114. https://doi.org/10.3390/cancers17132114
Chicago/Turabian StyleJumparway, Donlagon, Chiu-Wen Su, Amy Ming-Fang Yen, Yen-Tze Liu, Mu-Kuan Chen, Ko-Jiunn Liu, Pongdech Sarakarn, and Sam Li-Sheng Chen. 2025. "Quantifying Genetic and Environmental Factors Accounting for Multistage Progression of Precancerous Lesions and Oral Cancer: Applications to Risk-Guided Prevention" Cancers 17, no. 13: 2114. https://doi.org/10.3390/cancers17132114
APA StyleJumparway, D., Su, C.-W., Yen, A. M.-F., Liu, Y.-T., Chen, M.-K., Liu, K.-J., Sarakarn, P., & Chen, S. L.-S. (2025). Quantifying Genetic and Environmental Factors Accounting for Multistage Progression of Precancerous Lesions and Oral Cancer: Applications to Risk-Guided Prevention. Cancers, 17(13), 2114. https://doi.org/10.3390/cancers17132114