
Balancing Regeneration and Resistance: Targeting DCLK1 to Mitigate Gastrointestinal Radiation Injury and Oncogenesis


Simple Summary
Abstract
1. Introduction
2. DCLK1 Regulates Radiation Response in GI-ARS and Fractionated GI Injury
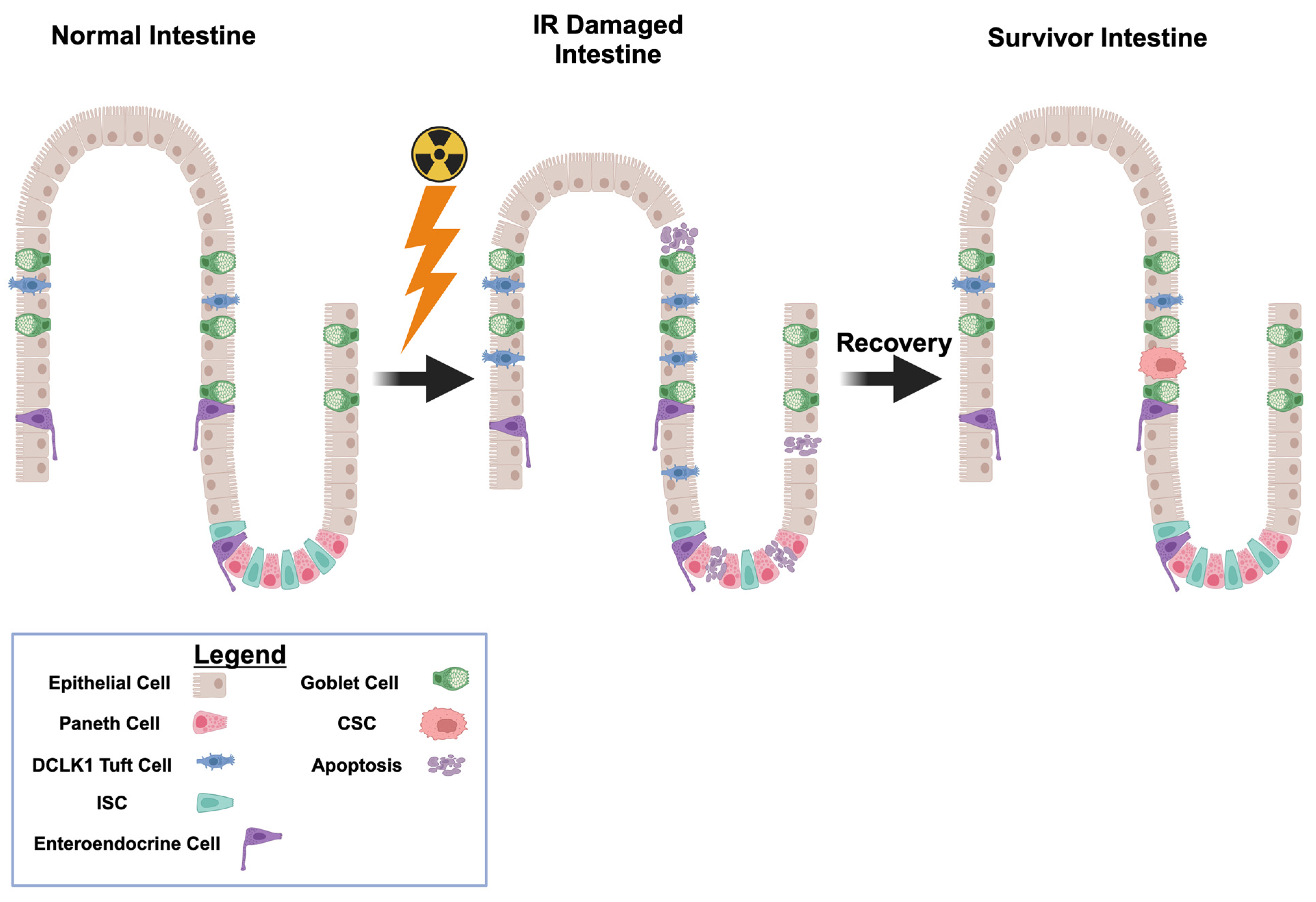
2.1. Radiation Induced GI Damages
2.2. RAS Amplifies GI-IR Injury
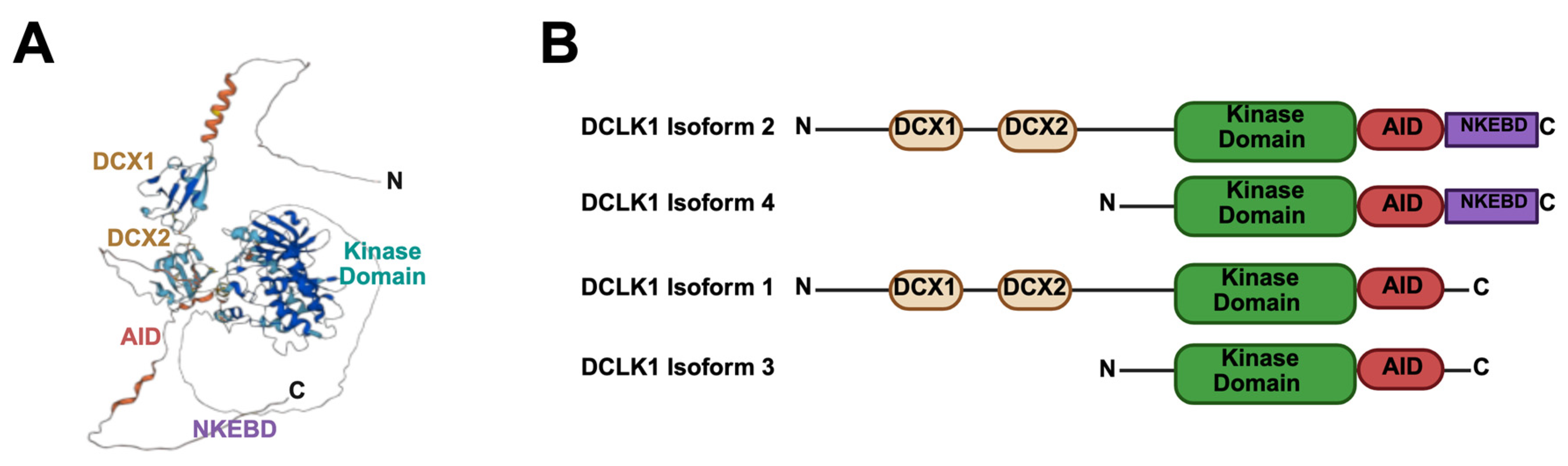
2.3. Gastrointestinal Damages and DCLK1-Mediated Repair
3. DCLK1 in Radiation Response Pathways
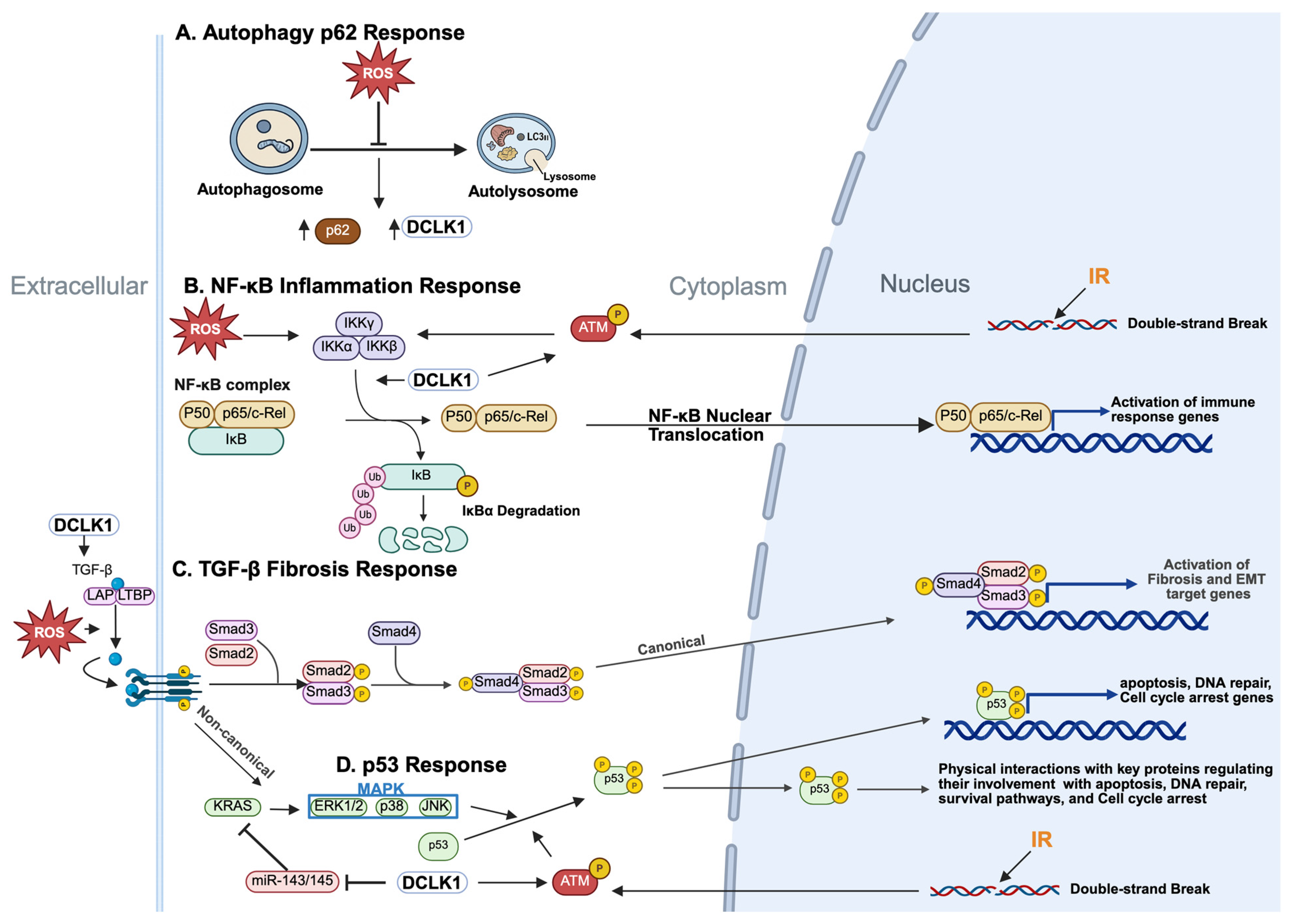
3.1. DCLK1, p62 and Autophagy
3.2. DCLK1 and Inflammatory Responses
3.3. DCLK1 and the MAPK Pathway
3.4. DCLK1 and the DNA Damage (p53) Response
3.5. GI Regeneration and Therapy Resistance Following IR
4. IR-Induced Microenvironment and the Role of DCLK1
4.1. The Microenvironment in Gastrointestinal Acute Radiation Syndrome
4.2. DCLK1 as a Modulator of the Radiation Microenvironment
4.3. Targeting DCLK1 to Overcome ARS-Induced Radioresistance and Secondary Cancer Risk
5. GI-ARS Radioprotection, Mitigation, and Treatment
5.1. Radioprotective and Radiomitigative Strategies
5.2. Therapeutic Interventions for Established GI-ARS and Radiation-Induced GI Injury
5.2.1. Senescence-Targeting Approaches
5.2.2. Anti-Fibrotic Strategies
5.2.3. Stem Cell-Based and Regenerative Therapies
5.2.4. DCLK1 and Radiation-Induced Senescence
6. Future Directions: Emerging Molecular Therapies
6.1. Nanoparticle-Based Delivery Systems
6.2. Modulating the Gut Microbiome and Innate Immunity
6.3. Emerging Immunomodulatory Approaches
6.4. Combined Drug Treatments
6.5. Integrating DCLK1 Inhibition with Fractionated Radiotherapy
6.6. Targeting DCLK1 to Enhance Radioprotection and Prevent Tumorigenesis
6.7. DCLK1 as a Biomarker of Radiation Exposure and Regeneration
7. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Shadad, A.K.; Sullivan, F.J.; Martin, J.D.; Egan, L.J. Gastrointestinal radiation injury: Symptoms, risk factors and mechanisms. World J. Gastroenterol. 2013, 19, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Grégoire, V.; Guckenberger, M.; Haustermans, K.; Lagendijk, J.J.W.; Ménard, C.; Pötter, R.; Slotman, B.J.; Tanderup, K.; Thorwarth, D.; van Herk, M.; et al. Image guidance in radiation therapy for better cure of cancer. Mol. Oncol. 2020, 14, 1470–1491. [Google Scholar] [CrossRef] [PubMed]
- Koukourakis, M.I. Radiation damage and radioprotectants: New concepts in the era of molecular medicine. Br. J. Radiol. 2012, 85, 313–330. [Google Scholar] [CrossRef]
- Talapko, J.; Talapko, D.; Katalinić, D.; Kotris, I.; Erić, I.; Belić, D.; Vasilj Mihaljević, M.; Vasilj, A.; Erić, S.; Flam, J.; et al. Health Effects of Ionizing Radiation on the Human Body. Medicina 2024, 60, 653. [Google Scholar] [CrossRef] [PubMed]
- ACUTE radiation syndrome. Br. Med. J. 1952, 2, 925–926. [CrossRef] [PubMed] [PubMed Central]
- Tanigawa, K. Case review of severe acute radiation syndrome from whole body exposure: Concepts of radiation-induced multi-organ dysfunction and failure. J. Radiat. Res. 2021, 62, i15–i20. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Futami, S.; Nishida, M.; Suzuki, T.; Sakamoto, T.; Suzuki, N.; Maekawa, K. Brief note and evaluation of acute-radiation syndrome and treatment of a Tokai-mura criticality accident patient. J. Radiat. Res. 2001, 42, S167–S182. [Google Scholar] [CrossRef]
- Parrish, J.S.; Seda, G. Disasters Resulting from Radiologic and Nuclear Events. Crit. Care Clin. 2019, 35, 619–631. [Google Scholar] [CrossRef]
- Kiang, J.G.; Olabisi, A.O. Radiation: A poly-traumatic hit leading to multi-organ injury. Cell Biosci. 2019, 9, 25. [Google Scholar] [CrossRef]
- Macia, I.G.M.; Lucas Calduch, A.; Lopez, E.C. Radiobiology of the acute radiation syndrome. Rep. Pract. Oncol. Radiother. 2011, 16, 123–130. [Google Scholar] [CrossRef]
- Durante, M.; Cucinotta, F.A. Physical basis of radiation protection in space travel. Rev. Mod. Phys. 2011, 83, 1245–1281. [Google Scholar] [CrossRef]
- Chancellor, J.C.; Scott, G.B.; Sutton, J.P. Space Radiation: The Number One Risk to Astronaut Health beyond Low Earth Orbit. Life 2014, 4, 491–510. [Google Scholar] [CrossRef] [PubMed]
- Montoro, A.; Obrador, E.; Mistry, D.; Forte, G.I.; Bravatà, V.; Minafra, L.; Calvaruso, M.; Cammarata, F.P.; Falk, M.; Schettino, G.; et al. Radioprotectors, Radiomitigators, and Radiosensitizers. In Radiobiology Textbook; Baatout, S., Ed.; Springer International Publishing: Cham, Switzerland, 2023; pp. 571–628. [Google Scholar]
- Grosu-Bularda, A.; Lita, F.-F.; Hodea, F.-V.; Bordeanu-Diaconescu, E.-M.; Cretu, A.; Dumitru, C.-S.; Cacior, S.; Marinescu, B.-M.; Lascar, I.; Hariga, C.-S. Navigating the Complexities of Radiation Injuries: Therapeutic Principles and Reconstructive Strategies. J. Pers. Med. 2024, 14, 1100. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.J.; Doll, R.; Goodhead, D.T.; Hall, E.J.; Land, C.E.; Little, J.B.; Lubin, J.H.; Preston, D.L.; Preston, R.J.; Puskin, J.S.; et al. Cancer risks attributable to low doses of ionizing radiation: Assessing what we really know. Proc. Natl. Acad. Sci. USA 2003, 100, 13761–13766. [Google Scholar] [CrossRef]
- Mettler, F.A. Medical effects and risks of exposure to ionising radiation. J. Radiol. Prot. 2012, 32, N9–N13. [Google Scholar] [CrossRef]
- Preston, D.L.; Ron, E.; Tokuoka, S.; Funamoto, S.; Nishi, N.; Soda, M.; Mabuchi, K.; Kodama, K. Solid cancer incidence in atomic bomb survivors: 1958–1998. Radiat. Res. 2007, 168, 1–64. [Google Scholar] [CrossRef]
- Gopinathan, L.; Gopinathan, C. Ionizing radiation-induced cancer: Perplexities of the bystander effect. Ecancermedicalscience 2023, 17, 1579. [Google Scholar] [CrossRef]
- Moore, L.L.; Houchen, C.W. Evolving insights into pancreatic tumor initiation and progression through DCLK1-expressing tuft cells. Dev. Cell 2025, 60, 817–818. [Google Scholar] [CrossRef]
- Salas-Escabillas, D.J.; Hoffman, M.T.; Brender, S.M.; Moore, J.S.; Wen, H.-J.; Benitz, S.; Davis, E.T.; Long, D.; Wombwell, A.M.; Chianis, E.R.D.; et al. Tuft cells transdifferentiate to neural-like progenitor cells in the progression of pancreatic cancer. Dev. Cell 2025, 60, 837–852.e3. [Google Scholar] [CrossRef]
- Burgess, H.A.; Martinez, S.; Reiner, O. KIAA0369, doublecortin-like kinase, is expressed during brain development. J. Neurosci. Res. 1999, 58, 567–575. [Google Scholar] [CrossRef]
- Matsumoto, N.; Pilz, D.T.; Ledbetter, D.H. Genomic structure, chromosomal mapping, and expression pattern of human DCAMKL1 (KIAA0369), a homologue of DCX (XLIS). Genomics 1999, 56, 179–183. [Google Scholar] [CrossRef]
- Omori, Y.; Suzuki, M.; Ozaki, K.; Harada, Y.; Nakamura, Y.; Takahashi, E.; Fujiwara, T. Expression and chromosomal localization of KIAA0369, a putative kinase structurally related to Doublecortin. J. Hum. Genet. 1998, 43, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Sossey-Alaoui, K.; Srivastava, A.K. DCAMKL1, a brain-specific transmembrane protein on 13q12.3 that is similar to doublecortin (DCX). Genomics 1999, 56, 121–126. [Google Scholar] [CrossRef]
- Cheng, L.; Yang, Z.; Guo, W.; Wu, C.; Liang, S.; Tong, A.; Cao, Z.; Thorne, R.F.; Yang, S.-Y.; Yu, Y.; et al. DCLK1 autoinhibition and activation in tumorigenesis. Innovation 2022, 3, 100191. [Google Scholar] [CrossRef] [PubMed]
- Venkat, A.; Watterson, G.; Byrne, D.P.; O’Boyle, B.; Shrestha, S.; Gravel, N.; Fairweather, E.E.; Daly, L.A.; Bunn, C.; Yeung, W.; et al. Mechanistic and evolutionary insights into isoform-specific ‘supercharging’ in DCLK family kinases. eLife 2023, 12, RP87958. [Google Scholar] [CrossRef]
- Sureban, S.M.; Berahovich, R.; Zhou, H.; Xu, S.; Wu, L.; Ding, K.; May, R.; Qu, D.; Bannerman-Menson, E.; Golubovskaya, V.; et al. DCLK1 Monoclonal Antibody-Based CAR-T Cells as a Novel Treatment Strategy against Human Colorectal Cancers. Cancers 2020, 12, 54. [Google Scholar] [CrossRef]
- Ge, Y.; Weygant, N.; Qu, D.; May, R.; Berry, W.L.; Yao, J.; Chandrakesan, P.; Zheng, W.; Zhao, L.; Zhao, K.L.; et al. Alternative splice variants of DCLK1 mark cancer stem cells, promote self-renewal and drug-resistance, and can be targeted to inhibit tumorigenesis in kidney cancer. Int. J. Cancer 2018, 143, 1162–1175. [Google Scholar] [CrossRef]
- Qu, D.; Weygant, N.; Yao, J.; Chandrakesan, P.; Berry, W.L.; May, R.; Pitts, K.; Husain, S.; Lightfoot, S.; Li, M.; et al. Overexpression of DCLK1-AL Increases Tumor Cell Invasion, Drug Resistance, and KRAS Activation and Can Be Targeted to Inhibit Tumorigenesis in Pancreatic Cancer. J. Oncol. 2019, 2019, 6402925. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Weygant, N.; Chandrakesan, P.; Houchen, C.W.; Peng, J.; Qu, D. Tuft and Cancer Stem Cell Marker DCLK1: A New Target to Enhance Anti-Tumor Immunity in the Tumor Microenvironment. Cancers 2020, 12, 3801. [Google Scholar] [CrossRef]
- Westphalen, C.B.; Asfaha, S.; Hayakawa, Y.; Takemoto, Y.; Lukin, D.J.; Nuber, A.H.; Brandtner, A.; Setlik, W.; Remotti, H.; Muley, A.; et al. Long-lived intestinal tuft cells serve as colon cancer-initiating cells. J. Clin. Investig. 2014, 124, 1283–1295. [Google Scholar] [CrossRef]
- Nakanishi, Y.; Seno, H.; Fukuoka, A.; Ueo, T.; Yamaga, Y.; Maruno, T.; Nakanishi, N.; Kanda, K.; Komekado, H.; Kawada, M.; et al. Dclk1 distinguishes between tumor and normal stem cells in the intestine. Nat. Genet. 2013, 45, 98–103. [Google Scholar] [CrossRef]
- Yi, J.; Bergstrom, K.; Fu, J.; Shan, X.; McDaniel, J.M.; McGee, S.; Qu, D.; Houchen, C.W.; Liu, X.; Xia, L. Dclk1 in tuft cells promotes inflammation-driven epithelial restitution and mitigates chronic colitis. Cell Death Differ. 2019, 26, 1656–1669. [Google Scholar] [CrossRef] [PubMed]
- May, R.; Qu, D.; Weygant, N.; Chandrakesan, P.; Ali, N.; Lightfoot, S.A.; Li, L.; Sureban, S.M.; Houchen, C.W. Brief report: Dclk1 deletion in tuft cells results in impaired epithelial repair after radiation injury. Stem. Cells 2014, 32, 822–827. [Google Scholar] [CrossRef]
- Westphalen, C.B.; Takemoto, Y.; Tanaka, T.; Macchini, M.; Jiang, Z.; Renz, B.W.; Chen, X.; Ormanns, S.; Nagar, K.; Tailor, Y.; et al. Dclk1 Defines Quiescent Pancreatic Progenitors that Promote Injury-Induced Regeneration and Tumorigenesis. Cell Stem. Cell 2016, 18, 441–455. [Google Scholar] [CrossRef]
- Ye, L.; Liu, B.; Huang, J.; Zhao, X.; Wang, Y.; Xu, Y.; Wang, S. DCLK1 and its oncogenic functions: A promising therapeutic target for cancers. Life Sci. 2024, 336, 122294. [Google Scholar] [CrossRef]
- Guo, S.; Yao, Y.; Tang, Y.; Xin, Z.; Wu, D.; Ni, C.; Huang, J.; Wei, Q.; Zhang, T. Radiation-induced tumor immune microenvironments and potential targets for combination therapy. Signal Transduct. Target. Ther. 2023, 8, 205. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Shao, X.; Zhang, Y.; Zhu, M.; Wang, F.X.C.; Mu, J.; Li, J.; Yao, H.; Chen, K. Role of tumor microenvironment in cancer progression and therapeutic strategy. Cancer Med. 2023, 12, 11149–11165. [Google Scholar] [CrossRef]
- Lopez, M.; Martin, M. Medical management of the acute radiation syndrome. Rep. Pract. Oncol. Radiother. 2011, 16, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shao, L.; Hendrickson, H.P.; Liu, L.; Chang, J.; Luo, Y.; Seng, J.; Pouliot, M.; Authier, S.; Zhou, D.; et al. Total Body Irradiation in the “Hematopoietic” Dose Range Induces Substantial Intestinal Injury in Non-Human Primates. Radiat. Res. 2015, 184, 545–553. [Google Scholar] [CrossRef]
- Potruch, A.; Schwartz, A.; Ilan, Y. The role of bacterial translocation in sepsis: A new target for therapy. Ther. Adv. Gastroenterol. 2022, 15, 17562848221094214. [Google Scholar] [CrossRef]
- Terry, N.H.; Travis, E.L. The influence of bone marrow depletion on intestinal radiation damage. Int. J. Radiat. Oncol. Biol. Phys. 1989, 17, 569–573. [Google Scholar] [CrossRef]
- Stolfi, C.; Maresca, C.; Monteleone, G.; Laudisi, F. Implication of Intestinal Barrier Dysfunction in Gut Dysbiosis and Diseases. Biomedicines 2022, 10, 289. [Google Scholar] [CrossRef] [PubMed]
- Winters, T.A.; Marzella, L.; Molinar-Inglis, O.; Price, P.W.; Han, N.C.; Cohen, J.E.; Wang, S.J.; Fotenos, A.F.; Sullivan, J.M.; Esker, J.I.; et al. Gastrointestinal Acute Radiation Syndrome: Mechanisms, Models, Markers, and Medical Countermeasures. Radiat. Res. 2024, 201, 628–646. [Google Scholar] [CrossRef] [PubMed]
- DiCarlo, A.L.; Maher, C.; Hick, J.L.; Hanfling, D.; Dainiak, N.; Chao, N.; Bader, J.L.; Coleman, C.N.; Weinstock, D.M. Radiation injury after a nuclear detonation: Medical consequences and the need for scarce resources allocation. Disaster Med. Public Health Prep. 2011, 5 (Suppl. S1), S32–S44. [Google Scholar] [CrossRef]
- Rübe, C.E.; Raid, S.; Palm, J.; Rübe, C. Radiation-Induced Brain Injury: Age Dependency of Neurocognitive Dysfunction Following Radiotherapy. Cancers 2023, 15, 2999. [Google Scholar] [CrossRef]
- Choi, W.H.; Cho, J. Evolving Clinical Cancer Radiotherapy: Concerns Regarding Normal Tissue Protection and Quality Assurance. J. Korean Med. Sci. 2016, 31 (Suppl. S1), S75–S87. [Google Scholar] [CrossRef]
- Chandrakesan, P.; May, R.; Weygant, N.; Qu, D.; Berry, W.L.; Sureban, S.M.; Ali, N.; Rao, C.; Huycke, M.; Bronze, M.S.; et al. Intestinal tuft cells regulate the ATM mediated DNA Damage response via Dclk1 dependent mechanism for crypt restitution following radiation injury. Sci. Rep. 2016, 6, 37667. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, C.; Mahdavinezhad, A.; Saidijam, M.; Bahreini, F.; Sedighi Pashaki, A.; Gholami, M.H.; Najafi, R. DCLK1 Inhibition Sensitizes Colorectal Cancer Cells to Radiation Treatment. Int. J. Mol. Cell Med. 2021, 10, 23–33. [Google Scholar] [CrossRef]
- Huang, R.-X.; Zhou, P.-K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct. Target. Ther. 2020, 5, 60. [Google Scholar] [CrossRef]
- Vaz, A.F.M.; Costa, R.M.P.B.; Coelho, L.C.B.B.; Oliva, M.L.V.; Santana, L.A.; Melo, A.M.M.A.; Correia, M.T.S. Gamma irradiation as an alternative treatment to abolish allergenicity of lectins in food. Food Chem. 2011, 124, 1289–1295. [Google Scholar] [CrossRef]
- Juan, C.A.; Pérez de la Lastra, J.M.; Plou, F.J.; Pérez-Lebeña, E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021, 22, 4642. [Google Scholar] [CrossRef]
- Reisz, J.A.; Bansal, N.; Qian, J.; Zhao, W.; Furdui, C.M. Effects of ionizing radiation on biological molecules—Mechanisms of damage and emerging methods of detection. Antioxid. Redox Signal 2014, 21, 260–292. [Google Scholar] [CrossRef]
- Leach, J.K.; Van Tuyle, G.; Lin, P.S.; Schmidt-Ullrich, R.; Mikkelsen, R.B. Ionizing radiation-induced, mitochondria-dependent generation of reactive oxygen/nitrogen. Cancer Res. 2001, 61, 3894–3901. [Google Scholar]
- Autsavapromporn, N.; de Toledo, S.M.; Little, J.B.; Jay-Gerin, J.P.; Harris, A.L.; Azzam, E.I. The role of gap junction communication and oxidative stress in the propagation of toxic effects among high-dose α-particle-irradiated human cells. Radiat. Res. 2011, 175, 347–357. [Google Scholar] [CrossRef]
- Tominaga, H.; Kodama, S.; Matsuda, N.; Suzuki, K.; Watanabe, M. Involvement of reactive oxygen species (ROS) in the induction of genetic instability by radiation. J. Radiat. Res. 2004, 45, 181–188. [Google Scholar] [CrossRef]
- Jomova, K.; Raptova, R.; Alomar, S.Y.; Alwasel, S.H.; Nepovimova, E.; Kuca, K.; Valko, M. Reactive oxygen species, toxicity, oxidative stress, and antioxidants: Chronic diseases and aging. Arch. Toxicol. 2023, 97, 2499–2574. [Google Scholar] [CrossRef]
- Hong, Y.; Boiti, A.; Vallone, D.; Foulkes, N.S. Reactive Oxygen Species Signaling and Oxidative Stress: Transcriptional Regulation and Evolution. Antioxidants 2024, 13, 312. [Google Scholar] [CrossRef]
- Kay, J.; Thadhani, E.; Samson, L.; Engelward, B. Inflammation-induced DNA damage, mutations and cancer. DNA Repair 2019, 83, 102673. [Google Scholar] [CrossRef]
- Li, L.; Peng, P.; Ding, N.; Jia, W.; Huang, C.; Tang, Y. Oxidative Stress, Inflammation, Gut Dysbiosis: What Can Polyphenols Do in Inflammatory Bowel Disease? Antioxidants 2023, 12, 967. [Google Scholar] [CrossRef]
- Lee, C.L.; Oh, P.; Xu, E.S.; Ma, Y.; Kim, Y.; Daniel, A.R.; Kirsch, D.G. Blocking Cyclin-Dependent Kinase 4/6 During Single Dose Versus Fractionated Radiation Therapy Leads to Opposite Effects on Acute Gastrointestinal Toxicity in Mice. Int. J. Radiat. Oncol. Biol. Phys. 2018, 102, 1569–1576. [Google Scholar] [CrossRef]
- Cao, S.; Wu, R. Expression of Angiotensin II and Aldosterone in Radiation-induced Lung Injury. Cancer Biol. Med. 2012, 9, 254–260. [Google Scholar] [CrossRef]
- Zhou, L.; Zhu, J.; Liu, Y.; Zhou, P.-K.; Gu, Y. Mechanisms of radiation-induced tissue damage and response. MedComm 2024, 5, e725. [Google Scholar] [CrossRef]
- Nguyen Dinh Cat, A.; Montezano, A.C.; Burger, D.; Touyz, R.M. Angiotensin II, NADPH oxidase, and redox signaling in the vasculature. Antioxid. Redox Signal 2013, 19, 1110–1120. [Google Scholar] [CrossRef]
- Fändriks, L. The angiotensin II type 2 receptor and the gastrointestinal tract. J. Renin Angiotensin Aldosterone Syst. 2010, 11, 43–48. [Google Scholar] [CrossRef]
- Benigni, A.; Cassis, P.; Remuzzi, G. Angiotensin II revisited: New roles in inflammation, immunology and aging. EMBO Mol. Med. 2010, 2, 247–257. [Google Scholar] [CrossRef]
- Yu, Y.; Liu, S.; Yang, L.; Song, P.; Liu, Z.; Liu, X.; Yan, X.; Dong, Q. Roles of reactive oxygen species in inflammation and cancer. MedComm 2024, 5, e519. [Google Scholar] [CrossRef]
- Fatima, N.; Patel, S.N.; Hussain, T. Angiotensin II Type 2 Receptor: A Target for Protection Against Hypertension, Metabolic Dysfunction, and Organ Remodeling. Hypertension 2021, 77, 1845–1856. [Google Scholar] [CrossRef]
- Padda, R.S.; Shi, Y.; Lo, C.S.; Zhang, S.L.; Chan, J.S. Angiotensin-(1-7): A Novel Peptide to Treat Hypertension and Nephropathy in Diabetes? J. Diabetes Metab. 2015, 6, 1000615. [Google Scholar] [CrossRef]
- Tikellis, C.; Thomas, M.C. Angiotensin-Converting Enzyme 2 (ACE2) Is a Key Modulator of the Renin Angiotensin System in Health and Disease. Int. J. Pept. 2012, 2012, 256294. [Google Scholar] [CrossRef]
- Murphy, A.M.; Wong, A.L.; Bezuhly, M. Modulation of angiotensin II signaling in the prevention of fibrosis. Fibrogenesis Tissue Repair. 2015, 8, 7. [Google Scholar] [CrossRef]
- Simões e Silva, A.C.; Silveira, K.D.; Ferreira, A.J.; Teixeira, M.M. ACE2, angiotensin-(1-7) and Mas receptor axis in inflammation and fibrosis. Br. J. Pharmacol. 2013, 169, 477–492. [Google Scholar] [CrossRef]
- Chittimalli, K.; Jahan, J.; Sakamuri, A.; McAdams, Z.L.; Ericsson, A.C.; Jarajapu, Y.P.R. Restoration of the gut barrier integrity and restructuring of the gut microbiome in aging by angiotensin-(1-7). Clin. Sci. 2023, 137, 913–930. [Google Scholar] [CrossRef]
- Moulder, J.E.; Cohen, E.P.; Medhora, M.; Fish, B.L. Angiotensin converting enzyme (ACE) inhibitors as radiation countermeasures for long-duration space flights. Life Sci. Space Res. 2022, 35, 60–68. [Google Scholar] [CrossRef]
- Turnquist, C.; Harris, B.T.; Harris, C.C. Radiation-induced brain injury: Current concepts and therapeutic strategies targeting neuroinflammation. Neuro-Oncol. Adv. 2020, 2, vdaa057. [Google Scholar] [CrossRef]
- Yan, R.; Li, J.; Xiao, Z.; Fan, X.; Liu, H.; Xu, Y.; Sun, R.; Liu, J.; Yao, J.; An, G.; et al. DCLK1 Suppresses Tumor-Specific Cytotoxic T Lymphocyte Function Through Recruitment of MDSCs via the CXCL1-CXCR2 Axis. Cell. Mol. Gastroenterol. Hepatol. 2023, 15, 463–485. [Google Scholar] [CrossRef]
- Liu, H.; Yan, R.; Xiao, Z.; Huang, X.; Yao, J.; Liu, J.; An, G.; Ge, Y. Targeting DCLK1 attenuates tumor stemness and evokes antitumor immunity in triple-negative breast cancer by inhibiting IL-6/STAT3 signaling. Breast Cancer Res. 2023, 25, 43. [Google Scholar] [CrossRef]
- Feakins, M.R. Radiation and the Gastrointestinal Tract. In Non-Neoplastic Pathology of the Gastrointestinal Tract: A Practical Guide to Biopsy Diagnosis; Feakins, R.M., Ed.; Cambridge University Press: Cambridge, UK, 2020; pp. 30–51. [Google Scholar]
- Otsuka, K.; Suzuki, K. Differences in Radiation Dose Response between Small and Large Intestinal Crypts. Radiat. Res. 2016, 186, 302–314. [Google Scholar] [CrossRef]
- Ahmed, M.; Ahmed, R. Radiation in Gastroenterology. Gastroenterol. Res. 2022, 15, 6. [Google Scholar] [CrossRef]
- Yariv, O.; Camphausen, K.; Krauze, A.V. Small Bowel Dose Constraints in Radiation Therapy—Where Omics-Driven Biomarkers and Bioinformatics Can Take Us in the Future. BioMedInformatics 2024, 4, 158–172. [Google Scholar] [CrossRef]
- Wang, K.; Tepper, J.E. Radiation therapy-associated toxicity: Etiology, management, and prevention. CA A Cancer J. Clin. 2021, 71, 437–454. [Google Scholar] [CrossRef]
- Beumer, J.; Clevers, H. Regulation and plasticity of intestinal stem cells during homeostasis and regeneration. Development 2016, 143, 3639–3649. [Google Scholar] [CrossRef]
- Li, L.; Clevers, H. Coexistence of quiescent and active adult stem cells in mammals. Science 2010, 327, 542–545. [Google Scholar] [CrossRef]
- Sheng, X.; Lin, Z.; Lv, C.; Shao, C.; Bi, X.; Deng, M.; Xu, J.; Guerrero-Juarez, C.F.; Li, M.; Wu, X.; et al. Cycling Stem Cells Are Radioresistant and Regenerate the Intestine. Cell Rep. 2020, 32, 107952. [Google Scholar] [CrossRef]
- Kim, C.K.; Yang, V.W.; Bialkowska, A.B. The Role of Intestinal Stem Cells in Epithelial Regeneration Following Radiation-Induced Gut Injury. Curr. Stem Cell Rep. 2017, 3, 320–332. [Google Scholar] [CrossRef]
- Meyer, A.R.; Brown, M.E.; McGrath, P.S.; Dempsey, P.J. Injury-Induced Cellular Plasticity Drives Intestinal Regeneration. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 843–856. [Google Scholar] [CrossRef]
- Roy, A.; Bera, S.; Saso, L.; Dwarakanath, B.S. Role of autophagy in tumor response to radiation: Implications for improving radiotherapy. Front. Oncol. 2022, 12, 957373. [Google Scholar] [CrossRef]
- Classen, F.; Kranz, P.; Riffkin, H.; Pompsch, M.; Wolf, A.; Göpelt, K.; Baumann, M.; Baumann, J.; Brockmeier, U.; Metzen, E. Autophagy induced by ionizing radiation promotes cell death over survival in human colorectal cancer cells. Exp. Cell Res. 2019, 374, 29–37. [Google Scholar] [CrossRef]
- He, X.; Li, X.; Tian, W.; Li, C.; Li, P.; Zhao, J.; Yang, S.; Li, S. The role of redox-mediated lysosomal dysfunction and therapeutic strategies. Biomed. Pharmacother. 2023, 165, 115121. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Interactions between reactive oxygen species and autophagy: Special issue: Death mechanisms in cellular homeostasis. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2021, 1868, 119041. [Google Scholar] [CrossRef]
- Roy, B.C.; Ahmed, I.; Ramalingam, S.; Jala, V.; Haribabu, B.; Ramamoorthy, P.; Ashcraft, J.; Valentino, J.; Anant, S.; Sampath, V.; et al. Co-localization of autophagy-related protein p62 with cancer stem cell marker dclk1 may hamper dclk1’s elimination during colon cancer development and progression. Oncotarget 2019, 10, 2340–2354. [Google Scholar] [CrossRef]
- Jain, A.; Lamark, T.; Sjøttem, E.; Larsen, K.B.; Awuh, J.A.; Øvervatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef]
- Kumar, A.V.; Mills, J.; Lapierre, L.R. Selective Autophagy Receptor p62/SQSTM1, a Pivotal Player in Stress and Aging. Front. Cell Dev. Biol. 2022, 10, 793328. [Google Scholar] [CrossRef]
- Tian, W.; Rojo de la Vega, M.; Schmidlin, C.J.; Ooi, A.; Zhang, D.D. Kelch-like ECH-associated protein 1 (KEAP1) differentially regulates nuclear factor erythroid-2-related factors 1 and 2 (NRF1 and NRF2). J. Biol. Chem. 2018, 293, 2029–2040. [Google Scholar] [CrossRef]
- Ngo, V.; Duennwald, M.L. Nrf2 and Oxidative Stress: A General Overview of Mechanisms and Implications in Human Disease. Antioxidants 2022, 11, 2345. [Google Scholar] [CrossRef]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef]
- Glorieux, C.; Buc Calderon, P. Targeting catalase in cancer. Redox Biol. 2024, 77, 103404. [Google Scholar] [CrossRef]
- Mapuskar, K.A.; Anderson, C.M.; Spitz, D.R.; Batinic-Haberle, I.; Allen, B.G.; Oberley-Deegan, R.E. Utilizing Superoxide Dismutase Mimetics to Enhance Radiation Therapy Response While Protecting Normal Tissues. Semin. Radiat. Oncol. 2019, 29, 72–80. [Google Scholar] [CrossRef]
- El-Mahdy, M.A.; Alzarie, Y.A.; Hemann, C.; Badary, O.A.; Nofal, S.; Zweier, J.L. The novel SOD mimetic GC4419 increases cancer cell killing with sensitization to ionizing radiation while protecting normal cells. Free Radic. Biol. Med. 2020, 160, 630–642. [Google Scholar] [CrossRef]
- Wang, W.; Mani, A.M.; Wu, Z.H. DNA damage-induced nuclear factor-kappa B activation and its roles in cancer progression. J. Cancer Metastasis Treat. 2017, 3, 45–59. [Google Scholar] [CrossRef]
- Shibata, A.; Jeggo, P.A. ATM’s Role in the Repair of DNA Double-Strand Breaks. Genes 2021, 12, 1370. [Google Scholar] [CrossRef]
- Singh, V.; Gupta, D.; Arora, R. NF-kB as a key player in regulation of cellular radiation responses and identification of radiation countermeasures. Discoveries 2015, 3, e35. [Google Scholar] [CrossRef]
- Ouellette, M.M.; Zhou, S.; Yan, Y. Cell Signaling Pathways That Promote Radioresistance of Cancer Cells. Diagnostics 2022, 12, 656. [Google Scholar] [CrossRef]
- Luo, W.; Jin, Y.; Jiang, Y.; Yang, L.; Xu, H.; Wu, D.; Zhang, Y.; Yin, L.; Khan, Z.A.; Liang, G.; et al. Doublecortin-like kinase 1 activates NF-κB to induce inflammatory responses by binding directly to IKKβ. Cell Death Differ. 2023, 30, 1184–1197. [Google Scholar] [CrossRef]
- Li, L.; Jones, K.; Mei, H. Doublecotin-Like Kinase 1 Increases Chemoresistance of Colorectal Cancer Cells through the Anti-Apoptosis Pathway. J. Stem Cell Res. Ther. 2019, 9, 447. [Google Scholar] [CrossRef]
- Vijai, M.; Baba, M.; Ramalingam, S.; Thiyagaraj, A. DCLK1 and its interaction partners: An effective therapeutic target for colorectal cancer. Oncol. Lett. 2021, 22, 850. [Google Scholar] [CrossRef]
- Chandrakesan, P.; Panneerselvam, J.; Qu, D.; Weygant, N.; May, R.; Bronze, M.S.; Houchen, C.W. Regulatory Roles of Dclk1 in Epithelial Mesenchymal Transition and Cancer Stem Cells. J. Carcinog. Mutagen. 2016, 7, 257. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, S.-H.; Jeong, H.; Kim, K.H.; Jeon, D.; Cho, Y.; Lee, D.; Nam, K.T. Role of Nox4 in Mitigating Inflammation and Fibrosis in Dextran Sulfate Sodium–Induced Colitis. Cell. Mol. Gastroenterol. Hepatol. 2023, 16, 411–429. [Google Scholar] [CrossRef]
- Mehr, R.N.M.; Kheirollah, A.; Seif, F.; Dayati, P.M.; Babaahmadi-Rezaei, H. Reactive Oxygen Species and p38MAPK Have a Role in the Smad2 Linker Region Phosphorylation Induced by TGF-β. Iran. J. Med. Sci. 2018, 43, 401–408. [Google Scholar]
- Jiang, J.; Wang, K.; Chen, Y.; Chen, H.; Nice, E.C.; Huang, C. Redox regulation in tumor cell epithelial–mesenchymal transition: Molecular basis and therapeutic strategy. Signal Transduct. Target. Ther. 2017, 2, 17036. [Google Scholar] [CrossRef]
- Liu, R.M.; Desai, L.P. Reciprocal regulation of TGF-β and reactive oxygen species: A perverse cycle for fibrosis. Redox Biol. 2015, 6, 565–577. [Google Scholar] [CrossRef]
- Sureban, S.M.; May, R.; Lightfoot, S.A.; Hoskins, A.B.; Lerner, M.; Brackett, D.J.; Postier, R.G.; Ramanujam, R.; Mohammed, A.; Rao, C.V.; et al. DCAMKL-1 regulates epithelial-mesenchymal transition in human pancreatic cells through a miR-200a-dependent mechanism. Cancer Res. 2011, 71, 2328–2338. [Google Scholar] [CrossRef]
- Moore, L.L.; Qu, D.; Sureban, S.; Mitchell, S.; Pitts, K.; Cooper, N.; Fazili, J.; Harty, R.; Oseini, A.; Ding, K.; et al. From Inflammation to Oncogenesis: Tracing Serum DCLK1 and miRNA Signatures in Chronic Liver Diseases. Int. J. Mol. Sci. 2024, 25, 6481. [Google Scholar] [CrossRef]
- Dent, P.; Yacoub, A.; Fisher, P.B.; Hagan, M.P.; Grant, S. MAPK pathways in radiation responses. Oncogene 2003, 22, 5885–5896. [Google Scholar] [CrossRef]
- Gui, T.; Sun, Y.; Shimokado, A.; Muragaki, Y. The Roles of Mitogen-Activated Protein Kinase Pathways in TGF-β-Induced Epithelial-Mesenchymal Transition. J. Signal Transduct. 2012, 2012, 289243. [Google Scholar] [CrossRef]
- Huang, G.; Shi, L.Z.; Chi, H. Regulation of JNK and p38 MAPK in the immune system: Signal integration, propagation and termination. Cytokine 2009, 48, 161–169. [Google Scholar] [CrossRef]
- Yue, J.; López, J.M. Understanding MAPK Signaling Pathways in Apoptosis. Int. J. Mol. Sci. 2020, 21, 2346. [Google Scholar] [CrossRef]
- Lee, Y.-J.; Cho, H.-N.; Soh, J.-W.; Jhon, G.J.; Cho, C.-K.; Chung, H.-Y.; Bae, S.; Lee, S.-J.; Lee, Y.-S. Oxidative stress-induced apoptosis is mediated by ERK1/2 phosphorylation. Exp. Cell Res. 2003, 291, 251–266. [Google Scholar] [CrossRef]
- Sugiura, R.; Satoh, R.; Takasaki, T. ERK: A Double-Edged Sword in Cancer. ERK-Dependent Apoptosis as a Potential Therapeutic Strategy for Cancer. Cells 2021, 10, 2509. [Google Scholar] [CrossRef]
- Rauf, A.; Khalil, A.A.; Awadallah, S.; Khan, S.A.; Abu-Izneid, T.; Kamran, M.; Hemeg, H.A.; Mubarak, M.S.; Khalid, A.; Wilairatana, P. Reactive oxygen species in biological systems: Pathways, associated diseases, and potential inhibitors—A review. Food Sci. Nutr. 2024, 12, 675–693. [Google Scholar] [CrossRef]
- Dhanasekaran, D.N.; Reddy, E.P. JNK signaling in apoptosis. Oncogene 2008, 27, 6245–6251. [Google Scholar] [CrossRef]
- Burton, J.C.; Antoniades, W.; Okalova, J.; Roos, M.M.; Grimsey, N.J. Atypical p38 Signaling, Activation, and Implications for Disease. Int. J. Mol. Sci. 2021, 22, 4183. [Google Scholar] [CrossRef]
- Canovas, B.; Nebreda, A.R. Diversity and versatility of p38 kinase signalling in health and disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 346–366. [Google Scholar] [CrossRef]
- Yan, H.; He, L.; Lv, D.; Yang, J.; Yuan, Z. The Role of the Dysregulated JNK Signaling Pathway in the Pathogenesis of Human Diseases and Its Potential Therapeutic Strategies: A Comprehensive Review. Biomolecules 2024, 14, 243. [Google Scholar] [CrossRef]
- Obsilova, V.; Honzejkova, K.; Obsil, T. Structural Insights Support Targeting ASK1 Kinase for Therapeutic Interventions. Int. J. Mol. Sci. 2021, 22, 13395. [Google Scholar] [CrossRef]
- Chandrakesan, P.; May, R.; Qu, D.; Weygant, N.; Taylor, V.E.; Li, J.D.; Ali, N.; Sureban, S.M.; Qante, M.; Wang, T.C.; et al. Dclk1+ small intestinal epithelial tuft cells display the hallmarks of quiescence and self-renewal. Oncotarget 2015, 6, 30876–30886. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef]
- Lee, C.L.; Blum, J.M.; Kirsch, D.G. Role of p53 in regulating tissue response to radiation by mechanisms independent of apoptosis. Transl. Cancer Res. 2013, 2, 412–421. [Google Scholar]
- Okazaki, R. Role of p53 in Regulating Radiation Responses. Life 2022, 12, 1099. [Google Scholar] [CrossRef]
- Al-Arafat, T.; Mao, A.; Katsube, T.; Wang, B. Exploring the Role of p53 in Radiosensitivity: A Key Player in Cancer Therapy. Radiation 2024, 4, 309–324. [Google Scholar] [CrossRef]
- Morral, C.; Ayyaz, A.; Kuo, H.C.; Fink, M.; Verginadis, I.I.; Daniel, A.R.; Burner, D.N.; Driver, L.M.; Satow, S.; Hasapis, S.; et al. p53 promotes revival stem cells in the regenerating intestine after severe radiation injury. Nat. Commun. 2024, 15, 3018. [Google Scholar] [CrossRef]
- Kirsch, D.G.; Santiago, P.M.; di Tomaso, E.; Sullivan, J.M.; Hou, W.-S.; Dayton, T.; Jeffords, L.B.; Sodha, P.; Mercer, K.L.; Cohen, R.; et al. p53 Controls Radiation-Induced Gastrointestinal Syndrome in Mice Independent of Apoptosis. Science 2010, 327, 593–596. [Google Scholar] [CrossRef]
- Badie, B.; Kramar, M.H.; Lau, R.; Boothman, D.A.; Economou, J.S.; Black, K.L. Adenovirus-mediated p53 gene delivery potentiates the radiation-induced growth inhibition of experimental brain tumors. J. Neurooncol. 1998, 37, 217–222. [Google Scholar] [CrossRef]
- Cannan, W.J.; Pederson, D.S. Mechanisms and Consequences of Double-Strand DNA Break Formation in Chromatin. J. Cell Physiol. 2016, 231, 3–14. [Google Scholar] [CrossRef]
- van de Kamp, G.; Heemskerk, T.; Kanaar, R.; Essers, J. DNA Double Strand Break Repair Pathways in Response to Different Types of Ionizing Radiation. Front. Genet. 2021, 12, 738230. [Google Scholar] [CrossRef]
- Udayakumar, D.; Horikoshi, N.; Mishra, L.; Hunt, C.; Pandita, T.K. Detecting ATM-dependent chromatin modification in DNA damage response. Methods Mol. Biol. 2015, 1288, 317–336. [Google Scholar] [CrossRef]
- Senturk, E.; Manfredi, J.J. p53 and cell cycle effects after DNA damage. Methods Mol. Biol. 2013, 962, 49–61. [Google Scholar] [CrossRef]
- Eriksson, S.E.; Ceder, S.; Bykov, V.J.N.; Wiman, K.G. p53 as a hub in cellular redox regulation and therapeutic target in cancer. J. Mol. Cell Biol. 2019, 11, 330–341. [Google Scholar] [CrossRef]
- Yu, J.; Wang, Z.; Kinzler, K.W.; Vogelstein, B.; Zhang, L. PUMA mediates the apoptotic response to p53 in colorectal cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 1931–1936. [Google Scholar] [CrossRef]
- Wang, H.; Guo, M.; Wei, H.; Chen, Y. Targeting p53 pathways: Mechanisms, structures and advances in therapy. Signal Transduct. Target. Ther. 2023, 8, 92. [Google Scholar] [CrossRef]
- Chen, J. The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104. [Google Scholar] [CrossRef]
- Liu, Y.; Su, Z.; Tavana, O.; Gu, W. Understanding the complexity of p53 in a new era of tumor suppression. Cancer Cell 2024, 42, 946–967. [Google Scholar] [CrossRef]
- Jiang, C.; Zhou, Q.; Yi, K.; Yuan, Y.; Xie, X. Colorectal cancer initiation: Understanding early-stage disease for intervention. Cancer Lett. 2024, 589, 216831. [Google Scholar] [CrossRef]
- Huang, L.; Bernink, J.H.; Giladi, A.; Krueger, D.; van Son, G.J.F.; Geurts, M.H.; Busslinger, G.; Lin, L.; Begthel, H.; Zandvliet, M.; et al. Tuft cells act as regenerative stem cells in the human intestine. Nature 2024, 634, 929–935. [Google Scholar] [CrossRef]
- Qu, D.; May, R.; Sureban, S.M.; Weygant, N.; Chandrakesan, P.; Ali, N.; Li, L.; Barrett, T.; Houchen, C.W. Inhibition of Notch signaling reduces the number of surviving Dclk1+ reserve crypt epithelial stem cells following radiation injury. Am. J. Physiol.-Gastrointest. Liver Physiol. 2014, 306, G404–G411. [Google Scholar] [CrossRef]
- Hammad, M.; Raftari, M.; Cesário, R.; Salma, R.; Godoy, P.; Emami, S.N.; Haghdoost, S. Roles of Oxidative Stress and Nrf2 Signaling in Pathogenic and Non-Pathogenic Cells: A Possible General Mechanism of Resistance to Therapy. Antioxidants 2023, 12, 1371. [Google Scholar] [CrossRef]
- Liu, Y.P.; Zheng, C.C.; Huang, Y.N.; He, M.L.; Xu, W.W.; Li, B. Molecular mechanisms of chemo- and radiotherapy resistance and the potential implications for cancer treatment. MedComm 2021, 2, 315–340. [Google Scholar] [CrossRef]
- Sadoughi, F.; Mirsafaei, L.; Dana, P.M.; Hallajzadeh, J.; Asemi, Z.; Mansournia, M.A.; Montazer, M.; Hosseinpour, M.; Yousefi, B. The role of DNA damage response in chemo- and radio-resistance of cancer cells: Can DDR inhibitors sole the problem? DNA Repair. 2021, 101, 103074. [Google Scholar] [CrossRef]
- Wu, Y.; Song, Y.; Wang, R.; Wang, T. Molecular mechanisms of tumor resistance to radiotherapy. Mol. Cancer 2023, 22, 96. [Google Scholar] [CrossRef]
- Zhou, H.-M.; Zhang, J.-G.; Zhang, X.; Li, Q. Targeting cancer stem cells for reversing therapy resistance: Mechanism, signaling, and prospective agents. Signal Transduct. Target. Ther. 2021, 6, 62. [Google Scholar] [CrossRef]
- Chhetri, D.; Vengadassalapathy, S.; Venkadassalapathy, S.; Balachandran, V.; Umapathy, V.R.; Veeraraghavan, V.P.; Jayaraman, S.; Patil, S.; Iyaswamy, A.; Palaniyandi, K.; et al. Pleiotropic effects of DCLK1 in cancer and cancer stem cells. Front. Mol. Biosci. 2022, 9, 965730. [Google Scholar] [CrossRef]
- Sureban, S.M.; May, R.; Qu, D.; Weygant, N.; Chandrakesan, P.; Ali, N.; Lightfoot, S.A.; Pantazis, P.; Rao, C.V.; Postier, R.G.; et al. DCLK1 regulates pluripotency and angiogenic factors via microRNA-dependent mechanisms in pancreatic cancer. PLoS ONE 2013, 8, e73940. [Google Scholar] [CrossRef]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef]
- Gao, T.; Wang, M.; Xu, L.; Wen, T.; Liu, J.; An, G. DCLK1 is up-regulated and associated with metastasis and prognosis in colorectal cancer. J. Cancer Res. Clin. Oncol. 2016, 142, 2131–2140. [Google Scholar] [CrossRef]
- Liu, D.; Zhong, Z.; Karin, M. NF-κB: A Double-Edged Sword Controlling Inflammation. Biomedicines 2022, 10, 1250. [Google Scholar] [CrossRef]
- Zhang, T.; Ma, C.; Zhang, Z.; Zhang, H.; Hu, H. NF-κB signaling in inflammation and cancer. MedComm 2021, 2, 618–653. [Google Scholar] [CrossRef]
- Lee, J.H.; Massagué, J. TGF-β in developmental and fibrogenic EMTs. Semin. Cancer Biol. 2022, 86, 136–145. [Google Scholar] [CrossRef]
- Giarratana, A.O.; Prendergast, C.M.; Salvatore, M.M.; Capaccione, K.M. TGF-β signaling: Critical nexus of fibrogenesis and cancer. J. Transl. Med. 2024, 22, 594. [Google Scholar] [CrossRef]
- Jensen, R.B.; Rothenberg, E. Preserving genome integrity in human cells via DNA double-strand break repair. Mol. Biol. Cell 2020, 31, 859–865. [Google Scholar] [CrossRef]
- Nakajima-Koyama, M.; Kabata, M.; Lee, J.; Sogabe, Y.; Sakurai, S.; Hirota, A.; Kimura, M.; Nakamura, T.; Imoto, Y.; Kometani, K.; et al. The balance between IFN-γ and ERK/MAPK signaling activities ensures lifelong maintenance of intestinal stem cells. Cell Rep. 2025, 44, 115286. [Google Scholar] [CrossRef]
- Azzam, E.I.; Jay-Gerin, J.P.; Pain, D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012, 327, 48–60. [Google Scholar] [CrossRef]
- Luissint, A.-C.; Parkos, C.A.; Nusrat, A. Inflammation and the Intestinal Barrier: Leukocyte-Epithelial Cell Interactions, Cell Junction Remodeling, and Mucosal Repair. Gastroenterology 2016, 151, 616–632. [Google Scholar] [CrossRef]
- Schaue, D.; McBride, W.H. Links between innate immunity and normal tissue radiobiology. Radiat. Res. 2010, 173, 406–417. [Google Scholar] [CrossRef]
- Rees, W.D.; Tandun, R.; Yau, E.; Zachos, N.C.; Steiner, T.S. Regenerative Intestinal Stem Cells Induced by Acute and Chronic Injury: The Saving Grace of the Epithelium? Front. Cell Dev. Biol. 2020, 8, 583919. [Google Scholar] [CrossRef]
- Carbonero, F.; Mayta-Apaza, A.C.; Yu, J.Z.; Lindeblad, M.; Lyubimov, A.; Neri, F.; Szilagyi, E.; Bartholomew, A. A comparative analysis of gut microbiota disturbances in the Gottingen minipig and rhesus macaque models of acute radiation syndrome following bioequivalent radiation exposures. Radiat. Environ. Biophys. 2018, 57, 419–426. [Google Scholar] [CrossRef]
- Hollingsworth, B.A.; Cassatt, D.R.; DiCarlo, A.L.; Rios, C.I.; Satyamitra, M.M.; Winters, T.A.; Taliaferro, L.P. Acute Radiation Syndrome and the Microbiome: Impact and Review. Front. Pharmacol. 2021, 12, 643283. [Google Scholar] [CrossRef]
- Horseman, T.S.; Parajuli, B.; Frank, A.M.; Weaver, A.; Schauer, D.A.; Moran, S.; Anderson, J.A.; Holmes-Hampton, G.P.; Burmeister, D.M. Microbiome and Inflammasome Alterations Found during Radiation Dose Finding in a Sinclair Minipig Model of Gastrointestinal Acute Radiation Syndrome. Shock 2024, 62, 556–564. [Google Scholar] [CrossRef]
- Kono, H.; Rock, K.L. How dying cells alert the immune system to danger. Nat. Rev. Immunol. 2008, 8, 279–289. [Google Scholar] [CrossRef]
- Di Maggio, F.M.; Minafra, L.; Forte, G.I.; Cammarata, F.P.; Lio, D.; Messa, C.; Gilardi, M.C.; Bravatà, V. Portrait of inflammatory response to ionizing radiation treatment. J. Inflamm. 2015, 12, 14. [Google Scholar] [CrossRef]
- Soliman, A.M.; Barreda, D.R. Acute Inflammation in Tissue Healing. Int. J. Mol. Sci. 2022, 24, 641. [Google Scholar] [CrossRef]
- Chen, W.; Dong, J.; Haiech, J.; Kilhoffer, M.C.; Zeniou, M. Cancer Stem Cell Quiescence and Plasticity as Major Challenges in Cancer Therapy. Stem Cells Int. 2016, 2016, 1740936. [Google Scholar] [CrossRef]
- Safa, A.R. Resistance to drugs and cell death in cancer stem cells (CSCs). J. Transl. Sci. 2020, 6, 341. [Google Scholar] [CrossRef]
- Chu, X.; Tian, W.; Ning, J.; Xiao, G.; Zhou, Y.; Wang, Z.; Zhai, Z.; Tanzhu, G.; Yang, J.; Zhou, R. Cancer stem cells: Advances in knowledge and implications for cancer therapy. Signal Transduct. Target. Ther. 2024, 9, 170. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Shen, L.; Fu, G.; Yao, Y.; Li, G.; Deng, Y.; Zhang, H.; Zhou, M.; Yang, W.; Hua, G.; et al. Regulation of the regeneration of intestinal stem cells after irradiation. Ann. Transl. Med. 2020, 8, 1063. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Chen, H.; Liang, J.; Li, Y.; Yang, J.; Luo, C.; Tang, Y.; Ding, Y.; Liu, X.; Yuan, Q.; et al. Dual Role of Reactive Oxygen Species and their Application in Cancer Therapy. J. Cancer 2021, 12, 5543–5561. [Google Scholar] [CrossRef]
- Gough, M.J.; Sharon, S.; Crittenden, M.R.; Young, K.H. Using Preclinical Data to Design Combination Clinical Trials of Radiation Therapy and Immunotherapy. Semin. Radiat. Oncol. 2020, 30, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.; Hong, J.; Go, S.; Kim, B.S. Immunomodulation for Tissue Repair and Regeneration. Tissue Eng. Regen. Med. 2023, 20, 389–409. [Google Scholar] [CrossRef]
- Lumniczky, K.; Impens, N.; Armengol, G.; Candéias, S.; Georgakilas, A.G.; Hornhardt, S.; Martin, O.A.; Rödel, F.; Schaue, D. Low dose ionizing radiation effects on the immune system. Environ. Int. 2021, 149, 106212. [Google Scholar] [CrossRef]
- Schaue, D.; Kachikwu, E.L.; McBride, W.H. Cytokines in radiobiological responses: A review. Radiat. Res. 2012, 178, 505–523. [Google Scholar] [CrossRef]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef]
- Haist, M.; Stege, H.; Grabbe, S.; Bros, M. The Functional Crosstalk between Myeloid-Derived Suppressor Cells and Regulatory T Cells within the Immunosuppressive Tumor Microenvironment. Cancers 2021, 13, 210. [Google Scholar] [CrossRef]
- Wu, Q.; Allouch, A.; Martins, I.; Modjtahedi, N.; Deutsch, E.; Perfettini, J.L. Macrophage biology plays a central role during ionizing radiation-elicited tumor response. Biomed. J. 2017, 40, 200–211. [Google Scholar] [CrossRef]
- Yu, Y.; Yue, Z.; Xu, M.; Zhang, M.; Shen, X.; Ma, Z.; Li, J.; Xie, X. Macrophages play a key role in tissue repair and regeneration. PeerJ 2022, 10, e14053. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, M.; Ji, C.; Liu, X.; Gu, B.; Dong, T. Macrophage polarization in the tumor microenvironment: Emerging roles and therapeutic potentials. Biomed. Pharmacother. 2024, 177, 116930. [Google Scholar] [CrossRef]
- Priceman, S.J.; Sung, J.L.; Shaposhnik, Z.; Burton, J.B.; Torres-Collado, A.X.; Moughon, D.L.; Johnson, M.; Lusis, A.J.; Cohen, D.A.; Iruela-Arispe, M.L.; et al. Targeting distinct tumor-infiltrating myeloid cells by inhibiting CSF-1 receptor: Combating tumor evasion of antiangiogenic therapy. Blood 2010, 115, 1461–1471. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, M.D.M.; Schlom, J.; Hamilton, D.H. Blockade of tumor-derived colony-stimulating factor 1 (CSF1) promotes an immune-permissive tumor microenvironment. Cancer Immunol. Immunother. 2023, 72, 3349–3362. [Google Scholar] [CrossRef]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Chandrakesan, P.; Panneerselvam, J.; May, R.; Weygant, N.; Qu, D.; Berry, W.R.; Pitts, K.; Stanger, B.Z.; Rao, C.V.; Bronze, M.S.; et al. DCLK1-Isoform2 Alternative Splice Variant Promotes Pancreatic Tumor Immunosuppressive M2-Macrophage Polarization. Mol. Cancer Ther. 2020, 19, 1539–1549. [Google Scholar] [CrossRef]
- Guo, R.; Wang, R.; Zhang, W.; Li, Y.; Wang, Y.; Wang, H.; Li, X.; Song, J. Macrophage Polarisation in the Tumour Microenvironment: Recent Research Advances and Therapeutic Potential of Different Macrophage Reprogramming. Cancer Control 2025, 32, 10732748251316604. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Song, M.Y.; Kim, E.H. Role of Oxidative Stress and Nrf2/KEAP1 Signaling in Colorectal Cancer: Mechanisms and Therapeutic Perspectives with Phytochemicals. Antioxidants 2021, 10, 743. [Google Scholar] [CrossRef]
- Nallasamy, P.; Nimmakayala, R.K.; Parte, S.; Are, A.C.; Batra, S.K.; Ponnusamy, M.P. Tumor microenvironment enriches the stemness features: The architectural event of therapy resistance and metastasis. Mol. Cancer 2022, 21, 225. [Google Scholar] [CrossRef]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem. Cell 2015, 16, 225–238. [Google Scholar] [CrossRef]
- Laviron, M.; Combadière, C.; Boissonnas, A. Tracking Monocytes and Macrophages in Tumors With Live Imaging. Front. Immunol. 2019, 10, 1201. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Song, Y.; Du, W.; Gong, L.; Chang, H.; Zou, Z. Tumor-associated macrophages: An accomplice in solid tumor progression. J. Biomed. Sci. 2019, 26, 78. [Google Scholar] [CrossRef]
- Xiao, M.; He, J.; Yin, L.; Chen, X.; Zu, X.; Shen, Y. Tumor-Associated Macrophages: Critical Players in Drug Resistance of Breast Cancer. Front. Immunol. 2021, 12, 799428. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef]
- Li, X.; Chen, L.; Peng, X.; Zhan, X. Progress of tumor-associated macrophages in the epithelial-mesenchymal transition of tumor. Front. Oncol. 2022, 12, 911410. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Zou, W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer 2005, 5, 263–274. [Google Scholar] [CrossRef]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef]
- Yin, G.; Zhao, C.; Pei, W. Crosstalk between macrophages and innate lymphoid cells (ILCs) in diseases. Int. Immunopharmacol. 2022, 110, 108937. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Marchesi, F.; Garlanda, C. Macrophages as tools and targets in cancer therapy. Nat. Rev. Drug Discov. 2022, 21, 799–820. [Google Scholar] [CrossRef] [PubMed]
- Bhol, N.K.; Bhanjadeo, M.M.; Singh, A.K.; Dash, U.C.; Ojha, R.R.; Majhi, S.; Duttaroy, A.K.; Jena, A.B. The interplay between cytokines, inflammation, and antioxidants: Mechanistic insights and therapeutic potentials of various antioxidants and anti-cytokine compounds. Biomed. Pharmacother. 2024, 178, 117177. [Google Scholar] [CrossRef]
- Voss, U.; Malipatlolla, D.K.; Patel, P.; Devarakonda, S.; Sjöberg, F.; Grandér, R.; Rascón, A.; Nyman, M.; Steineck, G.; Bull, C. Irradiation Induces Tuft Cell Hyperplasia and Myenteric Neuronal Loss in the Absence of Dietary Fiber in a Mouse Model of Pelvic Radiotherapy. Gastroenterol. Insights 2022, 13, 87–102. [Google Scholar] [CrossRef]
- Jang, B.; Kim, H.; Lee, S.H.; Won, Y.; Kaji, I.; Coffey, R.J.; Choi, E.; Goldenring, J.R. Dynamic tuft cell expansion during gastric metaplasia and dysplasia. J. Pathol. Clin. Res. 2024, 10, e352. [Google Scholar] [CrossRef] [PubMed]
- Citrin, D.; Cotrim, A.P.; Hyodo, F.; Baum, B.J.; Krishna, M.C.; Mitchell, J.B. Radioprotectors and mitigators of radiation-induced normal tissue injury. Oncologist 2010, 15, 360–371. [Google Scholar] [CrossRef]
- Liu, L.; Liang, Z.; Ma, S.; Li, L.; Liu, X. Radioprotective countermeasures for radiation injury (Review). Mol. Med. Rep. 2023, 27, 66. [Google Scholar] [CrossRef]
- Obrador, E.; Salvador, R.; Villaescusa, J.I.; Soriano, J.M.; Estrela, J.M.; Montoro, A. Radioprotection and Radiomitigation: From the Bench to Clinical Practice. Biomedicines 2020, 8, 461. [Google Scholar] [CrossRef] [PubMed]
- Nagesh, P.K.B.; Monette, S.; Shamu, T.; Giralt, S.; Jean, S.C.S.; Zhang, Z.; Fuks, Z.; Kolesnick, R. Anti-ceramide Single-Chain Variable Fragment Mitigates Gastrointestinal-Acute Radiation Syndrome and Improves Marrow Reconstitution, Rendering Near-Normal 90-Day Autopsies. Int. J. Radiat. Oncol. Biol. Phys. 2024, 120, 558–569. [Google Scholar] [CrossRef]
- U.S. Food & Durg Administration. FDA Approves Radiation Medical Countermeasure. Available online: https://www.fda.gov/emergency-preparedness-and-response/about-mcmi/fda-approves-radiation-medical-countermeasure (accessed on 30 April 2025).
- Fiocchi, C.; Lund, P.K. Themes in fibrosis and gastrointestinal inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G677–G683. [Google Scholar] [CrossRef]
- Kim, K.K.; Sheppard, D.; Chapman, H.A. TGF-β1 Signaling and Tissue Fibrosis. Cold Spring Harb. Perspect. Biol. 2018, 10, a022293. [Google Scholar] [CrossRef] [PubMed]
- Ku, J.C.; Raiten, J.; Li, Y. Understanding fibrosis: Mechanisms, clinical implications, current therapies, and prospects for future interventions. Biomed. Eng. Adv. 2024, 7, 100118. [Google Scholar] [CrossRef]
- AlQudah, M.; Hale, T.M.; Czubryt, M.P. Targeting the renin-angiotensin-aldosterone system in fibrosis. Matrix Biol. 2020, 91–92, 92–108. [Google Scholar] [CrossRef]
- Awad, K.; Zaki, M.M.; Mohammed, M.; Lewek, J.; Lavie, C.J.; Banach, M. Effect of the Renin-Angiotensin System Inhibitors on Inflammatory Markers: A Systematic Review and Meta-analysis of Randomized Controlled Trials. Mayo Clin. Proc. 2022, 97, 1808–1823. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, N.A.; Wang, J.K.; Zhu, S.M.; Zhu, H.L.; Liu, B.; Cui, Q.W.; Guan, G.C.; Tian, G. Telmisartan inhibited angiotensin II-induced collagen metabolic imbalance without directly targeting TGF-β 1/Smad signaling pathway in cardiac fibroblasts. Minerva Cardioangiol. 2015, 63, 507–514. [Google Scholar]
- Yu, Z.; Xu, C.; Song, B.; Zhang, S.; Chen, C.; Li, C.; Zhang, S. Tissue fibrosis induced by radiotherapy: Current understanding of the molecular mechanisms, diagnosis and therapeutic advances. J. Transl. Med. 2023, 21, 708. [Google Scholar] [CrossRef]
- Kuo, B.; Szabó, E.; Lee, S.C.; Balogh, A.; Norman, D.; Inoue, A.; Ono, Y.; Aoki, J.; Tigyi, G. The LPA(2) receptor agonist Radioprotectin-1 spares Lgr5-positive intestinal stem cells from radiation injury in murine enteroids. Cell Signal 2018, 51, 23–33. [Google Scholar] [CrossRef]
- Ahire, Y.S.; Bairagi, V.A.; Somavanshi, D.B.; Jadhav, S.R.; Jadhav, S.B.; Jagtap, S.D. Expanding telmisartan’s therapeutic horizon: Exploring its multifaceted mechanisms beyond cardiovascular disorders. Future J. Pharm. Sci. 2024, 10, 84. [Google Scholar] [CrossRef]
- Yang, J.; Yang, X.; Gao, L.; Zhang, J.; Yi, C.; Huang, Y. The role of the renin-angiotensin system inhibitors in malignancy: A review. Am. J. Cancer Res. 2021, 11, 884–897. [Google Scholar]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Arfin, S.; Jha, N.K.; Jha, S.K.; Kesari, K.K.; Ruokolainen, J.; Roychoudhury, S.; Rathi, B.; Kumar, D. Oxidative Stress in Cancer Cell Metabolism. Antioxidants 2021, 10, 642. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.A.; Rogoff, H.A. Implications of reactive oxygen species on cancer formation and its treatment. Semin. Oncol. 2021, 48, 238–245. [Google Scholar] [CrossRef]
- Kalyanaraman, B.; Cheng, G.; Hardy, M.; Ouari, O.; Bennett, B.; Zielonka, J. Teaching the basics of reactive oxygen species and their relevance to cancer biology: Mitochondrial reactive oxygen species detection, redox signaling, and targeted therapies. Redox Biol. 2018, 15, 347–362. [Google Scholar] [CrossRef]
- Egami, K.; Murohara, T.; Shimada, T.; Sasaki, K.; Shintani, S.; Sugaya, T.; Ishii, M.; Akagi, T.; Ikeda, H.; Matsuishi, T.; et al. Role of host angiotensin II type 1 receptor in tumor angiogenesis and growth. J. Clin. Investig. 2003, 112, 67–75. [Google Scholar] [CrossRef]
- Hassani, B.; Attar, Z.; Firouzabadi, N. The renin-angiotensin-aldosterone system (RAAS) signaling pathways and cancer: Foes versus allies. Cancer Cell Int. 2023, 23, 254. [Google Scholar] [CrossRef]
- Pinter, M.; Jain, R.K. Targeting the renin-angiotensin system to improve cancer treatment: Implications for immunotherapy. Sci. Transl. Med. 2017, 9, 410. [Google Scholar] [CrossRef]
- Zhu, J.; Li, X.; Huang, M.; Zhu, H.; Tan, Y.; He, X.; Sun, Z.; Cheng, H.; Li, F.; Jiang, P.; et al. Application of Recombinant Human Superoxide Dismutase in Radical Concurrent Chemoradiotherapy for Cervical Cancer to Prevent and Treat Radiation-induced Acute Rectal Injury: A Multicenter, Randomized, Open-label, Prospective Trial. Int. J. Radiat. Oncol. Biol. Phys. 2024, 120, 720–729. [Google Scholar] [CrossRef] [PubMed]
- Bhanja, P.; Norris, A.; Gupta-Saraf, P.; Hoover, A.; Saha, S. BCN057 induces intestinal stem cell repair and mitigates radiation-induced intestinal injury. Stem. Cell Res. Ther. 2018, 9, 26. [Google Scholar] [CrossRef] [PubMed]
- Simman, R.; Bach, K.; Abbas, F.; Klomparens, K.; Brickman, B.J. Management of Radiation-induced Tissue Injuries: A Review of Current Treatment Strategies. Plast. Reconstr. Surg. Glob. Open 2023, 11, e5043. [Google Scholar] [CrossRef]
- Legeza, V.I.; Grebenyuk, A.N.; Drachev, I.S. Radiomitigators: Classification, Pharmacological Properties, and Application Prospects. Biol. Bull. 2019, 46, 1625–1632. [Google Scholar] [CrossRef]
- Kim, M.H.; Kim, H. The Roles of Glutamine in the Intestine and Its Implication in Intestinal Diseases. Int. J. Mol. Sci. 2017, 18, 1051. [Google Scholar] [CrossRef] [PubMed]
- Finch, P.W.; Mark Cross, L.J.; McAuley, D.F.; Farrell, C.L. Palifermin for the protection and regeneration of epithelial tissues following injury: New findings in basic research and pre-clinical models. J. Cell Mol. Med. 2013, 17, 1065–1087. [Google Scholar] [CrossRef] [PubMed]
- Brubaker, P.L. Glucagon-like Peptide-2 and the Regulation of Intestinal Growth and Function. Compr. Physiol. 2018, 8, 1185–1210. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Han, J.; Elisseeff, J.H.; Demaria, M. The senescence-associated secretory phenotype and its physiological and pathological implications. Nat. Rev. Mol. Cell Biol. 2024, 25, 958–978. [Google Scholar] [CrossRef]
- Ibragimova, M.; Kussainova, A.; Aripova, A.; Bersimbaev, R.; Bulgakova, O. The Molecular Mechanisms in Senescent Cells Induced by Natural Aging and Ionizing Radiation. Cells 2024, 13, 550. [Google Scholar] [CrossRef]
- Robbins, P.D.; Jurk, D.; Khosla, S.; Kirkland, J.L.; LeBrasseur, N.K.; Miller, J.D.; Passos, J.F.; Pignolo, R.J.; Tchkonia, T.; Niedernhofer, L.J. Senolytic Drugs: Reducing Senescent Cell Viability to Extend Health Span. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 779–803. [Google Scholar] [CrossRef]
- Kumar, K.; Moon, B.-H.; Kumar, S.; Angdisen, J.; Kallakury, B.V.S.; AJ, F.; Suman, S. Senolytic agent ABT-263 mitigates low- and high-LET radiation-induced gastrointestinal cancer development in Apc1638N/+ mice. Aging 2025, 17, 97–115. [Google Scholar] [CrossRef]
- Wang, H.; Wang, Z.; Huang, Y.; Zhou, Y.; Sheng, X.; Jiang, Q.; Wang, Y.; Luo, P.; Luo, M.; Shi, C. Senolytics (DQ) Mitigates Radiation Ulcers by Removing Senescent Cells. Front. Oncol. 2020, 9, 1576. [Google Scholar] [CrossRef]
- Alqahtani, S.; Alqahtani, T.; Venkatesan, K.; Sivadasan, D.; Ahmed, R.; Sirag, N.; Elfadil, H.; Abdullah Mohamed, H.; Haseena, T.A.; Elsayed Ahmed, R.; et al. SASP Modulation for Cellular Rejuvenation and Tissue Homeostasis: Therapeutic Strategies and Molecular Insights. Cells 2025, 14, 608. [Google Scholar] [CrossRef]
- Hamama, S.; Gilbert-Sirieix, M.; Vozenin, M.-C.; Delanian, S. Radiation-induced enteropathy: Molecular basis of pentoxifylline–vitamin E anti-fibrotic effect involved TGF-β1 cascade inhibition. Radiother. Oncol. 2012, 105, 305–312. [Google Scholar] [CrossRef]
- Bennett, M.H.; Feldmeier, J.; Hampson, N.B.; Smee, R.; Milross, C. Hyperbaric oxygen therapy for late radiation tissue injury. Cochrane Database Syst. Rev. 2016, 4, Cd005005. [Google Scholar] [CrossRef] [PubMed]
- Haydont, V.r.; Bourgier, C.l.; Pocard, M.; Lusinchi, A.; Aigueperse, J.; Mathé, D.; Bourhis, J.; Vozenin-Brotons, M.-C. Pravastatin Inhibits the Rho/CCN2/Extracellular Matrix Cascade in Human Fibrosis Explants and Improves Radiation-Induced Intestinal Fibrosis in Rats. Clin. Cancer Res. 2007, 13, 5331–5340. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.; Fu, M.; Wang, M.; Wei, Y.; Wei, X. Targeting TGF-β signal transduction for fibrosis and cancer therapy. Mol. Cancer 2022, 21, 104. [Google Scholar] [CrossRef] [PubMed]
- Cai, T.; Jiang, J.; Yao, W.; Hu, Y.; Kong, S.; Fan, Q.; Yan, X.; Li, F.; Shi, Z. Pirfenidone inhibits stromal collagen deposition and improves intra-tumoral delivery and antitumor efficacy of Pegylated liposomal doxorubicin. Biomed. Pharmacother. 2023, 157, 114015. [Google Scholar] [CrossRef]
- Mett, V.; Kurnasov, O.V.; Bespalov, I.A.; Molodtsov, I.; Brackett, C.M.; Burdelya, L.G.; Purmal, A.A.; Gleiberman, A.S.; Toshkov, I.A.; Burkhart, C.A.; et al. A deimmunized and pharmacologically optimized Toll-like receptor 5 agonist for therapeutic applications. Commun. Biol. 2021, 4, 466. [Google Scholar] [CrossRef]
- Krivokrysenko, V.I.; Toshkov, I.A.; Gleiberman, A.S.; Krasnov, P.; Shyshynova, I.; Bespalov, I.; Maitra, R.K.; Narizhneva, N.V.; Singh, V.K.; Whitnall, M.H.; et al. The Toll-Like Receptor 5 Agonist Entolimod Mitigates Lethal Acute Radiation Syndrome in Non-Human Primates. PLoS ONE 2015, 10, e0135388. [Google Scholar] [CrossRef]
- Song, W.S.; Kim, J.-H.; Choi, C.-M.; Lee, W.-J.; Yoon, S.-i. TLR5 binding and activation by KMRC011, a flagellin-derived radiation countermeasure. Biochem. Biophys. Res. Commun. 2019, 508, 570–575. [Google Scholar] [CrossRef]
- Liu, Z.; Lei, X.; Li, X.; Cai, J.M.; Gao, F.; Yang, Y.Y. Toll-like receptors and radiation protection. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 31–39. [Google Scholar] [CrossRef]
- Kim, W.H.; Yoo, J.H.; Yoo, I.K.; Kwon, C.I.; Hong, S.P. Effects of Mesenchymal Stem Cells Treatment on Radiation-Induced Proctitis in Rats. Yonsei Med. J. 2023, 64, 167–174. [Google Scholar] [CrossRef]
- Linard, C.; Strup-Perrot, C.; Lacave-Lapalun, J.-V.; Benderitter, M. Flagellin preconditioning enhances the efficacy of mesenchymal stem cells in an irradiation-induced proctitis model. J. Leukoc. Biol. 2016, 100, 569–580. [Google Scholar] [CrossRef]
- Chaves-Pérez, A.; Santos-de-Frutos, K.; de la Rosa, S.; Herranz-Montoya, I.; Perna, C.; Djouder, N. Transit-amplifying cells control R-spondins in the mouse crypt to modulate intestinal stem cell proliferation. J. Exp. Med. 2022, 219, e20212405. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Kljavin, N.; Nguyen, T.T.T.; Storm, E.E.; Marsh, B.; Jiang, J.; Lin, W.; Menon, H.; Piskol, R.; de Sauvage, F.J. Adrenergic nerves regulate intestinal regeneration through IL-22 signaling from type 3 innate lymphoid cells. Cell Stem. Cell 2023, 30, 1166–1178.e1168. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yokoyama, Y.; Hirose, H.; Shimomura, Y.; Bonkobara, S.; Itakura, H.; Kouda, S.; Morimoto, Y.; Minami, K.; Takahashi, H.; et al. Functional assessment of miR-1291 in colon cancer cells. Int. J. Oncol. 2022, 60, 13. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Wu, Q.; Lu, F.; Lei, J.; Zhou, Y.; Liu, Y.; Zhu, N.; Yu, Y.; Ning, Z.; She, T.; et al. Nrf2 signaling pathway: Current status and potential therapeutic targetable role in human cancers. Front. Oncol. 2023, 13, 1184079. [Google Scholar] [CrossRef]
- Ebrahimi, N.; Abdulwahid, A.R.R.; Mansouri, A.; Karimi, N.; Bostani, R.J.; Beiranvand, S.; Adelian, S.; Khorram, R.; Vafadar, R.; Hamblin, M.R.; et al. Targeting the NF-κB pathway as a potential regulator of immune checkpoints in cancer immunotherapy. Cell. Mol. Life Sci. 2024, 81, 106. [Google Scholar] [CrossRef]
- Beamish, C.A.; Zawaski, J.A.; Inoue, T.; Sarkar, P.; Grosshans, D.R.; Sabek, O.M.; Gaber, M.W. NF-κB Blockade by NEMO Binding Domain Peptide Ameliorates Inflammation and Neurobehavioral Sequelae After Cranial Radiation Therapy in Juvenile Mice. Int. J. Radiat. Oncol. Biol. Phys. 2021, 109, 1508–1520. [Google Scholar] [CrossRef]
- Deng, Z.; Fan, T.; Xiao, C.; Tian, H.; Zheng, Y.; Li, C.; He, J. TGF-β signaling in health, disease, and therapeutics. Signal Transduct. Target. Ther. 2024, 9, 61. [Google Scholar] [CrossRef]
- Hu, J.; Cao, J.; Topatana, W.; Juengpanich, S.; Li, S.; Zhang, B.; Shen, J.; Cai, L.; Cai, X.; Chen, M. Targeting mutant p53 for cancer therapy: Direct and indirect strategies. J. Hematol. Oncol. 2021, 14, 157. [Google Scholar] [CrossRef]
- Nishikawa, S.; Iwakuma, T. Drugs Targeting p53 Mutations with FDA Approval and in Clinical Trials. Cancers 2023, 15, 429. [Google Scholar] [CrossRef]
- Boateng, F.; Ngwa, W. Delivery of Nanoparticle-Based Radiosensitizers for Radiotherapy Applications. Int. J. Mol. Sci. 2019, 21, 273. [Google Scholar] [CrossRef]
- Xiao, X.; Teng, F.; Shi, C.; Chen, J.; Wu, S.; Wang, B.; Meng, X.; Essiet Imeh, A.; Li, W. Polymeric nanoparticles-Promising carriers for cancer therapy. Front. Bioeng. Biotechnol. 2022, 10, 1024143. [Google Scholar] [CrossRef] [PubMed]
- Foglietta, F.; Serpe, L.; Canaparo, R. ROS-generating nanoplatforms as selective and tunable therapeutic weapons against cancer. Discov. Nano 2023, 18, 151. [Google Scholar] [CrossRef] [PubMed]
- Mitra, D.; Armijo, G.K.; Ober, E.H.; Baker, S.M.; Turner, H.C.; Broustas, C.G. MIIST305 mitigates gastrointestinal acute radiation syndrome injury and ameliorates radiation-induced gut microbiome dysbiosis. Gut Microbes 2024, 17, 2458189. [Google Scholar] [CrossRef] [PubMed]
- Sminia, P.; Guipaud, O.; Viktorsson, K.; Ahire, V.; Baatout, S.; Boterberg, T.; Cizkova, J.; Dostál, M.; Fernandez-Palomo, C.; Filipova, A.; et al. Clinical Radiobiology for Radiation Oncology. In Radiobiology Textbook; Baatout, S., Ed.; Springer International Publishing: Cham, Switzerland, 2023; pp. 237–309. [Google Scholar]
- Ahire, V.; Ahmadi Bidakhvidi, N.; Boterberg, T.; Chaudhary, P.; Chevalier, F.; Daems, N.; Delbart, W.; Baatout, S.; Deroose, C.M.; Fernandez-Palomo, C.; et al. Radiobiology of Combining Radiotherapy with Other Cancer Treatment Modalities. In Radiobiology Textbook; Baatout, S., Ed.; Springer International Publishing: Cham, Switzerland, 2023; pp. 311–386. [Google Scholar]
- Houchen, C.; Chandrakesan, P.; Panneerselvam, J.; Qu, D. DCLK1 and DNA Damage Response. In Genotoxicity and Mutagenicity—Mechanisms and Test Methods; Soloneski, S., Larramendy, M.L., Eds.; IntechOpen: Rijeka, Croatia, 2020. [Google Scholar]
- Wu, X.; Qu, D.; Weygant, N.; Peng, J.; Houchen, C.W. Cancer Stem Cell Marker DCLK1 Correlates with Tumorigenic Immune Infiltrates in the Colon and Gastric Adenocarcinoma Microenvironments. Cancers 2020, 12, 274. [Google Scholar] [CrossRef]
- Mohammadi, C.; Najafi, R. DCLK1 as a Promising Marker for Radioresistance in Colorectal Cancer. J. Gastrointest. Cancer 2020, 51, 714–715. [Google Scholar] [CrossRef]
- Carli, A.L.E.; Afshar-Sterle, S.; Rai, A.; Fang, H.; O’Keefe, R.; Tse, J.; Ferguson, F.M.; Gray, N.S.; Ernst, M.; Greening, D.W.; et al. Cancer stem cell marker DCLK1 reprograms small extracellular vesicles toward migratory phenotype in gastric cancer cells. Proteomics 2021, 21, e2000098. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dose Range (Gy) | Affected Syndrome | Organ-Specific Damage | Associated Symptoms | Overlapping Effects | Studies Summary |
|---|---|---|---|---|---|
| 1–2 Gy | Hematopoietic (H-ARS) | Mild bone marrow suppression, reduced blood cell counts | Fatigue, mild nausea, increased infection risk | Latent period may mask hematologic effects | Preclinical [13] |
| 2–7 Gy | Hematopoietic (H-ARS) | Severe bone marrow suppression, pancytopenia | Hemorrhage, infections, anemia, fever | GI symptoms (nausea, vomiting) may appear at higher end | Preclinical [13] Clinical [9] |
| 7–15 Gy | GI-ARS, H-ARS | Destruction of GI epithelial lining, crypt loss, mucosal barrier breakdown | Severe diarrhea, dehydration, abdominal pain, septicemia | Hematopoietic damage worsens GI injury, increasing lethality | Preclinical [13] Clinical [9] |
| 15–30 Gy | GI-ARS, CNS effects | Massive GI damage, early CNS dysfunction | Severe GI symptoms, neurovascular instability | Hematopoietic failure exacerbates multi-organ dysfunction | * Preclinical [13] Clinical [6] |
| >30 Gy | CNS-ARS, GI-ARS | Brain edema, neuronal death, blood–brain barrier disruption, irreversible GI damage | Headache, confusion, seizures, loss of consciousness, death within hours to days | GI and hematopoietic syndromes accelerate neurovascular instability | Clinical [6] |
| Pathway | Protective Effect | Oncogenic Effect |
|---|---|---|
| NF-κB | Transient NF-κB activation promotes pro-survival cytokines and regeneration [156] | Sustained NF-κB signaling drives inflammation, immune evasion, and tumor progression [157] |
| TGF-β | TGF-β–mediated EMT in repair phase facilitates wound closure and restitution [158] | Persistent TGF-β signaling leads to EMT-driven invasion, fibrosis, and metastatic potential [159] |
| p53 | Modulates DNA repair and cell-cycle checkpoints preserves genomic integrity in surviving cells [160] | Inhibition of p53–mediated apoptosis allows survival of cells with oncogenic lesions [143] |
| MAPK | Activation of ERK/JNK pathways drives proliferation of crypt progenitors and regeneration [161] | Chronic MAPK signaling leads to uncontrolled proliferation, survival, and resistance to apoptosis [118] |
| Combination Strategy | Drug Combination/Example | Mechanism/Rationale |
|---|---|---|
| Antioxidant + AT1 Receptor Blocker | Tempol (or MitoQ/SOD mimetics) + Telmisartan (or Losartan/Candesartan) | Antioxidants reduce ROS-induced damage while ARBs block angiotensin II–mediated inflammation and fibrosis, protecting both GI and hematopoietic tissues. |
| DCLK1 Inhibition + Antioxidant | DCLK1-targeting agent (experimental) + Tempol or MitoQ | Inhibiting DCLK1 prevents protection of cancer stem-like cells (CSCs) and, when combined with antioxidants, helps preserve normal tissue regeneration while reducing therapy resistance and tumor recurrence. |
| Senolytic Therapy + Radioprotective Agents | Dasatinib + Quercetin (or Fisetin/Navitoclax) combined with Tempol and/or Telmisartan | Senolytics clear radiation-induced senescent cells to reduce chronic inflammation and fibrosis; the addition of antioxidants/ARBs improves tissue repair and mitigates long-term complications. |
| Immunomodulatory Combination | CSF-1R inhibitor + Immune checkpoint inhibitors (e.g., anti-PD-1) + ARB | This approach reprograms tumor-associated macrophages (TAMs) from an M2 to M1 phenotype and restores anti-tumor immunity while protecting normal tissue repair mechanisms. |
| Supportive Hematopoietic Growth Factor Integration | Granulocyte Colony-Stimulating Factor (G-CSF) added to the above regimens | G-CSF aids in restoring bone marrow function and immune competence, which is critical given the interconnected hematopoietic and GI injuries observed in ARS. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moore, L.L.; Jaboin, J.; Brown, M.L.; Houchen, C.W. Balancing Regeneration and Resistance: Targeting DCLK1 to Mitigate Gastrointestinal Radiation Injury and Oncogenesis. Cancers 2025, 17, 2050. https://doi.org/10.3390/cancers17122050
Moore LL, Jaboin J, Brown ML, Houchen CW. Balancing Regeneration and Resistance: Targeting DCLK1 to Mitigate Gastrointestinal Radiation Injury and Oncogenesis. Cancers. 2025; 17(12):2050. https://doi.org/10.3390/cancers17122050
Chicago/Turabian StyleMoore, Landon L., Jerry Jaboin, Milton L. Brown, and Courtney W. Houchen. 2025. "Balancing Regeneration and Resistance: Targeting DCLK1 to Mitigate Gastrointestinal Radiation Injury and Oncogenesis" Cancers 17, no. 12: 2050. https://doi.org/10.3390/cancers17122050
APA StyleMoore, L. L., Jaboin, J., Brown, M. L., & Houchen, C. W. (2025). Balancing Regeneration and Resistance: Targeting DCLK1 to Mitigate Gastrointestinal Radiation Injury and Oncogenesis. Cancers, 17(12), 2050. https://doi.org/10.3390/cancers17122050