
Regulatory Mechanisms and Therapeutic Targeting of PD-L1 Trafficking and Stability in Cancer Immunotherapy


Simple Summary
Abstract
1. Introduction
2. Therapeutic Modulation of the PD-1 Pathway in Immune Regulation
3. Immune Checkpoint Therapy (ICT)
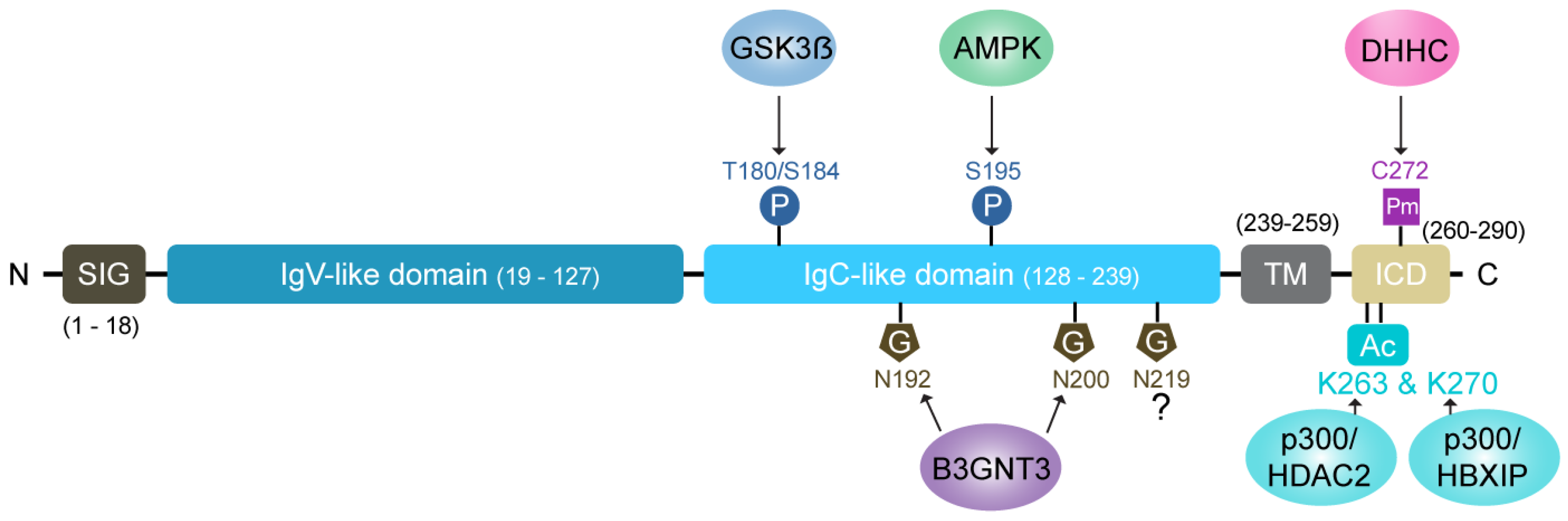
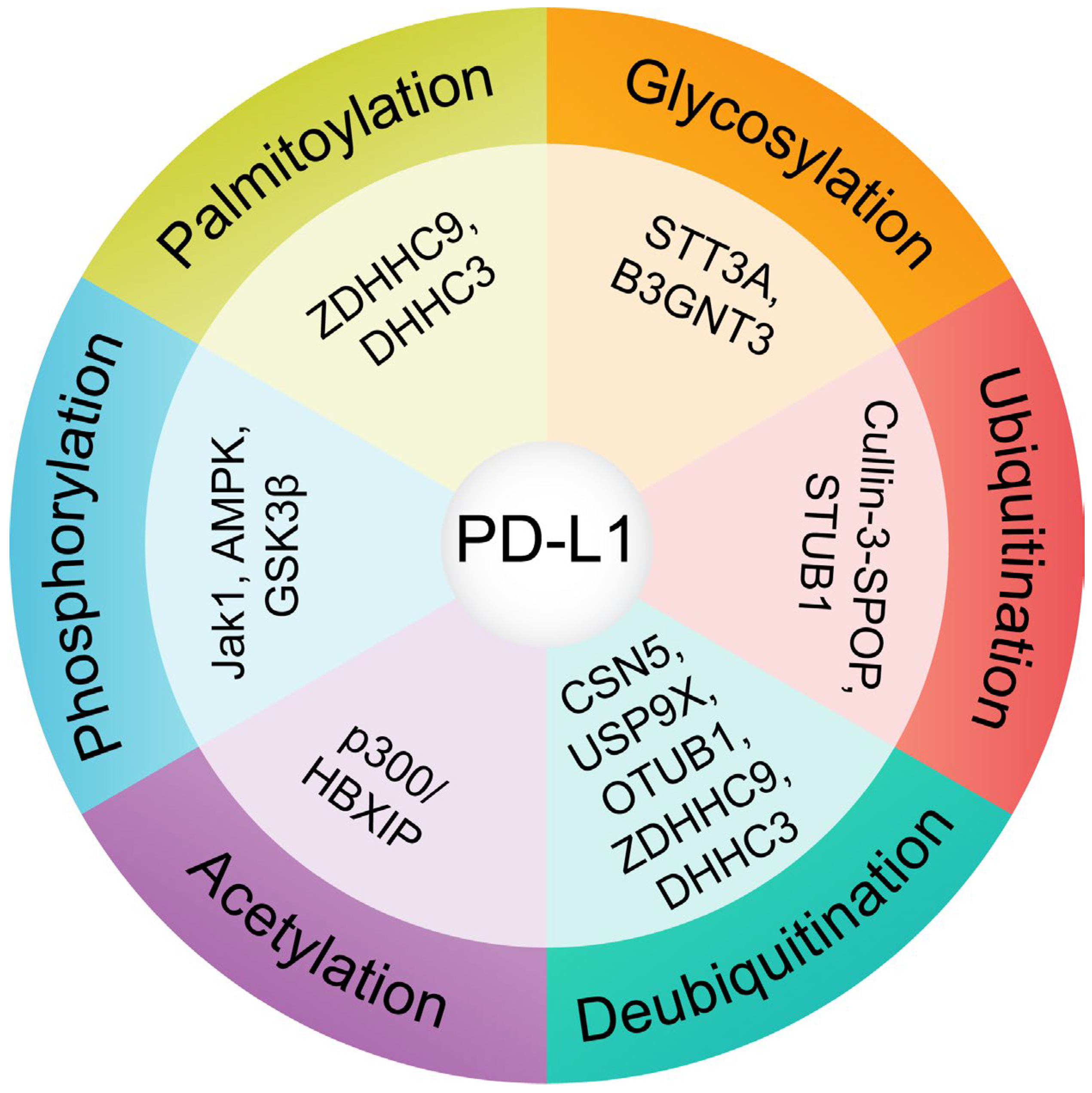
4. Significance of Post-Translational Modifications (PTMs) in PD-L1 Stability and Trafficking
4.1. Glycosylation: A Key Stabilizing Modification
4.2. Phosphorylation: A Double-Edged Sword
4.3. Ubiquitination and Deubiquitination: Balancing Stability and Degradation
4.4. Palmitoylation: Enhancing Surface Retention
4.5. Acetylation of PD-L1 Regulates Subcellular Localization and Accumulation
5. Integration and Therapeutic Implications
6. Intracellular Trafficking and Cell Surface Expression of PD-L1
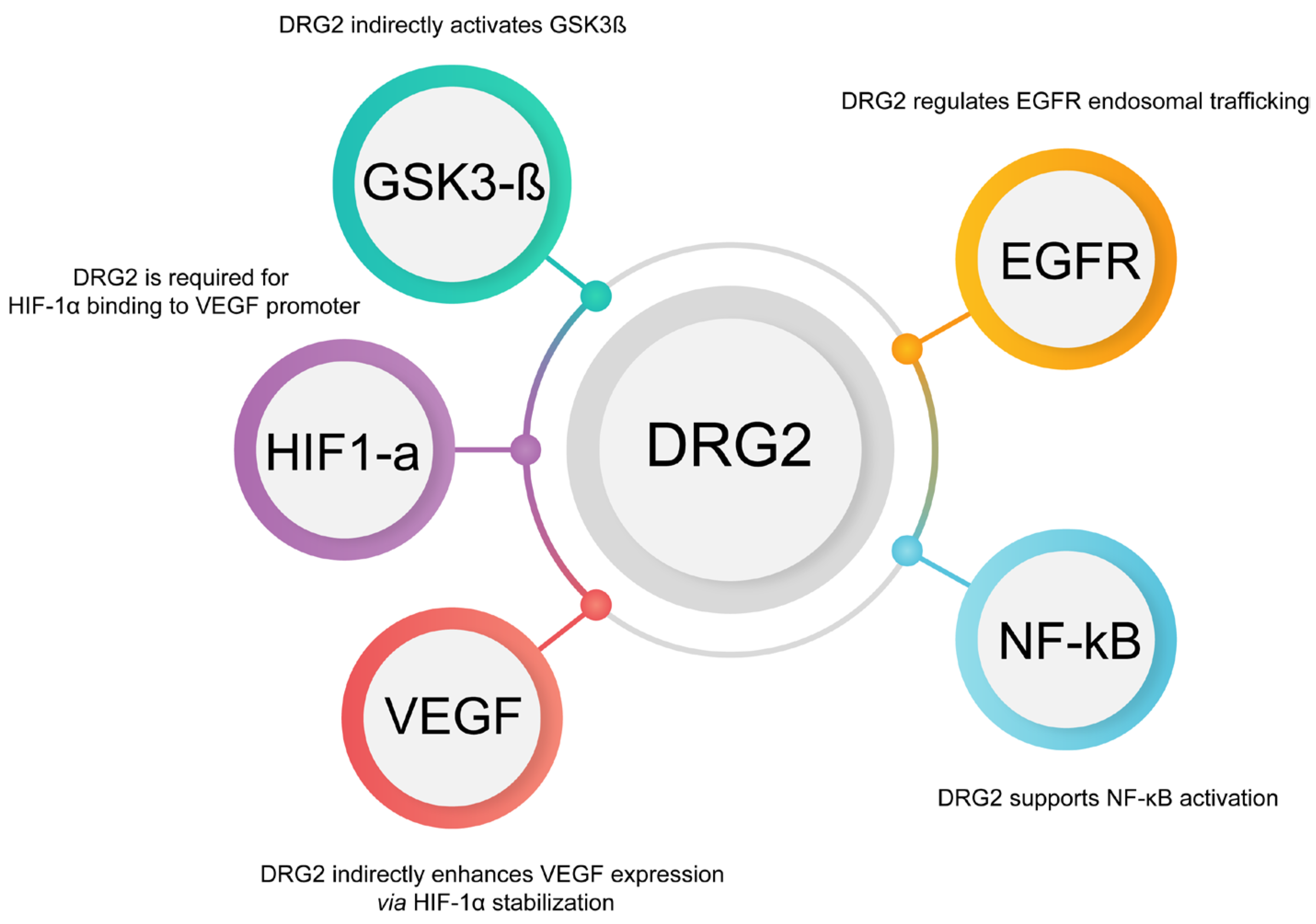
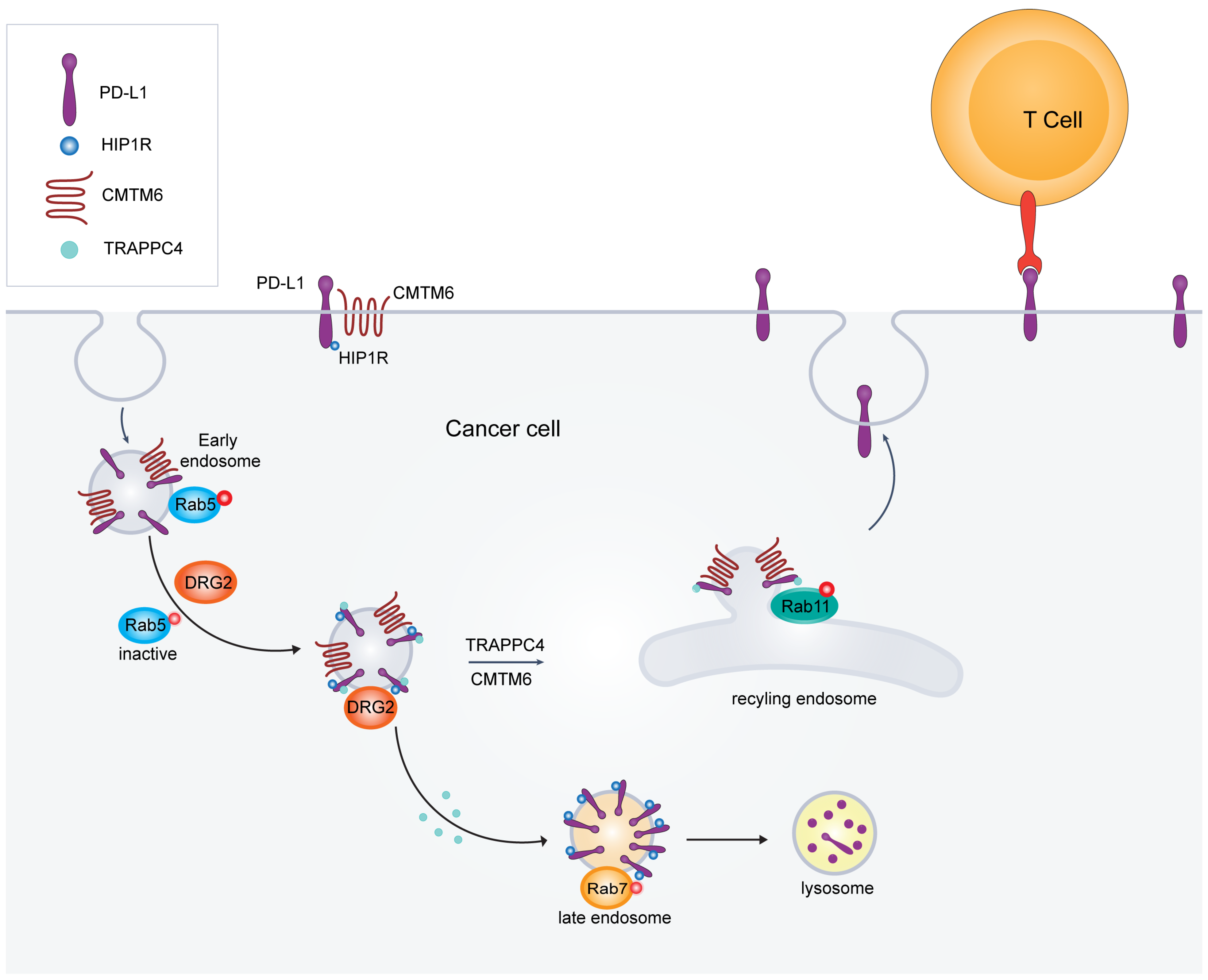
6.1. DRG2’s Role in Membrane Trafficking and PD-L1 Dynamics
6.1.1. Indirect Regulation of PD-L1 Expression by DRG2 Through EGFR and NF-κB
6.1.2. Interaction with HIF-1α and VEGF Pathways
6.2. HIP1R-Mediated Lysosomal Targeting of PD-L1
6.3. TRAPPC4: A Critical Regulator of PD-L1 Trafficking and Tumor Immune Evasion
6.4. CMTM6 and CMTM4: Key Regulators of PD-L1 Stability and Their Implications for Enhancing Cancer Immunotherapy
7. Conclusions and Future Directions for Targeting PD-L1 Trafficking
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar] [PubMed]
- Li, B.; Jin, J.; Guo, D.; Tao, Z.; Hu, X. Immune Checkpoint Inhibitors Combined with Targeted Therapy: The Recent Advances and Future Potentials. Cancers 2023, 15, 2858. [Google Scholar] [CrossRef]
- Ribas, A.; Hamid, O.; Daud, A.; Hodi, F.S.; Wolchok, J.D.; Kefford, R.; Joshua, A.M.; Patnaik, A.; Hwu, W.J.; Weber, J.S.; et al. Association of Pembrolizumab With Tumor Response and Survival Among Patients With Advanced Melanoma. JAMA 2016, 315, 1600–1609. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Wang, H.; Yao, H.; Li, C.; Shi, H.; Lan, J.; Li, Z.; Zhang, Y.; Liang, L.; Fang, J.Y.; Xu, J. HIP1R targets PD-L1 to lysosomal degradation to alter T cell-mediated cytotoxicity. Nat. Chem. Biol. 2019, 15, 42–50. [Google Scholar] [CrossRef]
- Burr, M.L.; Sparbier, C.E.; Chan, Y.C.; Williamson, J.C.; Woods, K.; Beavis, P.A.; Lam, E.Y.N.; Henderson, M.A.; Bell, C.C.; Stolzenburg, S.; et al. CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature 2017, 549, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Mani, M.; Kim, J.; Cho, W.J.; Martin, T.F.J.; Kim, J.H.; Chu, H.S.; Jeong, W.J.; Won, Y.W.; Lee, B.J.; et al. DRG2 is required for surface localization of PD-L1 and the efficacy of anti-PD-1 therapy. Cell Death Discov. 2024, 10, 260. [Google Scholar] [CrossRef]
- Ren, Y.; Qian, Y.; Ai, L.; Xie, Y.; Gao, Y.; Zhuang, Z.; Chen, J.; Chen, Y.X.; Fang, J.Y. TRAPPC4 regulates the intracellular trafficking of PD-L1 and antitumor immunity. Nat. Commun. 2021, 12, 5405. [Google Scholar] [CrossRef]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef]
- Anderson, M.S.; Bluestone, J.A. The NOD mouse: A model of immune dysregulation. Annu. Rev. Immunol. 2005, 23, 447–485. [Google Scholar] [CrossRef]
- Kroner, A.; Mehling, M.; Hemmer, B.; Rieckmann, P.; Toyka, K.V.; Maurer, M.; Wiendl, H. A PD-1 polymorphism is associated with disease progression in multiple sclerosis. Ann. Neurol. 2005, 58, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Honjo, T. PD-1 and PD-1 ligands: From discovery to clinical application. Int. Immunol. 2007, 19, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, Z.; Chen, W.; Zhang, Z.; Li, Y.; Shi, M.; Zhang, J.; Chen, L.; Wang, S.; Wang, F.S. B7-H1 up-regulation on myeloid dendritic cells significantly suppresses T cell immune function in patients with chronic hepatitis B. J. Immunol. 2007, 178, 6634–6641. [Google Scholar] [CrossRef] [PubMed]
- Day, C.L.; Kaufmann, D.E.; Kiepiela, P.; Brown, J.A.; Moodley, E.S.; Reddy, S.; Mackey, E.W.; Miller, J.D.; Leslie, A.J.; DePierres, C.; et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 2006, 443, 350–354. [Google Scholar] [CrossRef]
- Geng, L.; Jiang, G.; Fang, Y.; Dong, S.; Xie, H.; Chen, Y.; Shen, M.; Zheng, S. B7-H1 expression is upregulated in peripheral blood CD14+ monocytes of patients with chronic hepatitis B virus infection, which correlates with higher serum IL-10 levels. J. Viral Hepat. 2006, 13, 725–733. [Google Scholar] [CrossRef]
- Petrovas, C.; Casazza, J.P.; Brenchley, J.M.; Price, D.A.; Gostick, E.; Adams, W.C.; Precopio, M.L.; Schacker, T.; Roederer, M.; Douek, D.C.; et al. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J. Exp. Med. 2006, 203, 2281–2292. [Google Scholar] [CrossRef]
- Wherry, E.J.; Ahmed, R. Memory CD8 T-cell differentiation during viral infection. J. Virol. 2004, 78, 5535–5545. [Google Scholar] [CrossRef]
- Payandeh, Z.; Khalili, S.; Somi, M.H.; Mard-Soltani, M.; Baghbanzadeh, A.; Hajiasgharzadeh, K.; Samadi, N.; Baradaran, B. PD-1/PD-L1-dependent immune response in colorectal cancer. J. Cell Physiol. 2020, 235, 5461–5475. [Google Scholar] [CrossRef]
- Sun, J.Y.; Zhang, D.; Wu, S.; Xu, M.; Zhou, X.; Lu, X.J.; Ji, J. Resistance to PD-1/PD-L1 blockade cancer immunotherapy: Mechanisms, predictive factors, and future perspectives. Biomark. Res. 2020, 8, 35. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef] [PubMed]
- Odorizzi, P.M.; Pauken, K.E.; Paley, M.A.; Sharpe, A.; Wherry, E.J. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J. Exp. Med. 2015, 212, 1125–1137. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Li, M.O.; Rudensky, A.Y. T cell receptor signalling in the control of regulatory T cell differentiation and function. Nat. Rev. Immunol. 2016, 16, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, S.; Yuan, R.; Engleman, E.G. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu. Rev. Pathol. 2021, 16, 223–249. [Google Scholar] [CrossRef]
- Azuma, M.; Ito, D.; Yagita, H.; Okumura, K.; Phillips, J.H.; Lanier, L.L.; Somoza, C. B70 antigen is a second ligand for CTLA-4 and CD28. Nature 1993, 366, 76–79. [Google Scholar] [CrossRef]
- Ljunggren, H.G.; Jonsson, R.; Hoglund, P. Seminal immunologic discoveries with direct clinical implications: The 2018 Nobel Prize in Physiology or Medicine honours discoveries in cancer immunotherapy. Scand. J. Immunol. 2018, 88, e12731. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- Kluger, H.M.; Zito, C.R.; Turcu, G.; Baine, M.K.; Zhang, H.; Adeniran, A.; Sznol, M.; Rimm, D.L.; Kluger, Y.; Chen, L.; et al. PD-L1 Studies Across Tumor Types, Its Differential Expression and Predictive Value in Patients Treated with Immune Checkpoint Inhibitors. Clin. Cancer Res. 2017, 23, 4270–4279. [Google Scholar] [CrossRef] [PubMed]
- Strickler, J.H.; Hanks, B.A.; Khasraw, M. Tumor Mutational Burden as a Predictor of Immunotherapy Response: Is More Always Better? Clin. Cancer Res. 2021, 27, 1236–1241. [Google Scholar] [CrossRef] [PubMed]
- Kleinendorst, S.C.; Oosterwijk, E.; Bussink, J.; Westdorp, H.; Konijnenberg, M.W.; Heskamp, S. Combining Targeted Radionuclide Therapy and Immune Checkpoint Inhibition for Cancer Treatment. Clin. Cancer Res. 2022, 28, 3652–3657. [Google Scholar] [CrossRef]
- Varayathu, H.; Sarathy, V.; Thomas, B.E.; Mufti, S.S.; Naik, R. Combination Strategies to Augment Immune Check Point Inhibitors Efficacy—Implications for Translational Research. Front. Oncol. 2021, 11, 559161. [Google Scholar] [CrossRef]
- Cha, J.H.; Chan, L.C.; Li, C.W.; Hsu, J.L.; Hung, M.C. Mechanisms Controlling PD-L1 Expression in Cancer. Mol. Cell 2019, 76, 359–370. [Google Scholar] [CrossRef]
- Colombo, N.; Lorusso, D.; Monk, B.J.; Slomovitz, B.; Hasegawa, K.; Nogueira-Rodrigues, A.; Zale, M.; Okpara, C.E.; Barresi, G.; McKenzie, J.; et al. Characterization and Management of Adverse Reactions in Patients With Advanced Endometrial Cancer Receiving Lenvatinib Plus Pembrolizumab. Oncologist 2024, 29, 25–35. [Google Scholar] [CrossRef]
- Martins, F.; Sykiotis, G.P.; Maillard, M.; Fraga, M.; Ribi, C.; Kuntzer, T.; Michielin, O.; Peters, S.; Coukos, G.; Spertini, F.; et al. New therapeutic perspectives to manage refractory immune checkpoint-related toxicities. Lancet Oncol. 2019, 20, e54–e64. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Shoushtari, A.N.; Friedman, C.F.; Navid-Azarbaijani, P.; Postow, M.A.; Callahan, M.K.; Momtaz, P.; Panageas, K.S.; Wolchok, J.D.; Chapman, P.B. Measuring Toxic Effects and Time to Treatment Failure for Nivolumab Plus Ipilimumab in Melanoma. JAMA Oncol. 2018, 4, 98–101. [Google Scholar] [CrossRef]
- Weber, J.S.; Kahler, K.C.; Hauschild, A. Management of immune-related adverse events and kinetics of response with ipilimumab. J. Clin. Oncol. 2012, 30, 2691–2697. [Google Scholar] [CrossRef]
- Wang, R.; He, S.; Long, J.; Wang, Y.; Jiang, X.; Chen, M.; Wang, J. Emerging therapeutic frontiers in cancer: Insights into posttranslational modifications of PD-1/PD-L1 and regulatory pathways. Exp. Hematol. Oncol. 2024, 13, 46. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Zhang, L.; Chang, X.; Qin, D.; Zhang, T. Regulation of post-translational modification of PD-L1 and advances in tumor immunotherapy. Front. Immunol. 2023, 14, 1230135. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Li, W.; Young, K.H.; Li, Y. Posttranslational Modifications in PD-L1 Turnover and Function: From Cradle to Grave. Biomedicines 2021, 9, 1702. [Google Scholar] [CrossRef]
- Li, C.W.; Lim, S.O.; Xia, W.; Lee, H.H.; Chan, L.C.; Kuo, C.W.; Khoo, K.H.; Chang, S.S.; Cha, J.H.; Kim, T.; et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat. Commun. 2016, 7, 12632. [Google Scholar] [CrossRef]
- Chan, L.C.; Li, C.W.; Xia, W.; Hsu, J.M.; Lee, H.H.; Cha, J.H.; Wang, H.L.; Yang, W.H.; Yen, E.Y.; Chang, W.C.; et al. IL-6/JAK1 pathway drives PD-L1 Y112 phosphorylation to promote cancer immune evasion. J. Clin. Investig. 2019, 129, 3324–3338. [Google Scholar] [CrossRef]
- Cha, J.H.; Yang, W.H.; Xia, W.; Wei, Y.; Chan, L.C.; Lim, S.O.; Li, C.W.; Kim, T.; Chang, S.S.; Lee, H.H.; et al. Metformin Promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated Degradation of PD-L1. Mol. Cell 2018, 71, 606–620.e607. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Bu, X.; Wang, H.; Zhu, Y.; Geng, Y.; Nihira, N.T.; Tan, Y.; Ci, Y.; Wu, F.; Dai, X.; et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 2018, 553, 91–95. [Google Scholar] [CrossRef]
- Liu, Y.; Shah, S.V.; Xiang, X.; Wang, J.; Deng, Z.B.; Liu, C.; Zhang, L.; Wu, J.; Edmonds, T.; Jambor, C.; et al. COP9-associated CSN5 regulates exosomal protein deubiquitination and sorting. Am. J. Pathol. 2009, 174, 1415–1425. [Google Scholar] [CrossRef]
- Lim, S.O.; Li, C.W.; Xia, W.; Cha, J.H.; Chan, L.C.; Wu, Y.; Chang, S.S.; Lin, W.C.; Hsu, J.M.; Hsu, Y.H.; et al. Deubiquitination and Stabilization of PD-L1 by CSN5. Cancer Cell 2016, 30, 925–939. [Google Scholar] [CrossRef]
- Jingjing, W.; Wenzheng, G.; Donghua, W.; Guangyu, H.; Aiping, Z.; Wenjuan, W. Deubiquitination and stabilization of programmed cell death ligand 1 by ubiquitin-specific peptidase 9, X-linked in oral squamous cell carcinoma. Cancer Med. 2018, 7, 4004–4011. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Varthi, M.; Sykes, S.M.; Phillips, C.; Warzecha, C.; Zhu, W.; Wyce, A.; Thorne, A.W.; Berger, S.L.; McMahon, S.B. The putative cancer stem cell marker USP22 is a subunit of the human SAGA complex required for activated transcription and cell-cycle progression. Mol. Cell 2008, 29, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Xu, R.; Huang, X.; Tang, Z.; Tian, Y.; Zhang, J.; Zheng, X. Deubiquitinating enzyme OTUB1 promotes cancer cell immunosuppression via preventing ER-associated degradation of immune checkpoint protein PD-L1. Cell Death Differ. 2021, 28, 1773–1789. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.N.; Xia, B.R.; Deng, S.H.; Yang, C.; Pi, Y.N.; Cui, B.B.; Jin, W.L. Deubiquitinating Enzymes Orchestrate the Cancer Stem Cell-Immunosuppressive Niche Dialogue: New Perspectives and Therapeutic Potential. Front. Cell Dev. Biol. 2021, 9, 680100. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Sun, Y.; Wang, Z.; Huang, X.; Tang, L.; Jiang, K.; Jin, X. ZDHHC20-mediated S-palmitoylation of YTHDF3 stabilizes MYC mRNA to promote pancreatic cancer progression. Nat. Commun. 2024, 15, 4642. [Google Scholar] [CrossRef]
- Chen, Y.; Li, Y.; Wu, L. Protein S-palmitoylation modification: Implications in tumor and tumor immune microenvironment. Front. Immunol. 2024, 15, 1337478. [Google Scholar] [CrossRef]
- Horita, H.; Law, A.; Hong, S.; Middleton, K. Identifying Regulatory Posttranslational Modifications of PD-L1: A Focus on Monoubiquitinaton. Neoplasia 2017, 19, 346–353. [Google Scholar] [CrossRef]
- Gao, Y.; Nihira, N.T.; Bu, X.; Chu, C.; Zhang, J.; Kolodziejczyk, A.; Fan, Y.; Chan, N.T.; Ma, L.; Liu, J.; et al. Acetylation-dependent regulation of PD-L1 nuclear translocation dictates the efficacy of anti-PD-1 immunotherapy. Nat. Cell Biol. 2020, 22, 1064–1075. [Google Scholar] [CrossRef]
- Xu, F.F.; Sun, H.M.; Fang, R.P.; Zhang, L.; Shi, H.; Wang, X.; Fu, X.L.; Li, X.M.; Shi, X.H.; Wu, Y.; et al. The modulation of PD-L1 induced by the oncogenic HBXIP for breast cancer growth. Acta Pharmacol. Sin. 2022, 43, 429–445. [Google Scholar] [CrossRef]
- Munoz, L.E.; Huang, L.; Bommireddy, R.; Sharma, R.; Monterroza, L.; Guin, R.N.; Samaranayake, S.G.; Pack, C.D.; Ramachandiran, S.; Reddy, S.J.C.; et al. Metformin reduces PD-L1 on tumor cells and enhances the anti-tumor immune response generated by vaccine immunotherapy. J. Immunother. Cancer 2021, 9, e002614. [Google Scholar] [CrossRef]
- Leng, X.; Wei, S.; Mei, J.; Deng, S.; Yang, Z.; Liu, Z.; Guo, C.; Deng, Y.; Xia, L.; Cheng, J.; et al. Identifying the prognostic significance of B3GNT3 with PD-L1 expression in lung adenocarcinoma. Transl. Lung Cancer Res. 2021, 10, 965–980. [Google Scholar] [CrossRef]
- Mani, M.; Thao, D.T.; Kim, B.C.; Lee, U.H.; Kim, D.J.; Jang, S.H.; Back, S.H.; Lee, B.J.; Cho, W.J.; Han, I.S.; et al. DRG2 knockdown induces Golgi fragmentation via GSK3beta phosphorylation and microtubule stabilization. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1463–1474. [Google Scholar] [CrossRef] [PubMed]
- Ko, M.S.; Kim, H.J.; Kim, H.K.; Yoon, N.A.; Lee, U.H.; Lee, S.C.; Chung, D.K.; Lee, B.J.; Suh, J.H.; Cho, W.J.; et al. Developmentally regulated GTP-binding protein 2 ameliorates EAE by suppressing the development of TH17 cells. Clin. Immunol. 2014, 150, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Yoon, N.A.; Jung, S.J.; Choi, S.H.; Ryu, J.H.; Mani, M.; Lee, U.H.; Vo, M.T.; Jeon, D.Y.; Chung, S.W.; Ju Lee, B.; et al. DRG2 supports the growth of primary tumors and metastases of melanoma by enhancing VEGF-A expression. FEBS J. 2020, 287, 2070–2086. [Google Scholar] [CrossRef]
- Mani, M.; Lee, U.H.; Yoon, N.A.; Kim, H.J.; Ko, M.S.; Seol, W.; Joe, Y.; Chung, H.T.; Lee, B.J.; Moon, C.H.; et al. Developmentally regulated GTP-binding protein 2 coordinates Rab5 activity and transferrin recycling. Mol. Biol. Cell 2016, 27, 334–348. [Google Scholar] [CrossRef]
- Mani, M.; Lee, U.H.; Yoon, N.A.; Yoon, E.H.; Lee, B.J.; Cho, W.J.; Park, J.W. Developmentally regulated GTP-binding protein 2 is required for stabilization of Rac1-positive membrane tubules. Biochem. Biophys. Res. Commun. 2017, 493, 758–764. [Google Scholar] [CrossRef]
- Antonangeli, F.; Natalini, A.; Garassino, M.C.; Sica, A.; Santoni, A.; Di Rosa, F. Regulation of PD-L1 Expression by NF-kappaB in Cancer. Front. Immunol. 2020, 11, 584626. [Google Scholar] [CrossRef]
- Messai, Y.; Gad, S.; Noman, M.Z.; Le Teuff, G.; Couve, S.; Janji, B.; Kammerer, S.F.; Rioux-Leclerc, N.; Hasmim, M.; Ferlicot, S.; et al. Renal Cell Carcinoma Programmed Death-ligand 1, a New Direct Target of Hypoxia-inducible Factor-2 Alpha, is Regulated by von Hippel-Lindau Gene Mutation Status. Eur. Urol. 2016, 70, 623–632. [Google Scholar] [CrossRef]
- Banjade, S.; Zhu, L.; Jorgensen, J.R.; Suzuki, S.W.; Emr, S.D. Recruitment and organization of ESCRT-0 and ubiquitinated cargo via condensation. Sci. Adv. 2022, 8, eabm5149. [Google Scholar] [CrossRef]
- Sette, P.; Jadwin, J.A.; Dussupt, V.; Bello, N.F.; Bouamr, F. The ESCRT-associated protein Alix recruits the ubiquitin ligase Nedd4-1 to facilitate HIV-1 release through the LYPXnL L domain motif. J. Virol. 2010, 84, 8181–8192. [Google Scholar] [CrossRef]
- Yu, X.; Li, W.; Liu, H.; Wang, X.; Coarfa, C.; Cheng, C.; Yu, X.; Zeng, Z.; Cao, Y.; Young, K.H.; et al. PD-L1 translocation to the plasma membrane enables tumor immune evasion through MIB2 ubiquitination. J. Clin. Investig. 2023, 133, e160456. [Google Scholar] [CrossRef]
- Duan, Z.; Shi, R.; Gao, B.; Cai, J. N-linked glycosylation of PD-L1/PD-1: An emerging target for cancer diagnosis and treatment. J. Transl. Med. 2024, 22, 705. [Google Scholar] [CrossRef] [PubMed]
- Yaseen, M.M.; Abuharfeil, N.M.; Darmani, H. CMTM6 as a master regulator of PD-L1. Cancer Immunol. Immunother. 2022, 71, 2325–2340. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Chen, R.; Yu, X.; Tong, Y.; Peng, X.; Li, F.; Hu, C.; Sun, J.; Gong, L. Suppression of Tumor or Host Intrinsic CMTM6 Drives Antitumor Cytotoxicity in a PD-L1-Independent Manner. Cancer Immunol. Res. 2023, 11, 241–260. [Google Scholar] [CrossRef] [PubMed]
- Mezzadra, R.; Sun, C.; Jae, L.T.; Gomez-Eerland, R.; de Vries, E.; Wu, W.; Logtenberg, M.E.W.; Slagter, M.; Rozeman, E.A.; Hofland, I.; et al. Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature 2017, 549, 106–110. [Google Scholar] [CrossRef]
- Koikawa, K.; Kibe, S.; Suizu, F.; Sekino, N.; Kim, N.; Manz, T.D.; Pinch, B.J.; Akshinthala, D.; Verma, A.; Gaglia, G.; et al. Targeting Pin1 renders pancreatic cancer eradicable by synergizing with immunochemotherapy. Cell 2021, 184, 4753–4771.e4727. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Post-Translational Modification (PTM) | Regulator/Enzyme | Effect on PD-L1 | Therapeutic Implications |
|---|---|---|---|
| Glycosylation | STT3A [45] | Enhances PD-L1 stability and proper folding. Protects against degradation by preventing GSK3β binding. | Disruption of glycosylation destabilizes PD-L1 and reduces its immune-suppressive function. |
| B3GNT3 [60] | Facilitates PD-L1 interaction with PD-1. | N-linked glycosylation inhibitors impair PD-L1/PD-1 binding, enhancing anti-tumor immunity. | |
| Phosphorylation | JAK1 [4,45] | Facilitates glycosylation and trafficking to the cell surface. | Essential for maintaining PD-L1 surface expression. |
| AMPK [46] | Promotes abnormal glycosylation and ER retention, leading to degradation. | AMPK activators (e.g., metformin) promote PD-L1 degradation and enhance T-cell-mediated killing. | |
| GSK3β [44] | Targets PD-L1 for degradation via phosphorylation. | Potential target to modulate PD-L1 levels in tumor cells. | |
| Ubiquitination | Cullin-3-SPOP [47] | Marks PD-L1 for proteasomal degradation. | Enhancing this pathway can reduce PD-L1 stability and improve immunotherapy efficacy. |
| STUB1 [47] | Promotes PD-L1 ubiquitination and degradation. | Prevents PD-L1 accumulation in tumor cells. | |
| Deubiquitination | CSN5 [49], USP9X [50], OTUB1 [52] | Stabilizes PD-L1 by removing ubiquitin chains. | Inhibition of DUBs can reduce PD-L1 levels and enhance immune checkpoint blockade therapies. |
| Palmitoylation | ZDHHC9, ZDHHC3 [54,55] | Maintains PD-L1 stability and surface expression. Prevents lysosomal degradation. | Inhibitors like 2-bromopalmitate destabilize PD-L1, sensitizing tumor cells to immune responses. |
| Acetylation | p300 [57] | Decrease the stability | Blocks nuclear translocation. Increase membrane retention |
| HBXIP/p300 | Increase the stability | Cytoplasmic accumulation. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mani, M.; Park, J.W.; Martin, T.F.J. Regulatory Mechanisms and Therapeutic Targeting of PD-L1 Trafficking and Stability in Cancer Immunotherapy. Cancers 2025, 17, 1747. https://doi.org/10.3390/cancers17111747
Mani M, Park JW, Martin TFJ. Regulatory Mechanisms and Therapeutic Targeting of PD-L1 Trafficking and Stability in Cancer Immunotherapy. Cancers. 2025; 17(11):1747. https://doi.org/10.3390/cancers17111747
Chicago/Turabian StyleMani, Muralidharan, Jeong Woo Park, and Thomas F. J. Martin. 2025. "Regulatory Mechanisms and Therapeutic Targeting of PD-L1 Trafficking and Stability in Cancer Immunotherapy" Cancers 17, no. 11: 1747. https://doi.org/10.3390/cancers17111747
APA StyleMani, M., Park, J. W., & Martin, T. F. J. (2025). Regulatory Mechanisms and Therapeutic Targeting of PD-L1 Trafficking and Stability in Cancer Immunotherapy. Cancers, 17(11), 1747. https://doi.org/10.3390/cancers17111747