
LMP7-Specific Inhibitor M3258 Modulates the Tumor Microenvironment of Triple-Negative Breast Cancer and Inflammatory Breast Cancer

 , , , , , , and
, , , , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Immunohistochemistry (IHC) Staining of Human TNBC Tumors
2.2. Digital Image Analysis of Immunostained TNBC Tumor Samples
2.3. Cell Lines and Reagents
2.4. Assessment of LMP7 Proteolytic Activity
2.5. In Vitro Cell Viability Assay
2.6. Caspase 3/7 Activity Measurement
2.7. Western Blotting
2.8. Humanized SUM-149 PT Xenograft Mouse Model
2.9. Single-Cell RNA Sequencing (scRNA-seq) from SUM-149 PT Tumors
2.10. Flow Cytometry Analysis of THP-1 Cell–Derived M1 and M2 Macrophages
2.11. In Vitro Cell Migration and Invasion Assays Under Co-Culture Conditions
2.12. Statistical Analysis
3. Results
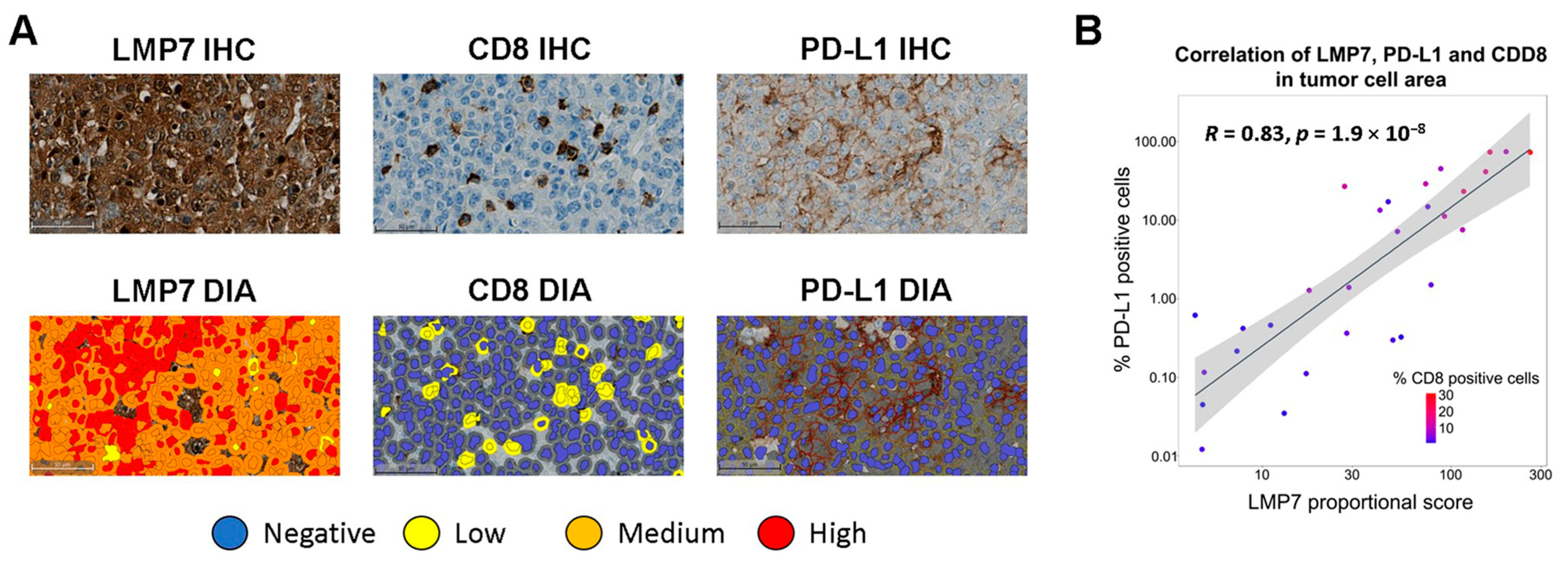
3.1. LMP7 Expression Correlates with CD8+ T Cell Infiltration and PD-L1 Expression in TNBC
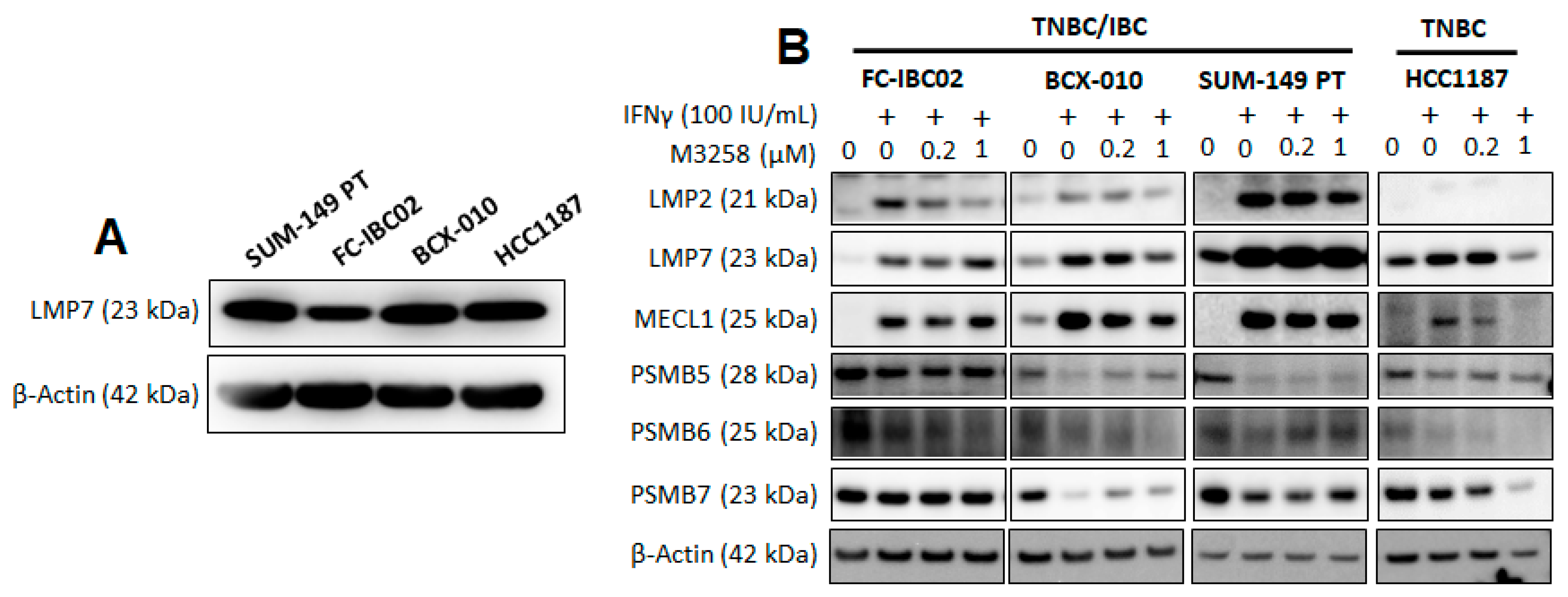
3.2. Effects of IFNγ and M3258 on Expression of IP and Constitutive Proteasome Subunits
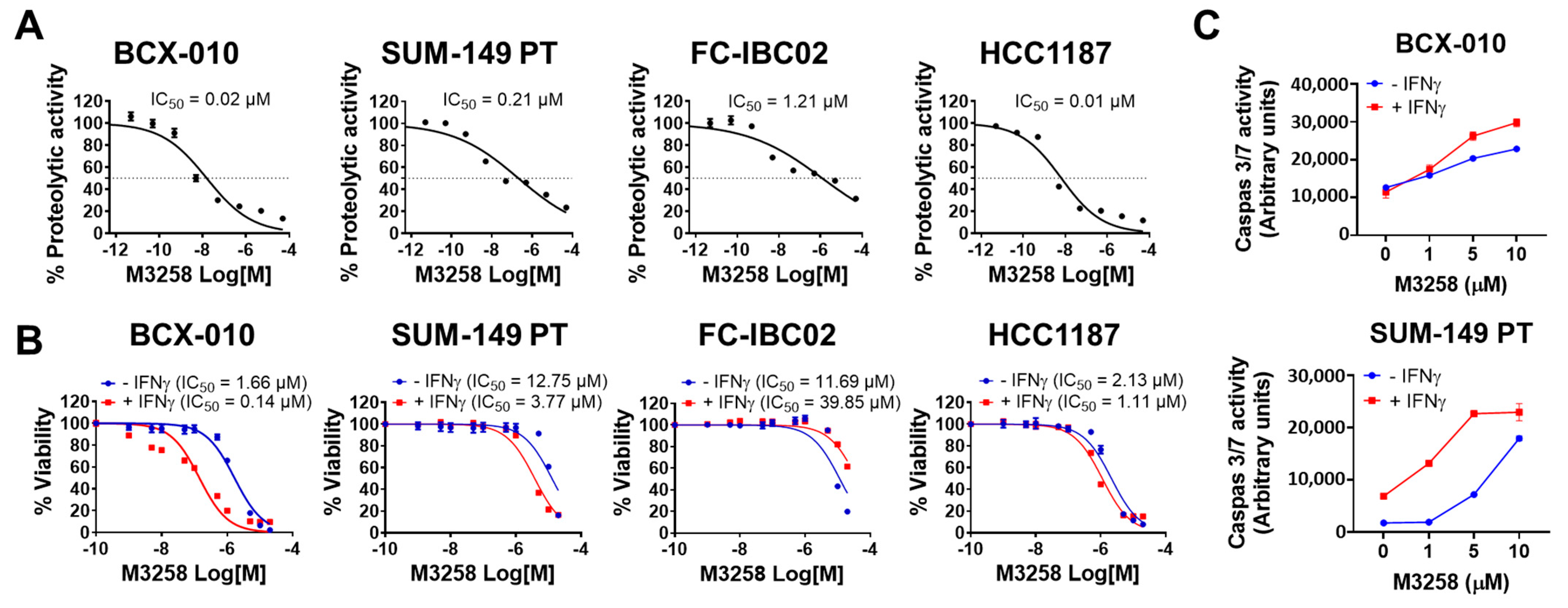
3.3. Suppression of LMP7 Activity by M3258 Reduces Viability and Induces Apoptosis in TNBC/IBC Cells In Vitro
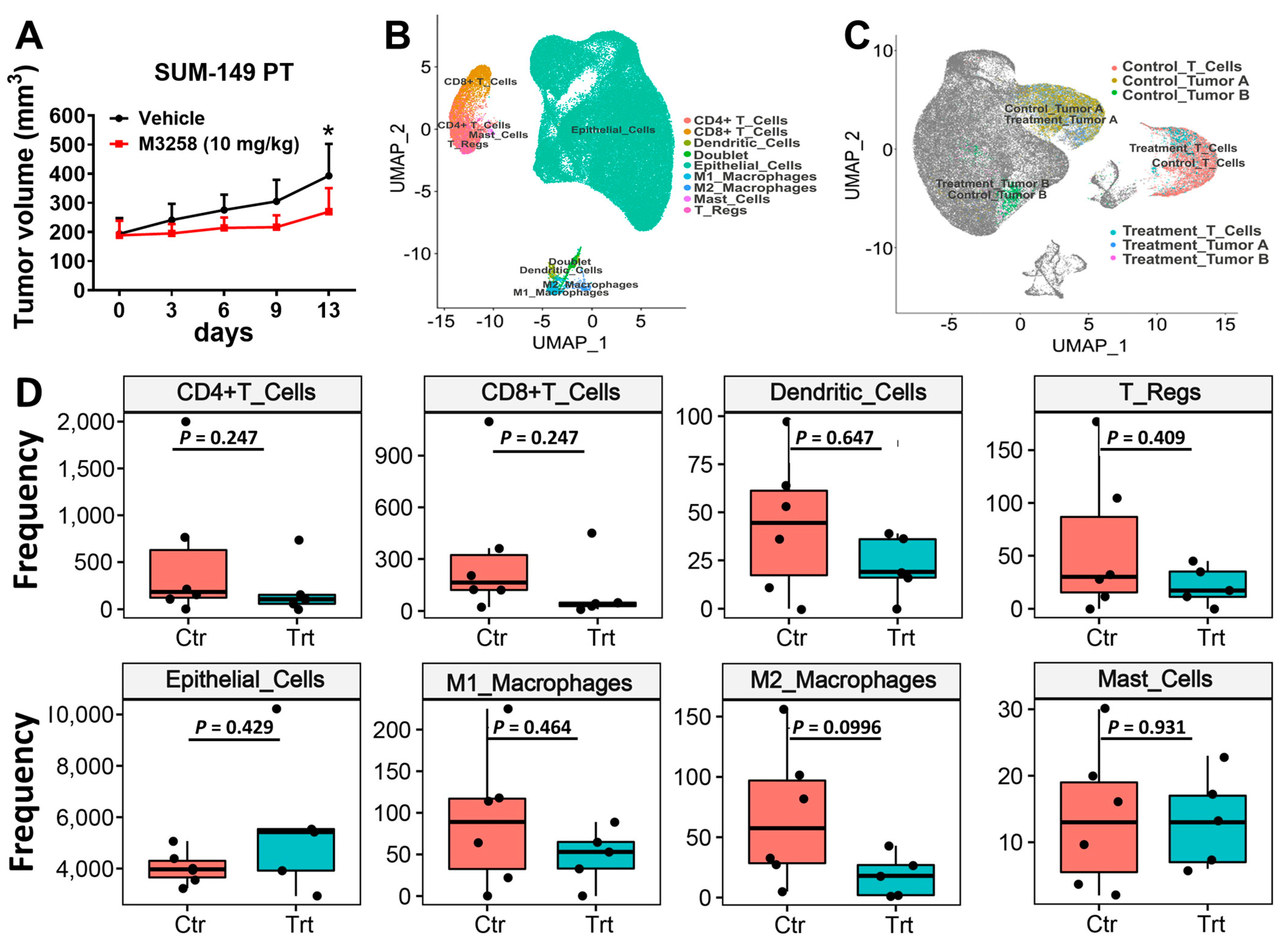
3.4. M3258 Inhibits Tumor Growth in a SUM-149 PT Xenograft Model In Vivo
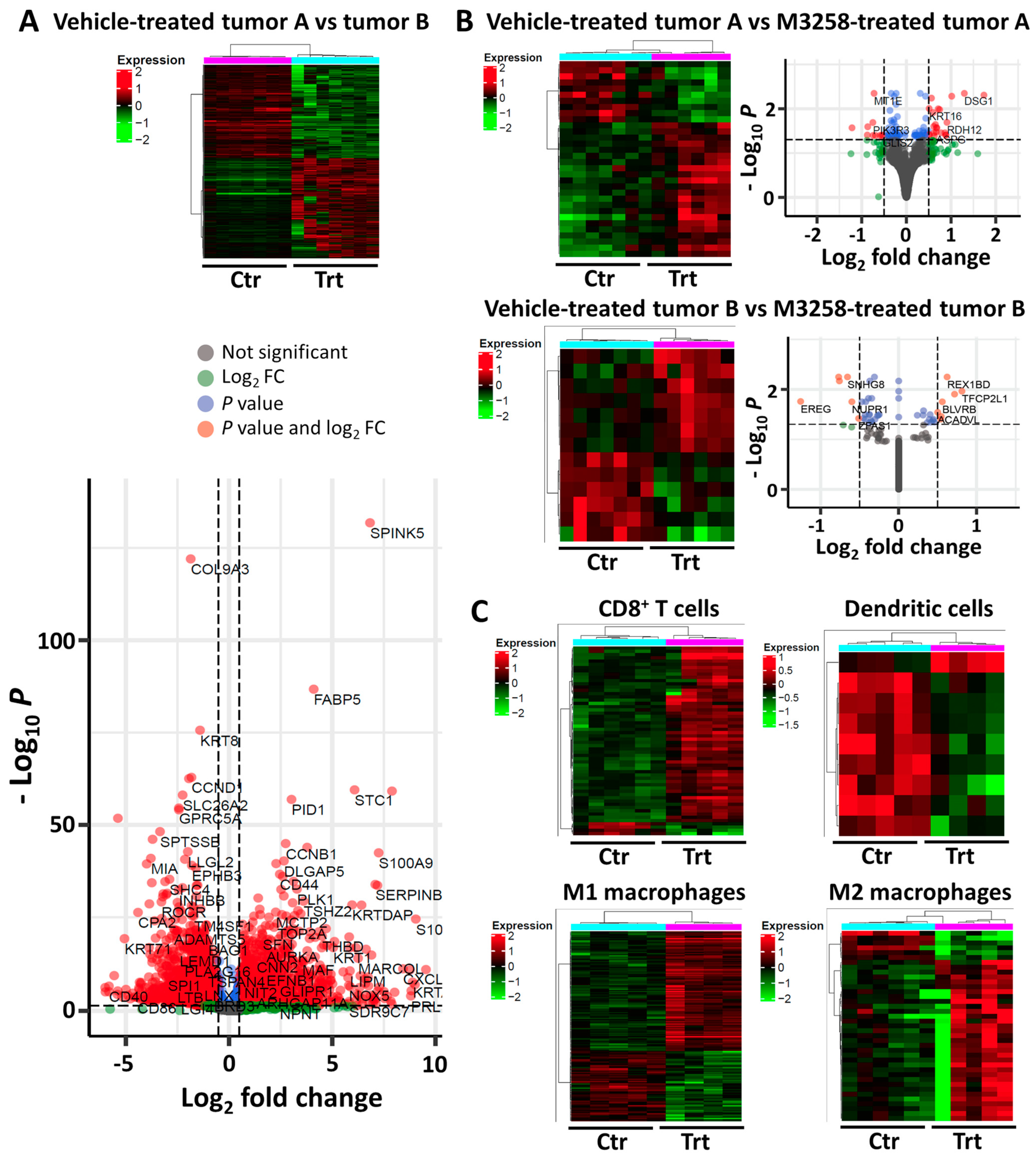
3.5. Effects of M3258 on Immune Cell Composition of SUM-149 PT Tumors
3.6. Tumor and Immune Cell Pathways Affected by M3258 Treatment in SUM-149 PT Tumors
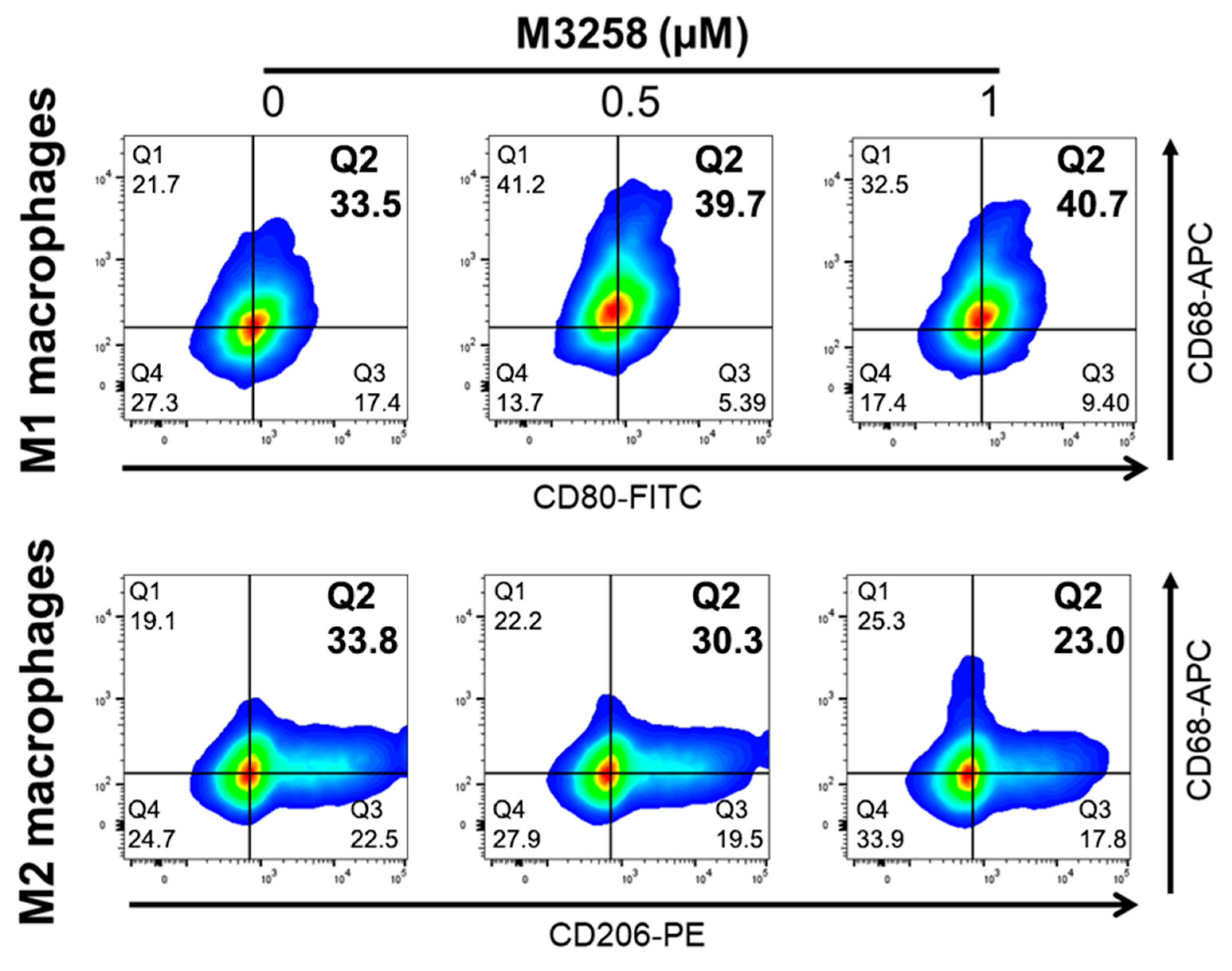
3.7. M3258 Enhances THP-1 Differentiation toward M1 Macrophages In Vitro
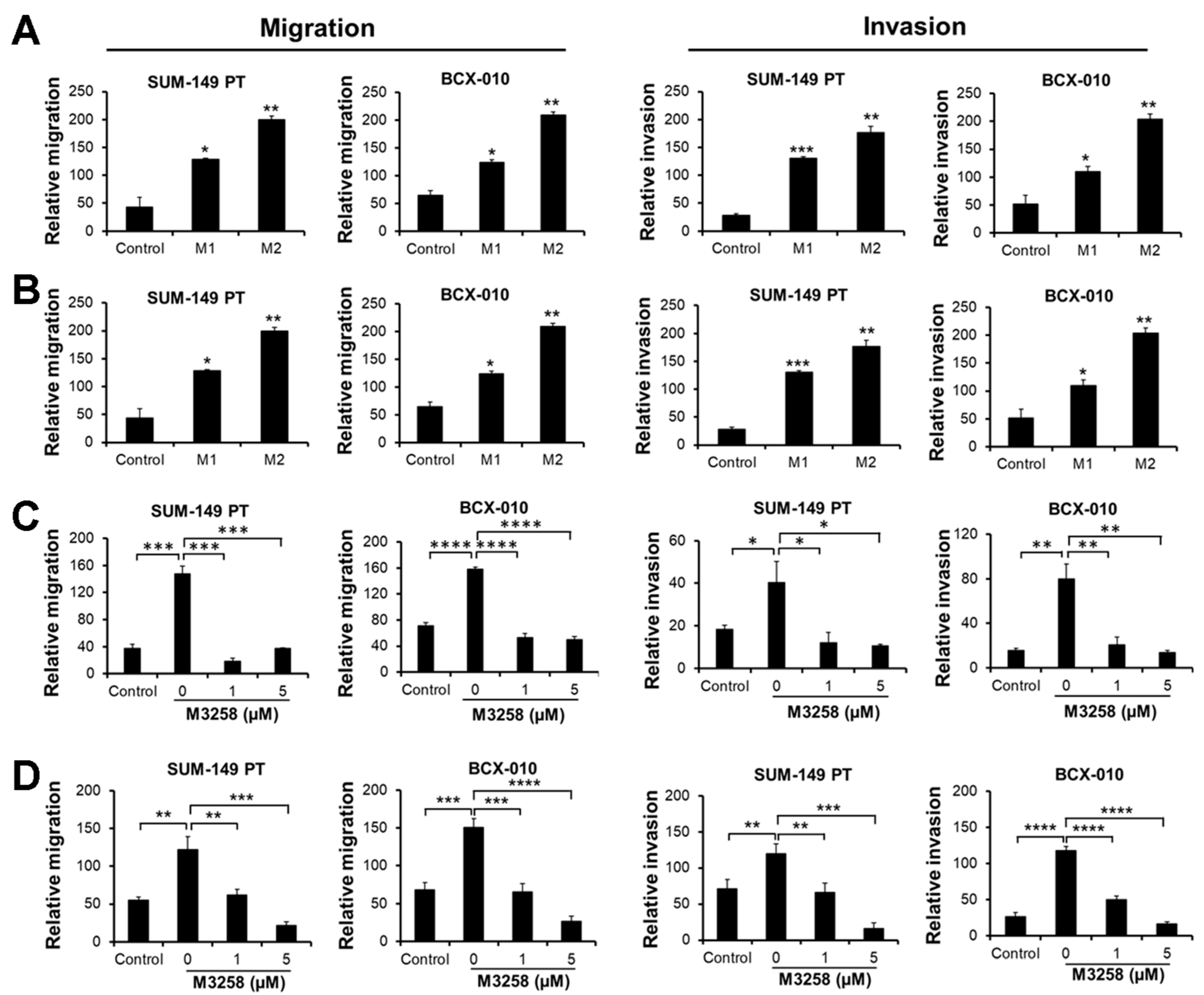
3.8. M3258 Inhibits M2 Macrophage-Induced TNBC/IBC Cell Migration and Invasion In Vitro
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, W.; Norbury, C.C.; Cho, Y.; Yewdell, J.W.; Bennink, J.R. Immunoproteasomes shape immunodominance hierarchies of antiviral CD8(+) T cells at the levels of T cell repertoire and presentation of viral antigens. J. Exp. Med. 2001, 193, 1319–1326. [Google Scholar] [CrossRef] [PubMed]
- Nathan, J.A.; Spinnenhirn, V.; Schmidtke, G.; Basler, M.; Groettrup, M.; Goldberg, A.L. Immuno- and constitutive proteasomes do not differ in their abilities to degrade ubiquitinated proteins. Cell 2013, 152, 1184–1194. [Google Scholar] [CrossRef] [PubMed]
- Eskandari, S.K.; Seelen, M.A.J.; Lin, G.; Azzi, J.R. The immunoproteasome: An old player with a novel and emerging role in alloimmunity. Am. J. Transplant. 2017, 17, 3033–3039. [Google Scholar] [CrossRef] [PubMed]
- Kloetzel, P.M. Antigen processing by the proteasome. Nat. Rev. Mol. Cell Biol. 2001, 2, 179–187. [Google Scholar] [CrossRef]
- Fehling, H.J.; Swat, W.; Laplace, C.; Kuhn, R.; Rajewsky, K.; Muller, U.; von Boehmer, H. MHC class I expression in mice lacking the proteasome subunit LMP-7. Science 1994, 265, 1234–1237. [Google Scholar] [CrossRef]
- Zhou, X.; Xu, R.; Wu, Y.; Zhou, L.; Xiang, T. The role of proteasomes in tumorigenesis. Genes Dis. 2024, 11, 101070. [Google Scholar] [CrossRef]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef]
- Huang, Z.; Wu, Y.; Zhou, X.; Xu, J.; Zhu, W.; Shu, Y.; Liu, P. Efficacy of therapy with bortezomib in solid tumors: A review based on 32 clinical trials. Future Oncol. 2014, 10, 1795–1807. [Google Scholar] [CrossRef]
- Kubiczkova, L.; Pour, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. Proteasome inhibitors—Molecular basis and current perspectives in multiple myeloma. J. Cell Mol. Med. 2014, 18, 947–961. [Google Scholar] [CrossRef]
- Schlafer, D.; Shah, K.S.; Panjic, E.H.; Lonial, S. Safety of proteasome inhibitors for treatment of multiple myeloma. Expert Opin. Drug Saf. 2017, 16, 167–183. [Google Scholar] [CrossRef]
- Kirk, C.J.; Muchamuel, T.; Wang, J.; Fan, R.A. Discovery and Early Clinical Development of Selective Immunoproteasome Inhibitors. Cells 2021, 11, 9. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Caturegli, P.; Takahashi, M.; Suzuki, K. New Insights into the Function of the Immunoproteasome in Immune and Nonimmune Cells. J. Immunol. Res. 2015, 2015, 541984. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, M.P.; Friese-Hamim, M.; Walter-Bausch, G.; Busch, M.; Gaus, S.; Musil, D.; Rohdich, F.; Zanelli, U.; Downey-Kopyscinski, S.L.; Mitsiades, C.S.; et al. M3258 Is a Selective Inhibitor of the Immunoproteasome Subunit LMP7 (beta5i) Delivering Efficacy in Multiple Myeloma Models. Mol. Cancer Ther. 2021, 20, 1378–1387. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.; Busch, M.; Friese-Hamim, M.; Crosignani, S.; Fuchss, T.; Musil, D.; Rohdich, F.; Sanderson, M.P.; Seenisamy, J.; Walter-Bausch, G.; et al. Structure-Based Optimization and Discovery of M3258, a Specific Inhibitor of the Immunoproteasome Subunit LMP7 (beta5i). J. Med. Chem. 2021, 64, 10230–10245. [Google Scholar] [CrossRef] [PubMed]
- Sloot, W.; Glaser, N.; Hansen, A.; Hellmann, J.; Jaeckel, S.; Johannes, S.; Knippel, A.; Lai, V.; Onidi, M. Improved nonclinical safety profile of a novel, highly selective inhibitor of the immunoproteasome subunit LMP7 (M3258). Toxicol. Appl. Pharmacol. 2021, 429, 115695. [Google Scholar] [CrossRef]
- Johnson, H.W.B.; Lowe, E.; Anderl, J.L.; Fan, A.; Muchamuel, T.; Bowers, S.; Moebius, D.C.; Kirk, C.; McMinn, D.L. Required Immunoproteasome Subunit Inhibition Profile for Anti-Inflammatory Efficacy and Clinical Candidate KZR-616 ((2 S,3 R)- N-((S)-3-(Cyclopent-1-en-1-yl)-1-((R)-2-methyloxiran-2-yl)-1-oxopropan-2-yl)-3-hydroxy-3-(4-methoxyphenyl)-2-((S)-2-(2-morpholinoacetamido)propanamido)propenamide). J. Med. Chem. 2018, 61, 11127–11143. [Google Scholar] [CrossRef]
- Muchamuel, T.; Fan, R.A.; Anderl, J.L.; Bomba, D.J.; Johnson, H.W.B.; Lowe, E.; Tuch, B.B.; McMinn, D.L.; Millare, B.; Kirk, C.J. Zetomipzomib (KZR-616) attenuates lupus in mice via modulation of innate and adaptive immune responses. Front. Immunol. 2023, 14, 1043680. [Google Scholar] [CrossRef]
- Koerner, J.; Brunner, T.; Groettrup, M. Inhibition and deficiency of the immunoproteasome subunit LMP7 suppress the development and progression of colorectal carcinoma in mice. Oncotarget 2017, 8, 50873–50888. [Google Scholar] [CrossRef]
- Li, J.; Liu, N.; Zhou, H.; Xian, P.; Song, Y.; Tang, X.; Li, Y.; Basler, M. Immunoproteasome inhibition prevents progression of castration-resistant prostate cancer. Br. J. Cancer 2023, 128, 1377–1390. [Google Scholar] [CrossRef]
- Morris, G.J.; Naidu, S.; Topham, A.K.; Guiles, F.; Xu, Y.; McCue, P.; Schwartz, G.F.; Park, P.K.; Rosenberg, A.L.; Brill, K.; et al. Differences in breast carcinoma characteristics in newly diagnosed African-American and Caucasian patients: A single-institution compilation compared with the National Cancer Institute’s Surveillance, Epidemiology, and End Results database. Cancer 2007, 110, 876–884. [Google Scholar] [CrossRef]
- Heitz, F.; Harter, P.; Lueck, H.J.; Fissler-Eckhoff, A.; Lorenz-Salehi, F.; Scheil-Bertram, S.; Traut, A.; du Bois, A. Triple-negative and HER2-overexpressing breast cancers exhibit an elevated risk and an earlier occurrence of cerebral metastases. Eur. J. Cancer 2009, 45, 2792–2798. [Google Scholar] [CrossRef] [PubMed]
- Anders, C.K.; Carey, L.A. Biology, metastatic patterns, and treatment of patients with triple-negative breast cancer. Clin. Breast Cancer 2009, 9 (Suppl. 2), S73–S81. [Google Scholar] [CrossRef] [PubMed]
- Haffty, B.G.; Yang, Q.; Reiss, M.; Kearney, T.; Higgins, S.A.; Weidhaas, J.; Harris, L.; Hait, W.; Toppmeyer, D. Locoregional relapse and distant metastasis in conservatively managed triple negative early-stage breast cancer. J. Clin. Oncol. 2006, 24, 5652–5657. [Google Scholar] [CrossRef] [PubMed]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef]
- Yamauchi, H.; Woodward, W.A.; Valero, V.; Alvarez, R.H.; Lucci, A.; Buchholz, T.A.; Iwamoto, T.; Krishnamurthy, S.; Yang, W.; Reuben, J.M.; et al. Inflammatory breast cancer: What we know and what we need to learn. Oncologist 2012, 17, 891–899. [Google Scholar] [CrossRef]
- Hance, K.W.; Anderson, W.F.; Devesa, S.S.; Young, H.A.; Levine, P.H. Trends in inflammatory breast carcinoma incidence and survival: The surveillance, epidemiology, and end results program at the National Cancer Institute. J. Natl. Cancer Inst. 2005, 97, 966–975. [Google Scholar] [CrossRef]
- Labidi, S.I.; Mrad, K.; Mezlini, A.; Ouarda, M.A.; Combes, J.D.; Ben Abdallah, M.; Ben Romdhane, K.; Viens, P.; Ben Ayed, F. Inflammatory breast cancer in Tunisia in the era of multimodality therapy. Ann. Oncol. 2008, 19, 473–480. [Google Scholar] [CrossRef]
- Dawood, S.; Cristofanilli, M. Inflammatory breast cancer: What progress have we made? Oncology 2011, 25, 264–270, 273. [Google Scholar]
- Gonzalez-Angulo, A.M.; Hennessy, B.T.; Broglio, K.; Meric-Bernstam, F.; Cristofanilli, M.; Giordano, S.H.; Buchholz, T.A.; Sahin, A.; Singletary, S.E.; Buzdar, A.U.; et al. Trends for inflammatory breast cancer: Is survival improving? Oncologist 2007, 12, 904–912. [Google Scholar] [CrossRef]
- Geoffroy, K.; Araripe Saraiva, B.; Viens, M.; Beland, D.; Bourgeois-Daigneault, M.C. Increased expression of the immunoproteasome subunits PSMB8 and PSMB9 by cancer cells correlate with better outcomes for triple-negative breast cancers. Sci. Rep. 2023, 13, 2129. [Google Scholar] [CrossRef]
- Adwal, A.; Kalita-de Croft, P.; Shakya, R.; Lim, M.; Kalaw, E.; Taege, L.D.; McCart Reed, A.E.; Lakhani, S.R.; Callen, D.F.; Saunus, J.M. Tradeoff between metabolic i-proteasome addiction and immune evasion in triple-negative breast cancer. Life Sci. Alliance 2020, 3, e201900562. [Google Scholar] [CrossRef] [PubMed]
- Engel, R.H.; Brown, J.A.; Von Roenn, J.H.; O’Regan, R.M.; Bergan, R.; Badve, S.; Rademaker, A.; Gradishar, W.J. A phase II study of single agent bortezomib in patients with metastatic breast cancer: A single institution experience. Cancer Investig. 2007, 25, 733–737. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Gonzalez-Angulo, A.M.; Reuben, J.M.; Booser, D.J.; Pusztai, L.; Krishnamurthy, S.; Esseltine, D.; Stec, J.; Broglio, K.R.; Islam, R.; et al. Bortezomib (VELCADE) in metastatic breast cancer: Pharmacodynamics, biological effects, and prediction of clinical benefits. Ann. Oncol. 2006, 17, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M., 3rd; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- McInnes, L.; Healy, J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv 2018, arXiv:1802.03426v3. [Google Scholar]
- Korotkevich, G.; Sukhov, V.; Budin, N.; Shpak, B.; Artyomov, M.N.; Sergushichev, A. Fast gene set enrichment analysis. bioRxiv 2019. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Chen, D.; Jin, C.; Dong, X.; Wen, J.; Xia, E.; Wang, Q.; Wang, O. Pan-cancer analysis of the prognostic and immunological role of PSMB8. Sci. Rep. 2021, 11, 20492. [Google Scholar] [CrossRef]
- Kalaora, S.; Lee, J.S.; Barnea, E.; Levy, R.; Greenberg, P.; Alon, M.; Yagel, G.; Bar Eli, G.; Oren, R.; Peri, A.; et al. Immunoproteasome expression is associated with better prognosis and response to checkpoint therapies in melanoma. Nat. Commun. 2020, 11, 896. [Google Scholar] [CrossRef]
- Oerlemans, R.; Franke, N.E.; Assaraf, Y.G.; Cloos, J.; van Zantwijk, I.; Berkers, C.R.; Scheffer, G.L.; Debipersad, K.; Vojtekova, K.; Lemos, C.; et al. Molecular basis of bortezomib resistance: Proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood 2008, 112, 2489–2499. [Google Scholar] [CrossRef] [PubMed]
- Raskov, H.; Orhan, A.; Christensen, J.P.; Gogenur, I. Cytotoxic CD8(+) T cells in cancer and cancer immunotherapy. Br. J. Cancer 2021, 124, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Merkenschlager, M.; Beverley, P.C. Evidence for differential expression of CD45 isoforms by precursors for memory-dependent and independent cytotoxic responses: Human CD8 memory CTLp selectively express CD45RO (UCHL1). Int. Immunol. 1989, 1, 450–459. [Google Scholar] [CrossRef]
- Miyawaki, T.; Kasahara, Y.; Kanegane, H.; Ohta, K.; Yokoi, T.; Yachie, A.; Taniguchi, N. Expression of CD45R0 (UCHL1) by CD4+ and CD8+ T cells as a sign of in vivo activation in infectious mononucleosis. Clin. Exp. Immunol. 1991, 83, 447–451. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Song, Y.; Du, W.; Gong, L.; Chang, H.; Zou, Z. Tumor-associated macrophages: An accomplice in solid tumor progression. J. Biomed. Sci. 2019, 26, 78. [Google Scholar] [CrossRef]
- Fricker, L.D. Proteasome Inhibitor Drugs. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 457–476. [Google Scholar] [CrossRef]
- Csizmar, C.M.; Kim, D.H.; Sachs, Z. The role of the proteasome in AML. Blood Cancer J. 2016, 6, e503. [Google Scholar] [CrossRef]
- Kambhampati, S.; Wiita, A.P. Lessons Learned from Proteasome Inhibitors, the Paradigm for Targeting Protein Homeostasis in Cancer. Adv. Exp. Med. Biol. 2020, 1243, 147–162. [Google Scholar] [CrossRef]
- Lee, M.; Song, I.H.; Heo, S.H.; Kim, Y.A.; Park, I.A.; Bang, W.S.; Park, H.S.; Gong, G.; Lee, H.J. Expression of Immunoproteasome Subunit LMP7 in Breast Cancer and Its Association with Immune-Related Markers. Cancer Res. Treat. 2019, 51, 80–89. [Google Scholar] [CrossRef]
- Roeten, M.S.; van Meerloo, J.; Kwidama, Z.J.; Ter Huizen, G.; Segerink, W.H.; Zweegman, S.; Kaspers, G.J.L.; Jansen, G.; Cloos, J. Pre-Clinical Evaluation of the Proteasome Inhibitor Ixazomib against Bortezomib-Resistant Leukemia Cells and Primary Acute Leukemia Cells. Cells 2021, 10, 665. [Google Scholar] [CrossRef]
- Wolfe, A.R.; Trenton, N.J.; Debeb, B.G.; Larson, R.; Ruffell, B.; Chu, K.; Hittelman, W.; Diehl, M.; Reuben, J.M.; Ueno, N.T.; et al. Mesenchymal stem cells and macrophages interact through IL-6 to promote inflammatory breast cancer in pre-clinical models. Oncotarget 2016, 7, 82482–82492. [Google Scholar] [CrossRef] [PubMed]
- Semba, T.; Wang, X.; Xie, X.; Cohen, E.N.; Reuben, J.M.; Dalby, K.N.; Long, J.P.; Phi, L.T.H.; Tripathy, D.; Ueno, N.T. Identification of the JNK-Active Triple-Negative Breast Cancer Cluster Associated With an Immunosuppressive Tumor Microenvironment. J. Natl. Cancer Inst. 2022, 114, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Lee, J.; Iwase, T.; Kai, M.; Ueno, N.T. Emerging drug targets for triple-negative breast cancer: A guided tour of the preclinical landscape. Expert Opin. Ther. Targets 2022, 26, 405–425. [Google Scholar] [CrossRef]
- Du, S.H.; Xiang, Y.J.; Liu, L.; Nie, M.; Hou, Y.; Wang, L.; Li, B.B.; Xu, M.; Teng, Q.L.; Peng, J.; et al. Co-Inhibition of the Immunoproteasome Subunits LMP2 and LMP7 Ameliorates Immune Thrombocytopenia. Front. Immunol. 2020, 11, 603278. [Google Scholar] [CrossRef] [PubMed]
- Basler, M.; Lindstrom, M.M.; LaStant, J.J.; Bradshaw, J.M.; Owens, T.D.; Schmidt, C.; Maurits, E.; Tsu, C.; Overkleeft, H.S.; Kirk, C.J.; et al. Co-inhibition of immunoproteasome subunits LMP2 and LMP7 is required to block autoimmunity. EMBO Rep. 2018, 19, e46512. [Google Scholar] [CrossRef]
- Duan, Z.; Luo, Y. Targeting macrophages in cancer immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 127. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, X.; Lee, J.; Manyam, G.C.; Pearson, T.; Walter-Bausch, G.; Friese-Hamim, M.; Zhao, S.; Jabs, J.; Manginelli, A.A.; Piske, N.; et al. LMP7-Specific Inhibitor M3258 Modulates the Tumor Microenvironment of Triple-Negative Breast Cancer and Inflammatory Breast Cancer. Cancers 2025, 17, 1887. https://doi.org/10.3390/cancers17111887
Xie X, Lee J, Manyam GC, Pearson T, Walter-Bausch G, Friese-Hamim M, Zhao S, Jabs J, Manginelli AA, Piske N, et al. LMP7-Specific Inhibitor M3258 Modulates the Tumor Microenvironment of Triple-Negative Breast Cancer and Inflammatory Breast Cancer. Cancers. 2025; 17(11):1887. https://doi.org/10.3390/cancers17111887
Chicago/Turabian StyleXie, Xuemei, Jangsoon Lee, Ganiraju C. Manyam, Troy Pearson, Gina Walter-Bausch, Manja Friese-Hamim, Sheng Zhao, Julia Jabs, Angela A. Manginelli, Nadine Piske, and et al. 2025. "LMP7-Specific Inhibitor M3258 Modulates the Tumor Microenvironment of Triple-Negative Breast Cancer and Inflammatory Breast Cancer" Cancers 17, no. 11: 1887. https://doi.org/10.3390/cancers17111887
APA StyleXie, X., Lee, J., Manyam, G. C., Pearson, T., Walter-Bausch, G., Friese-Hamim, M., Zhao, S., Jabs, J., Manginelli, A. A., Piske, N., Mrowiec, T., Wolf, C. M., Kuntal, B. S., Tripathy, D., Wang, J., Sanderson, M. P., & Ueno, N. T. (2025). LMP7-Specific Inhibitor M3258 Modulates the Tumor Microenvironment of Triple-Negative Breast Cancer and Inflammatory Breast Cancer. Cancers, 17(11), 1887. https://doi.org/10.3390/cancers17111887