ADAM Proteases in Cancer: Biological Roles, Therapeutic Challenges, and Emerging Opportunities




Simple Summary
Abstract
1. Introduction
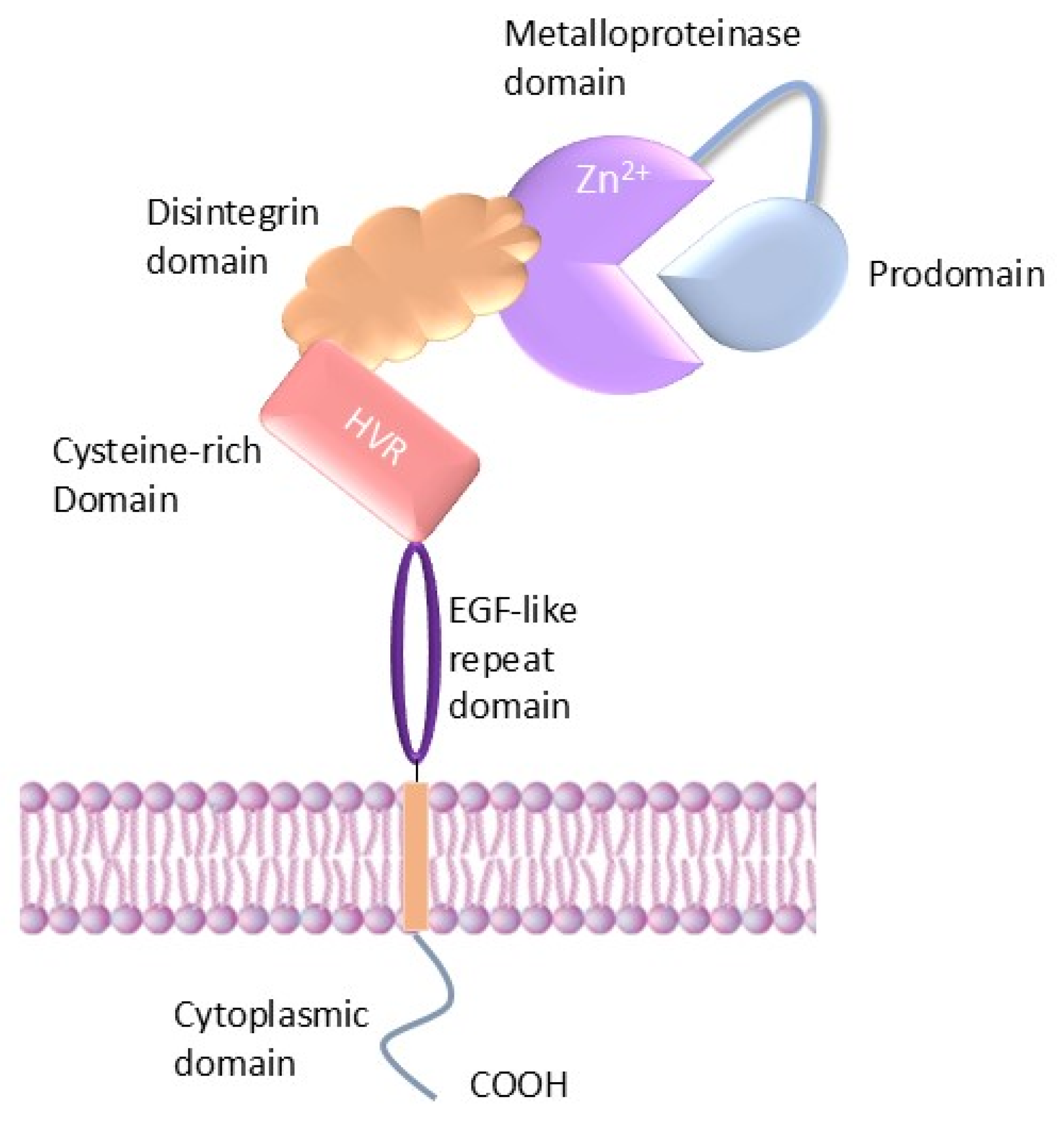
2. Structure of ADAMs
3. Mechanisms Regulating ADAM Metalloproteases
3.1. Modulation by Accessory Proteins and Membrane Trafficking
3.2. Prodomain Cleavage and Activation
3.3. Dimerization, TIMPs, and Intracellular Signalling
4. Major ADAM Family Members in Cancer
4.1. ADAM8
4.2. ADAM9
4.3. ADAM12
4.4. ADAM10
4.5. ADAM17
5. Recent Advances to Target ADAMs
5.1. ADAM8
5.2. ADAM9
5.3. ADAM12
5.4. ADAMs 10 and 17
6. Conclusions, Challenges, and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Spano, D.; Heck, C.; De Antonellis, P.; Christofori, G.; Zollo, M. Molecular networks that regulate cancer metastasis. Semin. Cancer Biol. 2012, 22, 234–249. [Google Scholar] [CrossRef]
- Murphy, G. The ADAMs: Signalling scissors in the tumour microenvironment. Nat. Rev. Cancer 2008, 8, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Fingleton, B.; Matrisian, L.M. Matrix Metalloproteinase Inhibitors and Cancer—Trials and Tribulations. Science 2002, 295, 2387–2392. [Google Scholar] [CrossRef]
- Gialeli, C.; Theocharis, A.D.; Karamanos, N.K. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 2011, 278, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Avila, G.; Sommer, B.; Mendoza-Posada, D.A.; Ramos, C.; Garcia-Hernandez, A.A.; Falfan-Valencia, R. Matrix metalloproteinases participation in the metastatic process and their diagnostic and therapeutic applications in cancer. Crit. Rev. Oncol./Hematol. 2019, 137, 57–83. [Google Scholar] [CrossRef]
- Edwards, D.R.; Handsley, M.M.; Pennington, C.J. The ADAM metalloproteinases. Mol. Asp. Med. 2008, 29, 258–289. [Google Scholar] [CrossRef]
- Takeda, S. ADAM and ADAMTS Family Proteins and Snake Venom Metalloproteinases: A Structural Overview. Toxins 2016, 8, 155. [Google Scholar] [CrossRef]
- Mullooly, M.; McGowan, P.M.; Crown, J.; Duffy, M.J. The ADAMs family of proteases as targets for the treatment of cancer. Cancer Biol. Ther. 2016, 17, 870–880. [Google Scholar] [CrossRef]
- Seegar, T.C.M.; Blacklow, S.C. Domain integration of ADAM family proteins: Emerging themes from structural studies. Exp. Biol. Med. 2019, 244, 1510–1519. [Google Scholar] [CrossRef]
- Smith, T.M.; Tharakan, A.; Martin, R.K. Targeting ADAM10 in Cancer and Autoimmunity. Front. Immunol. 2020, 11, 499. [Google Scholar] [CrossRef] [PubMed]
- Van Wart, H.E.; Birkedal-Hansen, H. The cysteine switch: A principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc. Natl. Acad. Sci. USA 1990, 87, 5578–5582. [Google Scholar] [CrossRef]
- Leonard, J.D.; Lin, F.; Milla, M.E. Chaperone-like properties of the prodomain of TNFalpha-converting enzyme (TACE) and the functional role of its cysteine switch. Biochem. J. 2005, 387, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Anders, L.; Mertins, P.; Lammich, S.; Murgia, M.; Hartmann, D.; Saftig, P.; Haass, C.; Ullrich, A. Furin-, ADAM 10-, and gamma-secretase-mediated cleavage of a receptor tyrosine phosphatase and regulation of beta-catenin’s transcriptional activity. Mol. Cell. Biol. 2006, 26, 3917–3934. [Google Scholar] [CrossRef]
- Le Gall, S.M.; Maretzky, T.; Issuree, P.D.; Niu, X.D.; Reiss, K.; Saftig, P.; Khokha, R.; Lundell, D.; Blobel, C.P. ADAM17 is regulated by a rapid and reversible mechanism that controls access to its catalytic site. J. Cell Sci. 2010, 123, 3913–3922. [Google Scholar] [CrossRef]
- Hsia, H.-E.; Tüshaus, J.; Brummer, T.; Zheng, Y.; Scilabra, S.D.; Lichtenthaler, S.F. Functions of ‘A disintegrin and metalloproteases (ADAMs)’ in the mammalian nervous system. Cell. Mol. Life Sci. 2019, 76, 3055–3081. [Google Scholar] [CrossRef] [PubMed]
- Schlomann, U.; Wildeboer, D.; Webster, A.; Antropova, O.; Zeuschner, D.; Knight, C.G.; Docherty, A.J.P.; Lambert, M.; Skelton, L.; Jockusch, H.; et al. The Metalloprotease Disintegrin ADAM8: Processing by autocatalysis is required for proteolytic activity and cell adhesion. J. Biol. Chem. 2002, 277, 48210–48219. [Google Scholar] [CrossRef]
- Rocks, N.; Paulissen, G.; El Hour, M.; Quesada, F.; Crahay, C.; Gueders, M.; Foidart, J.M.; Noel, A.; Cataldo, D. Emerging roles of ADAM and ADAMTS metalloproteinases in cancer. Biochimie 2008, 90, 369–379. [Google Scholar] [CrossRef]
- Janes, P.W.; Saha, N.; Barton, W.A.; Kolev, M.V.; Wimmer-Kleikamp, S.H.; Nievergall, E.; Blobel, C.P.; Himanen, J.-P.; Lackmann, M.; Nikolov, D.B. Adam Meets Eph: An ADAM Substrate Recognition Module Acts as a Molecular Switch for Ephrin Cleavage In trans. Cell 2005, 123, 291–304. [Google Scholar] [CrossRef]
- Seegar, T.C.M.; Killingsworth, L.B.; Saha, N.; Meyer, P.A.; Patra, D.; Zimmerman, B.; Janes, P.W.; Rubinstein, E.; Nikolov, D.B.; Skiniotis, G.; et al. Structural Basis for Regulated Proteolysis by the α-Secretase ADAM10. Cell 2017, 171, 1638–1648.e1637. [Google Scholar] [CrossRef]
- Reddy, P.; Slack, J.L.; Davis, R.; Cerretti, D.P.; Kozlosky, C.J.; Blanton, R.A.; Shows, D.; Peschon, J.J.; Black, R.A. Functional Analysis of the Domain Structure of Tumor Necrosis Factor-α Converting Enzyme*. J. Biol. Chem. 2000, 275, 14608–14614. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Cho, S.; Su, P.C.; Berger, B.W.; Li, R. Membrane-enabled dimerization of the intrinsically disordered cytoplasmic domain of ADAM10. Proc. Natl. Acad. Sci. USA 2014, 111, 15987–15992. [Google Scholar] [CrossRef] [PubMed]
- Tosetti, F.; Alessio, M.; Poggi, A.; Zocchi, M.R. ADAM10 Site-Dependent Biology: Keeping Control of a Pervasive Protease. Int. J. Mol. Sci. 2021, 22, 4969. [Google Scholar] [CrossRef] [PubMed]
- Gharzia, F.G.; Aljohmani, A.; Beck, A.; Philipp, S.E.; Yildiz, D. Regulation of ADAM10 activity through microdomain-dependent intracellular calcium changes. Cell Commun. Signal. 2024, 22, 531. [Google Scholar] [CrossRef]
- Lambrecht, B.N.; Vanderkerken, M.; Hammad, H. The emerging role of ADAM metalloproteinases in immunity. Nat. Rev. Immunol. 2018, 18, 745–758. [Google Scholar] [CrossRef]
- Xu, P.; Derynck, R. Direct Activation of TACE-Mediated Ectodomain Shedding by p38 MAP Kinase Regulates EGF Receptor-Dependent Cell Proliferation. Mol. Cell 2010, 37, 551–566. [Google Scholar] [CrossRef]
- Kleino, I.; Järviluoma, A.; Hepojoki, J.; Huovila, A.P.; Saksela, K. Preferred SH3 domain partners of ADAM metalloproteases include shared and ADAM-specific SH3 interactions. PLoS ONE 2015, 10, e0121301. [Google Scholar] [CrossRef]
- Harrison, N.; Koo, C.Z.; Tomlinson, M.G. Regulation of ADAM10 by the TspanC8 Family of Tetraspanins and Their Therapeutic Potential. Int. J. Mol. Sci. 2021, 22, 6707. [Google Scholar] [CrossRef]
- Susa, K.J.; Kruse, A.C.; Blacklow, S.C. Tetraspanins: Structure, dynamics, and principles of partner-protein recognition. Trends Cell Biol. 2024, 34, 509–522. [Google Scholar] [CrossRef]
- Hemler, M.E. Tetraspanin proteins promote multiple cancer stages. Nat. Rev. Cancer 2014, 14, 49–60. [Google Scholar]
- Noy, P.J.; Yang, J.; Reyat, J.S.; Matthews, A.L.; Charlton, A.E.; Furmston, J.; Rogers, D.A.; Rainger, G.E.; Tomlinson, M.G. TspanC8 Tetraspanins and A Disintegrin and Metalloprotease 10 (ADAM10) Interact via Their Extracellular Regions: Evidence for distinct binding mechanisms for different TspanC8 proteins. J. Biol. Chem. 2016, 291, 3145–3157. [Google Scholar] [CrossRef] [PubMed]
- Koo, C.Z.; Harrison, N.; Noy, P.J.; Szyroka, J.; Matthews, A.L.; Hsia, H.E.; Müller, S.A.; Tüshaus, J.; Goulding, J.; Willis, K.; et al. The tetraspanin Tspan15 is an essential subunit of an ADAM10 scissor complex. J. Biol. Chem. 2020, 295, 12822–12839. [Google Scholar] [CrossRef]
- Lipper, C.H.; Egan, E.D.; Gabriel, K.H.; Blacklow, S.C. Structural basis for membrane-proximal proteolysis of substrates by ADAM10. Cell 2023, 186, 3632–3641.e3610. [Google Scholar] [CrossRef] [PubMed]
- Geesala, R.; Issuree, P.D.; Maretzky, T. Novel functions of inactive rhomboid proteins in immunity and disease. J. Leukoc. Biol. 2019, 106, 823–835. [Google Scholar] [CrossRef]
- Giese, A.A.; Babendreyer, A.; Krappen, P.; Gross, A.; Strnad, P.; Düsterhöft, S.; Ludwig, A. Inflammatory activation of surface molecule shedding by upregulation of the pseudoprotease iRhom2 in colon epithelial cells. Sci. Rep. 2021, 11, 24230. [Google Scholar] [CrossRef]
- Brooke, M.A.; Etheridge, S.L.; Kaplan, N.; Simpson, C.; O’Toole, E.A.; Ishida-Yamamoto, A.; Marches, O.; Getsios, S.; Kelsell, D.P. iRHOM2-dependent regulation of ADAM17 in cutaneous disease and epidermal barrier function. Hum. Mol. Genet. 2014, 23, 4064–4076. [Google Scholar] [CrossRef] [PubMed]
- Weskamp, G.; Tüshaus, J.; Li, D.; Feederle, R.; Maretzky, T.; Swendemann, S.; Falck-Pedersen, E.; McIlwain, D.R.; Mak, T.W.; Salmon, J.E.; et al. ADAM17 stabilizes its interacting partner inactive Rhomboid 2 (iRhom2) but not inactive Rhomboid 1 (iRhom1). J. Biol. Chem. 2020, 295, 4350–4358. [Google Scholar] [CrossRef]
- Lu, F.; Zhao, H.; Dai, Y.; Wang, Y.; Lee, C.-H.; Freeman, M. Cryo-EM reveals that iRhom2 restrains ADAM17 protease activity to control the release of growth factor and inflammatory signals. Mol. Cell 2024, 84, 2152–2165.e2155. [Google Scholar] [CrossRef]
- Sieber, B.; Lu, F.; Stribbling, S.M.; Grieve, A.G.; Ryan, A.J.; Freeman, M. iRhom2 regulates ERBB signalling to promote KRAS-driven tumour growth of lung cancer cells. J. Cell Sci. 2022, 135, jcs259949. [Google Scholar] [CrossRef]
- Bläsius, K.; Ludwig, L.; Knapp, S.; Flaßhove, C.; Sonnabend, F.; Keller, D.; Tacken, N.; Gao, X.; Kahveci-Türköz, S.; Grannemann, C.; et al. Pathological mutations reveal the key role of the cytosolic iRhom2 N-terminus for phosphorylation-independent 14-3-3 interaction and ADAM17 binding, stability, and activity. Cell. Mol. Life Sci. 2024, 81, 102. [Google Scholar] [CrossRef]
- Endres, K.; Anders, A.; Kojro, E.; Gilbert, S.; Fahrenholz, F.; Postina, R. Tumor necrosis factor-α converting enzyme is processed by proprotein-convertases to its mature form which is degraded upon phorbol ester stimulation. Eur. J. Biochem. 2003, 270, 2386–2393. [Google Scholar] [CrossRef]
- Seals, D.F.; Courtneidge, S.A. The ADAMs family of metalloproteases: Multidomain proteins with multiple functions. Genes Dev. 2003, 17, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Anders, A.; Gilbert, S.; Garten, W.; Postina, R.; Fahrenholz, F. Regulation of the α-secretase ADAM10 by its prodomain and proprotein convertases. FASEB J. 2001, 15, 1837–1839. [Google Scholar] [CrossRef] [PubMed]
- Loechel, F.; Fox, J.W.; Murphy, G.; Albrechtsen, R.; Wewer, U.M. ADAM 12-S Cleaves IGFBP-3 and IGFBP-5 and Is Inhibited by TIMP-3. Biochem. Biophys. Res. Commun. 2000, 278, 511–515. [Google Scholar] [CrossRef]
- Roghani, M.; Becherer, J.D.; Moss, M.L.; Atherton, R.E.; Erdjument-Bromage, H.; Arribas, J.; Blackburn, R.K.; Weskamp, G.; Tempst, P.; Blobel, C.P. Metalloprotease-disintegrin MDC9: Intracellular maturation and catalytic activity. J. Biol. Chem. 1999, 274, 3531–3540. [Google Scholar] [CrossRef]
- Atapattu, L.; Saha, N.; Chheang, C.; Eissman, M.F.; Xu, K.; Vail, M.E.; Hii, L.; Llerena, C.; Liu, Z.; Horvay, K.; et al. An activated form of ADAM10 is tumor selective and regulates cancer stem-like cells and tumor growth. J. Exp. Med. 2016, 213, 1741–1757. [Google Scholar] [CrossRef]
- Willems, S.H.; Tape, C.J.; Stanley, P.L.; Taylor, N.A.; Mills, I.G.; Neal, D.E.; McCafferty, J.; Murphy, G. Thiol isomerases negatively regulate the cellular shedding activity of ADAM17. Biochem. J. 2010, 428, 439–450. [Google Scholar] [CrossRef]
- Wang, Y.; Herrera, A.H.; Li, Y.; Belani, K.K.; Walcheck, B. Regulation of mature ADAM17 by redox agents for L-selectin shedding. J. Immunol. 2009, 182, 2449–2457. [Google Scholar] [CrossRef] [PubMed]
- Düsterhöft, S.; Jung, S.; Hung, C.W.; Tholey, A.; Sönnichsen, F.D.; Grötzinger, J.; Lorenzen, I. Membrane-proximal domain of a disintegrin and metalloprotease-17 represents the putative molecular switch of its shedding activity operated by protein-disulfide isomerase. J. Am. Chem. Soc. 2013, 135, 5776–5781. [Google Scholar] [CrossRef]
- Xu, P.; Liu, J.; Sakaki-Yumoto, M.; Derynck, R. TACE activation by MAPK-mediated regulation of cell surface dimerization and TIMP3 association. Sci. Signal. 2012, 5, ra34. [Google Scholar] [CrossRef]
- Brew, K.; Nagase, H. The tissue inhibitors of metalloproteinases (TIMPs): An ancient family with structural and functional diversity. Biochim. Biophys. Acta 2010, 1803, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.M.; Kaur, A.; Montague, S.J.; Hicks, S.M.; Andrews, R.K.; Gardiner, E.E. Tissue inhibitors of metalloproteinases (TIMPs) modulate platelet ADAM10 activity. Platelets 2023, 34, 2288213. [Google Scholar] [CrossRef]
- Liao, S.; Lin, Y.; Liu, L.; Yang, S.; Lin, Y.; He, J.; Shao, Y. ADAM10-a “multitasker” in sepsis: Focus on its posttranslational target. Inflamm. Res. 2023, 72, 395–423. [Google Scholar] [CrossRef]
- Maretzky, T.; Evers, A.; Le Gall, S.; Alabi, R.O.; Speck, N.; Reiss, K.; Blobel, C.P. The cytoplasmic domain of a disintegrin and metalloproteinase 10 (ADAM10) regulates its constitutive activity but is dispensable for stimulated ADAM10-dependent shedding. J. Biol. Chem. 2015, 290, 7416–7425. [Google Scholar] [CrossRef]
- Janes, P.W.; Wimmer-Kleikamp, S.H.; Frangakis, A.S.; Treble, K.; Griesshaber, B.; Sabet, O.; Grabenbauer, M.; Ting, A.Y.; Saftig, P.; Bastiaens, P.I.; et al. Cytoplasmic relaxation of active Eph controls ephrin shedding by ADAM10. PLoS Biol. 2009, 7, e1000215. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, M.; Herrlich, A.; Herrlich, P. Who decides when to cleave an ectodomain? Trends Biochem. Sci. 2013, 38, 111–120. [Google Scholar] [CrossRef]
- Arai, J.; Otoyama, Y.; Nozawa, H.; Kato, N.; Yoshida, H. The immunological role of ADAMs in the field of gastroenterological chronic inflammatory diseases and cancers: A review. Oncogene 2023, 42, 549–558. [Google Scholar] [CrossRef]
- Conrad, C.; Benzel, J.; Dorzweiler, K.; Cook, L.; Schlomann, U.; Zarbock, A.; Slater, E.P.; Nimsky, C.; Bartsch Jörg, W. ADAM8 in invasive cancers: Links to tumor progression, metastasis, and chemoresistance. Clin. Sci. 2019, 133, 83–99. [Google Scholar] [CrossRef] [PubMed]
- Fritzsche, F.R.; Jung, M.; Xu, C.; Rabien, A.; Schicktanz, H.; Stephan, C.; Dietel, M.; Jung, K.; Kristiansen, G. ADAM8 expression in prostate cancer is associated with parameters of unfavorable prognosis. Virchows Arch. 2006, 449, 628–636. [Google Scholar] [CrossRef]
- Hernández, I.; Moreno, J.L.; Zandueta, C.; Montuenga, L.; Lecanda, F. Novel alternatively spliced ADAM8 isoforms contribute to the aggressive bone metastatic phenotype of lung cancer. Oncogene 2010, 29, 3758–3769. [Google Scholar] [CrossRef]
- Jaworek, C.; Verel-Yilmaz, Y.; Driesch, S.; Ostgathe, S.; Cook, L.; Wagner, S.; Bartsch, D.K.; Slater, E.P.; Bartsch, J.W. Cohort Analysis of ADAM8 Expression in the PDAC Tumor Stroma. J. Pers. Med. 2021, 11, 113. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, E.T.; Benton, R.L.; Maddie, M.A.; Whittemore, S.R.; Hagg, T. ADAM8 is selectively up-regulated in endothelial cells and is associated with angiogenesis after spinal cord injury in adult mice. J. Comp. Neurol. 2009, 512, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Naus, S.; Richter, M.; Wildeboer, D.; Moss, M.; Schachner, M.; Bartsch, J.W. Ectodomain Shedding of the Neural Recognition Molecule CHL1 by the Metalloprotease-disintegrin ADAM8 Promotes Neurite Outgrowth and Suppresses Neuronal Cell Death. J. Biol. Chem. 2004, 279, 16083–16090. [Google Scholar] [CrossRef]
- Pianetti, S.; Miller, K.D.; Chen, H.H.; Althouse, S.; Cao, S.; Michael, S.J.; Sonenshein, G.E.; Mineva, N.D. ADAM8 is expressed widely in breast cancer and predicts poor outcome in hormone receptor positive, HER-2 negative patients. Cancer Cell Int. 2023, 23, 165. [Google Scholar] [CrossRef]
- Romagnoli, M.; Mineva, N.D.; Polmear, M.; Conrad, C.; Srinivasan, S.; Loussouarn, D.; Barillé-Nion, S.; Georgakoudi, I.; Dagg, Á.; McDermott, E.W.; et al. ADAM8 expression in invasive breast cancer promotes tumor dissemination and metastasis. EMBO Mol. Med. 2014, 6, 278–294. [Google Scholar] [CrossRef]
- Zack, M.D.; Malfait, A.-M.; Skepner, A.P.; Yates, M.P.; Griggs, D.W.; Hall, T.; Hills, R.L.; Alston, J.T.; Nemirovskiy, O.V.; Radabaugh, M.R.; et al. ADAM-8 isolated from human osteoarthritic chondrocytes cleaves fibronectin at Ala271. Arthritis Rheum. 2009, 60, 2704–2713. [Google Scholar] [CrossRef]
- Zhao, K.; Calero-Pérez, P.; Bopp, M.H.A.; Möschl, V.; Pagenstecher, A.; Mulero-Acevedo, M.; Vázquez, M.; Barcia, C.; Arús, C.; Nimsky, C.; et al. Correlation of MR-Based Metabolomics and Molecular Profiling in the Tumor Microenvironment of Temozolomide-Treated Orthotopic GL261 Glioblastoma in Mice. Int. J. Mol. Sci. 2023, 24, 17628. [Google Scholar] [CrossRef]
- Fritzsche, F.R.; Jung, M.; Tölle, A.; Wild, P.; Hartmann, A.; Wassermann, K.; Rabien, A.; Lein, M.; Dietel, M.; Pilarsky, C.; et al. ADAM9 Expression is a Significant and Independent Prognostic Marker of PSA Relapse in Prostate Cancer. Eur. Urol. 2008, 54, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Fritzsche, F.R.; Wassermann, K.; Jung, M.; Tölle, A.; Kristiansen, I.; Lein, M.; Johannsen, M.; Dietel, M.; Jung, K.; Kristiansen, G. ADAM9 is highly expressed in renal cell cancer and is associated with tumour progression. BMC Cancer 2008, 8, 179. [Google Scholar] [CrossRef]
- Horiuchi, K.; Zhou, H.-M.; Kelly, K.; Manova, K.; Blobel, C.P. Evaluation of the contributions of ADAMs 9, 12, 15, 17, and 19 to heart development and ectodomain shedding of neuregulins β1 and β2. Dev. Biol. 2005, 283, 459–471. [Google Scholar] [CrossRef]
- Huang, Y.-K.; Cheng, W.-C.; Kuo, T.-T.; Yang, J.-C.; Wu, Y.-C.; Wu, H.-H.; Lo, C.-C.; Hsieh, C.-Y.; Wong, S.-C.; Lu, C.-H.; et al. Inhibition of ADAM9 promotes the selective degradation of KRAS and sensitizes pancreatic cancers to chemotherapy. Nat. Cancer 2024, 5, 400–419. [Google Scholar] [CrossRef] [PubMed]
- Micocci, K.C.; Martin, A.C.B.M.; Montenegro, C.d.F.; Durante, A.C.; Pouliot, N.; Cominetti, M.R.; Selistre-de-Araujo, H.S. ADAM9 silencing inhibits breast tumor cell invasion in vitro. Biochimie 2013, 95, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Oria, V.O.; Lopatta, P.; Schmitz, T.; Preca, B.-T.; Nyström, A.; Conrad, C.; Bartsch, J.W.; Kulemann, B.; Hoeppner, J.; Maurer, J.; et al. ADAM9 contributes to vascular invasion in pancreatic ductal adenocarcinoma. Mol. Oncol. 2019, 13, 456–479. [Google Scholar] [CrossRef]
- Sammel, M.; Peters, F.; Lokau, J.; Scharfenberg, F.; Werny, L.; Linder, S.; Garbers, C.; Rose-John, S.; Becker-Pauly, C. Differences in Shedding of the Interleukin-11 Receptor by the Proteases ADAM9, ADAM10, ADAM17, Meprin α, Meprin β and MT1-MMP. Int. J. Mol. Sci. 2019, 20, 3677. [Google Scholar] [CrossRef]
- Tanasubsinn, P.; Aung, W.P.P.; Pata, S.; Laopajon, W.; Makeudom, A.; Sastraruji, T.; Kasinrerk, W.; Krisanaprakornkit, S. Overexpression of ADAM9 in oral squamous cell carcinoma. Oncol. Lett. 2018, 15, 495–502. [Google Scholar] [CrossRef]
- Weskamp, G.; Cai, H.; Brodie, T.A.; Higashyama, S.; Manova, K.; Ludwig, T.; Blobel, C.P. Mice lacking the metalloprotease-disintegrin MDC9 (ADAM9) have no evident major abnormalities during development or adult life. Mol. Cell. Biol. 2002, 22, 1537–1544. [Google Scholar] [CrossRef]
- Du, S.; Sun, L.; Wang, Y.; Zhu, W.; Gao, J.; Pei, W.; Zhang, Y. ADAM12 is an independent predictor of poor prognosis in liver cancer. Sci. Rep. 2022, 12, 6634. [Google Scholar] [CrossRef] [PubMed]
- Duhachek-Muggy, S.; Qi, Y.; Wise, R.; Alyahya, L.; Li, H.; Hodge, J.; Zolkiewska, A. Metalloprotease-disintegrin ADAM12 actively promotes the stem cell-like phenotype in claudin-low breast cancer. Mol. Cancer 2017, 16, 32. [Google Scholar] [CrossRef]
- Huang, Z.; Lai, H.; Liao, J.; Cai, J.; Li, B.; Meng, L.; Wang, W.; Mo, X.; Qin, H. Upregulation of ADAM12 Is Associated With a Poor Survival and Immune Cell Infiltration in Colon Adenocarcinoma. Front. Oncol. 2021, 11, 729230. [Google Scholar] [CrossRef]
- Mendaza, S.; Ulazia-Garmendia, A.; Monreal-Santesteban, I.; Córdoba, A.; Azúa, Y.R.; Aguiar, B.; Beloqui, R.; Armendáriz, P.; Arriola, M.; Martín-Sánchez, E.; et al. ADAM12 is A Potential Therapeutic Target Regulated by Hypomethylation in Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2020, 21, 903. [Google Scholar] [CrossRef]
- Piotrowski, K.B.; Blasco, L.P.; Samsøe-Petersen, J.; Eefsen, R.L.; Illemann, M.; Oria, V.O.; Campos, K.I.A.; Lopresti, A.M.; Albrechtsen, R.; Sørensen, C.S.; et al. ADAM12 expression is upregulated in cancer cells upon radiation and constitutes a prognostic factor in rectal cancer patients following radiotherapy. Cancer Gene Ther. 2023, 30, 1369–1381. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Dagher, A.; Butterfield, C.; Moses, M.A. ADAM12 Is a Novel Regulator of Tumor Angiogenesis via STAT3 Signaling. Mol. Cancer Res. 2017, 15, 1608–1622. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Godet, I.; Yang, Y.; Salman, S.; Lu, H.; Lyu, Y.; Zuo, Q.; Wang, Y.; Zhu, Y.; Chen, C.; et al. Hypoxia-inducible factor-dependent ADAM12 expression mediates breast cancer invasion and metastasis. Proc. Natl. Acad. Sci. USA 2021, 118, e2020490118. [Google Scholar] [CrossRef]
- Zahnow, C.A. ErbB receptors and their ligands in the breast. Expert Rev. Mol. Med. 2006, 8, 1–21. [Google Scholar] [CrossRef]
- Álvarez-Fernández, S.M.; Barbariga, M.; Cannizzaro, L.; Cannistraci, C.V.; Hurley, L.; Zanardi, A.; Conti, A.; Sanvito, F.; Innocenzi, A.; Pecorelli, N.; et al. Serological immune response against ADAM10 pro-domain is associated with favourable prognosis in stage III colorectal cancer patients. Oncotarget 2016, 7, 80059–80076. [Google Scholar] [CrossRef]
- Blobel, C.P. ADAMs: Key components in EGFR signalling and development. Nat. Rev. Mol. Cell Biol. 2005, 6, 32–43. [Google Scholar] [CrossRef]
- Cai, C.; Zhang, M.; Liu, L.; Zhang, H.; Guo, Y.; Lan, T.; Xu, Y.; Ma, P.; Li, S. ADAM10-cleaved ephrin-A5 contributes to prostate cancer metastasis. Cell Death Dis. 2022, 13, 453. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Lin, L.; Li, X.; Lu, A.; Hou, C.; Wu, Q.; Hu, X.; Zhou, Z.; Chen, Z.; Tang, F. ADAM10 is involved in the oncogenic process and chemo-resistance of triple-negative breast cancer via regulating Notch1 signaling pathway, CD44 and PrPc. Cancer Cell Int. 2021, 21, 32. [Google Scholar] [CrossRef]
- Gaida, M.M.; Haag, N.; Günther, F.; Tschaharganeh, D.F.; Schirmacher, P.; Friess, H.; Giese, N.A.; Schmidt, J.; Wente, M.N. Expression of A disintegrin and metalloprotease 10 in pancreatic carcinoma. Int. J. Mol. Med. 2010, 26, 281–288. [Google Scholar] [CrossRef]
- Guo, J.; He, L.; Yuan, P.; Wang, P.; Lu, Y.; Tong, F.; Wang, Y.; Yin, Y.; Tian, J.; Sun, J. ADAM10 overexpression in human non-small cell lung cancer correlates with cell migration and invasion through the activation of the Notch1 signaling pathway. Oncol. Rep. 2012, 28, 1709–1718. [Google Scholar] [CrossRef]
- Hartmann, D.; de Strooper, B.; Serneels, L.; Craessaerts, K.; Herreman, A.; Annaert, W.; Umans, L.; Lübke, T.; Lena Illert, A.; von Figura, K.; et al. The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for alpha-secretase activity in fibroblasts. Hum. Mol. Genet. 2002, 11, 2615–2624. [Google Scholar] [CrossRef] [PubMed]
- Mueller, A.C.; Piper, M.; Goodspeed, A.; Bhuvane, S.; Williams, J.S.; Bhatia, S.; Phan, A.V.; Van Court, B.; Zolman, K.L.; Peña, B.; et al. Induction of ADAM10 by Radiation Therapy Drives Fibrosis, Resistance, and Epithelial-to-Mesenchyal Transition in Pancreatic Cancer. Cancer Res. 2021, 81, 3255–3269. [Google Scholar] [CrossRef] [PubMed]
- Mullooly, M.; McGowan, P.M.; Kennedy, S.A.; Madden, S.F.; Crown, J.; O’ Donovan, N.; Duffy, M.J. ADAM10: A new player in breast cancer progression? Br. J. Cancer 2015, 113, 945–951. [Google Scholar] [CrossRef]
- Orme, J.J.; Jazieh, K.A.; Xie, T.; Harrington, S.; Liu, X.; Ball, M.; Madden, B.; Charlesworth, M.C.; Azam, T.U.; Lucien, F.; et al. ADAM10 and ADAM17 cleave PD-L1 to mediate PD-(L)1 inhibitor resistance. OncoImmunology 2020, 9, 1744980. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Ye, Z.Y.; Li, L.; Zhao, Z.S.; Shao, Q.S.; Tao, H.Q. ADAM 10 is associated with gastric cancer progression and prognosis of patients. J. Surg. Oncol. 2011, 103, 116–123. [Google Scholar] [CrossRef]
- Yoneyama, T.; Gorry, M.; Sobo-Vujanovic, A.; Lin, Y.; Vujanovic, L.; Gaither-Davis, A.; Moss, M.L.; Miller, M.A.; Griffith, L.G.; Lauffenburger, D.A.; et al. ADAM10 Sheddase Activity is a Potential Lung-Cancer Biomarker. J. Cancer 2018, 9, 2559–2570. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Yu, H.; Chen, J.; Song, X.; Sun, L. ADAM10 promotes cell growth, migration, and invasion in osteosarcoma via regulating E-cadherin/β-catenin signaling pathway and is regulated by miR-122-5p. Cancer Cell Int. 2020, 20, 99. [Google Scholar] [CrossRef]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef]
- Duffy, M.J.; McKiernan, E.; O’Donovan, N.; McGowan, P.M. Role of ADAMs in Cancer Formation and Progression. Clin. Cancer Res. 2009, 15, 1140–1144. [Google Scholar] [CrossRef]
- Jones, J.C.; Rustagi, S.; Dempsey, P.J. ADAM Proteases and Gastrointestinal Function. Annu. Rev. Physiol. 2016, 78, 243–276. [Google Scholar] [CrossRef]
- Li, K.; Xue, W.; Lu, Z.; Wang, S.; Zheng, J.; Lu, K.; Li, M.; Zong, Y.; Xu, F.; Dai, J.; et al. Tumor-derived exosomal ADAM17 promotes pre-metastatic niche formation by enhancing vascular permeability in colorectal cancer. J. Exp. Clin. Cancer Res. 2024, 43, 59. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Li, Y.; Qian, M.; Ma, C.; Jing, H.; Wen, Z.; Qian, D. ADAM17 silencing suppresses the migration and invasion of non-small cell lung cancer. Mol. Med. Rep. 2014, 9, 1935–1940. [Google Scholar] [CrossRef]
- Saad, M.I.; Alhayyani, S.; McLeod, L.; Yu, L.; Alanazi, M.; Deswaerte, V.; Tang, K.; Jarde, T.; Smith, J.A.; Prodanovic, Z.; et al. ADAM17 selectively activates the IL-6 trans-signaling/ERK MAPK axis in KRAS-addicted lung cancer. EMBO Mol. Med. 2019, 11, e9976. [Google Scholar] [CrossRef]
- Saad, M.I.; Rose-John, S.; Jenkins, B.J. ADAM17: An Emerging Therapeutic Target for Lung Cancer. Cancers 2019, 11, 1218. [Google Scholar] [CrossRef]
- Schumacher, N.; Rose-John, S. ADAM17 Activity and IL-6 Trans-Signaling in Inflammation and Cancer. Cancers 2019, 11, 1736. [Google Scholar] [CrossRef]
- Shi, W.; Men, L.; Pi, X.; Jiang, T.; Peng, D.; Huo, S.; Luo, P.; Wang, M.; Guo, J.; Jiang, Y.; et al. Shikonin suppresses colon cancer cell growth and exerts synergistic effects by regulating ADAM17 and the IL-6/STAT3 signaling pathway. Int. J. Oncol. 2021, 59, 99. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.-J.; Lin, P.; Lin, F.; Liu, X.; Qin, W.; Zou, H.-F.; Guo, L.; Liu, W.; Wang, S.-J.; Yu, X.-G. ADAM17 targets MMP-2 and MMP-9 via EGFR-MEK-ERK pathway activation to promote prostate cancer cell invasion. Int. J. Oncol. 2012, 40, 1714–1724. [Google Scholar] [CrossRef]
- Zheng, X.; Jiang, F.; Katakowski, M.; Lu, Y.; Chopp, M. ADAM17 promotes glioma cell malignant phenotype. Mol. Carcinog. 2012, 51, 150–164. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jiang, X.; Duan, Y.; Long, J.; Bartsch, J.W.; Deng, L. ADAM8 in asthma. Friend or foe to airway inflammation? Am. J. Respir. Cell Mol. Biol. 2013, 49, 875–884. [Google Scholar] [CrossRef]
- Qu, H.; Mao, M.; Wang, K.; Mu, Z.; Hu, B. Knockdown of ADAM8 inhibits the proliferation, migration, invasion, and tumorigenesis of renal clear cell carcinoma cells to enhance the immunotherapy efficacy. Transl. Res. 2024, 266, 32–48. [Google Scholar] [CrossRef]
- Chou, C.-W.; Huang, Y.-K.; Kuo, T.-T.; Liu, J.-P.; Sher, Y.-P. An Overview of ADAM9: Structure, Activation, and Regulation in Human Diseases. Int. J. Mol. Sci. 2020, 21, 7790. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekera, P.; Perfetto, M.; Lu, C.; Zhuo, M.; Bahudhanapati, H.; Li, J.; Chen, W.-C.; Kulkarni, P.; Christian, L.; Liu, J.; et al. Metalloprotease ADAM9 cleaves ephrin-B ligands and differentially regulates Wnt and mTOR signaling downstream of Akt kinase in colorectal cancer cells. J. Biol. Chem. 2022, 298, 102225. [Google Scholar] [CrossRef]
- Albrechtsen, R.; Wewer Albrechtsen, N.J.; Gnosa, S.; Schwarz, J.; Dyrskjøt, L.; Kveiborg, M. Identification of ADAM12 as a Novel Basigin Sheddase. Int. J. Mol. Sci. 2019, 20, 1957. [Google Scholar] [CrossRef]
- Lendeckel, U.; Wolke, C.; Bernstein, H.G.; Keilhoff, G. Effects of nitric oxide synthase deficiency on a disintegrin and metalloproteinase domain-containing protein 12 expression in mouse brain samples. Mol. Med. Rep. 2015, 12, 2253–2262. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, H.-G.; Keilhoff, G.; Dobrowolny, H.; Lendeckel, U.; Steiner, J. From putative brain tumor marker to high cognitive abilities: Emerging roles of a disintegrin and metalloprotease (ADAM) 12 in the brain. J. Chem. Neuroanat. 2020, 109, 101846. [Google Scholar] [CrossRef]
- Romero, Y.; Wise, R.; Zolkiewska, A. Proteolytic processing of PD-L1 by ADAM proteases in breast cancer cells. Cancer Immunol. Immunother. CII 2020, 69, 43–55. [Google Scholar] [CrossRef] [PubMed]
- McGowan, P.M.; Ryan, B.d.M.; Hill, A.D.K.; McDermott, E.; O’Higgins, N.; Duffy, M.J. ADAM-17 Expression in Breast Cancer Correlates with Variables of Tumor Progression. Clin. Cancer Res. 2007, 13, 2335–2343. [Google Scholar] [CrossRef]
- Ringel, J.r.; Jesnowski, R.; Moniaux, N.; Lüttges, J.; Ringel, J.; Choudhury, A.; Batra, S.K.; Klöppel, G.n.; Löhr, M. Aberrant Expression of a Disintegrin and Metalloproteinase 17/Tumor Necrosis Factor-α Converting Enzyme Increases the Malignant Potential in Human Pancreatic Ductal Adenocarcinoma. Cancer Res. 2006, 66, 9045–9053. [Google Scholar] [CrossRef]
- Ye, J.; Yuen, S.M.; Murphy, G.; Xie, R.; Kwok, H.F. Anti-tumor effects of a ‘human & mouse cross-reactive’ anti-ADAM17 antibody in a pancreatic cancer model in vivo. Eur. J. Pharm. Sci. 2017, 110, 62–69. [Google Scholar] [CrossRef]
- Ardito, C.M.; Grüner, B.M.; Takeuchi, K.K.; Lubeseder-Martellato, C.; Teichmann, N.; Mazur, P.K.; DelGiorno, K.E.; Carpenter, E.S.; Halbrook, C.J.; Hall, J.C.; et al. EGF Receptor Is Required for KRAS-Induced Pancreatic Tumorigenesis. Cancer Cell 2012, 22, 304–317. [Google Scholar] [CrossRef]
- Shin, D.H.; Kim, S.H.; Choi, M.; Bae, Y.-K.; Han, C.; Choi, B.K.; Kim, S.S.; Han, J.-Y. Oncogenic KRAS promotes growth of lung cancer cells expressing SLC3A2-NRG1 fusion via ADAM17-mediated shedding of NRG1. Oncogene 2022, 41, 280–292. [Google Scholar] [CrossRef]
- Saad, M.I.; Weng, T.; Lundy, J.; Gearing, L.J.; West, A.C.; Harpur, C.M.; Alanazi, M.; Hodges, C.; Croagh, D.; Kumar, B.; et al. Blockade of the protease ADAM17 ameliorates experimental pancreatitis. Proc. Natl. Acad. Sci. USA 2022, 119, e2213744119. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Mullooly, M.; O’Donovan, N.; Sukor, S.; Crown, J.; Pierce, A.; McGowan, P.M. The ADAMs family of proteases: New biomarkers and therapeutic targets for cancer? Clin. Proteom. 2011, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Schlomann, U.; Koller, G.; Conrad, C.; Ferdous, T.; Golfi, P.; Garcia, A.M.; Höfling, S.; Parsons, M.; Costa, P.; Soper, R.; et al. ADAM8 as a drug target in pancreatic cancer. Nat. Commun. 2015, 6, 6175. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhou, Y.; Wang, C.; Sample, K.M.; Yu, X.; Ben-David, Y. Propofol mediates pancreatic cancer cell activity through the repression of ADAM8 via SP1. Oncol. Rep. 2021, 46, 249. [Google Scholar] [CrossRef]
- Arai, J.; Goto, K.; Otoyama, Y.; Nakajima, Y.; Sugiura, I.; Kajiwara, A.; Tojo, M.; Ichikawa, Y.; Uozumi, S.; Shimozuma, Y.; et al. Leukotriene receptor antagonists enhance HCC treatment efficacy by inhibiting ADAMs and suppressing MICA shedding. Cancer Immunol. Immunother. CII 2021, 70, 203–213. [Google Scholar] [CrossRef]
- Arai, J.; Goto, K.; Stephanou, A.; Tanoue, Y.; Ito, S.; Muroyama, R.; Matsubara, Y.; Nakagawa, R.; Morimoto, S.; Kaise, Y.; et al. Predominance of regorafenib over sorafenib: Restoration of membrane-bound MICA in hepatocellular carcinoma cells. J. Gastroenterol. Hepatol. 2018, 33, 1075–1081. [Google Scholar] [CrossRef]
- Kohga, K.; Takehara, T.; Tatsumi, T.; Ishida, H.; Miyagi, T.; Hosui, A.; Hayashi, N. Sorafenib inhibits the shedding of major histocompatibility complex class I-related chain A on hepatocellular carcinoma cells by down-regulating a disintegrin and metalloproteinase 9. Hepatology 2010, 51, 1264–1273. [Google Scholar] [CrossRef]
- Chen, C.-M.; Hsieh, Y.-H.; Hwang, J.-M.; Jan, H.-J.; Hsieh, S.-C.; Lin, S.-H.; Lai, C.-Y. Fisetin suppresses ADAM9 expression and inhibits invasion of glioma cancer cells through increased phosphorylation of ERK1/2. Tumor Biol. 2015, 36, 3407–3415. [Google Scholar] [CrossRef]
- Hsieh, M.H.; Tsai, J.P.; Yang, S.F.; Chiou, H.L.; Lin, C.L.; Hsieh, Y.H.; Chang, H.R. Fisetin Suppresses the Proliferation and Metastasis of Renal Cell Carcinoma through Upregulation of MEK/ERK-Targeting CTSS and ADAM9. Cells 2019, 8, 948. [Google Scholar] [CrossRef]
- Scribner, J.A.; Hicks, S.W.; Sinkevicius, K.W.; Yoder, N.C.; Diedrich, G.; Brown, J.G.; Lucas, J.; Fuller, M.E.; Son, T.; Dastur, A.; et al. Preclinical Evaluation of IMGC936, a Next-Generation Maytansinoid-based Antibody-drug Conjugate Targeting ADAM9-expressing Tumors. Mol. Cancer Ther. 2022, 21, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Asakura, M.; Kitakaze, M.; Takashima, S.; Liao, Y.; Ishikura, F.; Yoshinaka, T.; Ohmoto, H.; Node, K.; Yoshino, K.; Ishiguro, H.; et al. Cardiac hypertrophy is inhibited by antagonism of ADAM12 processing of HB-EGF: Metalloproteinase inhibitors as a new therapy. Nat. Med. 2002, 8, 35–40. [Google Scholar] [CrossRef]
- Ludwig, A.; Hundhausen, C.; Lambert, M.H.; Broadway, N.; Andrews, R.C.; Bickett, D.M.; Leesnitzer, M.A.; Becherer, J.D. Metalloproteinase inhibitors for the disintegrin-like metalloproteinases ADAM10 and ADAM17 that differentially block constitutive and phorbol ester-inducible shedding of cell surface molecules. Comb. Chem. High Throughput Screen. 2005, 8, 161–171. [Google Scholar] [CrossRef]
- Babendreyer, A.; Kieselhorst, J.; Rinkens, C.; Lyashenko, A.M.; Düsterhöft, S.; Jahr, H.; Craveiro, R.B.; Wolf, M.; Ludwig, A. Downregulation of the metalloproteinases ADAM10 or ADAM17 promotes osteoclast differentiation. Cell Commun. Signal. 2024, 22, 322. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, S.; Seipold, L.; Saftig, P. The metalloproteinase ADAM10: A useful therapeutic target? Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2017, 1864, 2071–2081. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, D.; Saftig, P. New insights into the function and pathophysiology of the ectodomain sheddase A Disintegrin And Metalloproteinase 10 (ADAM10). FEBS J. 2024, 291, 2733–2766. [Google Scholar] [CrossRef]
- Zhou, B.B.; Peyton, M.; He, B.; Liu, C.; Girard, L.; Caudler, E.; Lo, Y.; Baribaud, F.; Mikami, I.; Reguart, N.; et al. Targeting ADAM-mediated ligand cleavage to inhibit HER3 and EGFR pathways in non-small cell lung cancer. Cancer Cell 2006, 10, 39–50. [Google Scholar] [CrossRef]
- Newton, R.C.; Bradley, E.C.; Levy, R.S.; Doval, D.; Bondarde, S.; Sahoo, T.P.; Lokanatha, D.; Julka, P.K.; Nagarkar, R.; Friedman, S.M. Clinical benefit of INCB7839, a potent and selective ADAM inhibitor, in combination with trastuzumab in patients with metastatic HER2+ breast cancer. J. Clin. Oncol. 2010, 28, 3025. [Google Scholar] [CrossRef]
- Richards, F.M.; Tape, C.J.; Jodrell, D.I.; Murphy, G. Anti-tumour effects of a specific anti-ADAM17 antibody in an ovarian cancer model in vivo. PLoS ONE 2012, 7, e40597. [Google Scholar] [CrossRef]
- Tape, C.J.; Willems, S.H.; Dombernowsky, S.L.; Stanley, P.L.; Fogarasi, M.; Ouwehand, W.; McCafferty, J.; Murphy, G. Cross-domain inhibition of TACE ectodomain. Proc. Natl. Acad. Sci. USA 2011, 108, 5578–5583. [Google Scholar] [CrossRef]
- Dosch, J.; Ziemke, E.; Wan, S.; Luker, K.; Welling, T.; Hardiman, K.; Fearon, E.; Thomas, S.; Flynn, M.; Rios-Doria, J.; et al. Targeting ADAM17 inhibits human colorectal adenocarcinoma progression and tumor-initiating cell frequency. Oncotarget 2017, 8, 65090–65099. [Google Scholar] [CrossRef] [PubMed]
- Mishra, H.K.; Pore, N.; Michelotti, E.F.; Walcheck, B. Anti-ADAM17 monoclonal antibody MEDI3622 increases IFNγ production by human NK cells in the presence of antibody-bound tumor cells. Cancer Immunol. Immunother. 2018, 67, 1407–1416. [Google Scholar] [CrossRef]
- Peng, L.; Cook, K.; Xu, L.; Cheng, L.; Damschroder, M.; Gao, C.; Wu, H.; Dall’Acqua, W.F. Molecular basis for the mechanism of action of an anti-TACE antibody. mAbs 2016, 8, 1598–1605. [Google Scholar] [CrossRef]
- Rios-Doria, J.; Sabol, D.; Chesebrough, J.; Stewart, D.; Xu, L.; Tammali, R.; Cheng, L.; Du, Q.; Schifferli, K.; Rothstein, R.; et al. A Monoclonal Antibody to ADAM17 Inhibits Tumor Growth by Inhibiting EGFR and Non-EGFR-Mediated Pathways. Mol. Cancer therapeutics 2015, 14, 1637–1649. [Google Scholar] [CrossRef] [PubMed]
- Trad, A.; Hansen, H.P.; Shomali, M.; Peipp, M.; Klausz, K.; Hedemann, N.; Yamamoto, K.; Mauermann, A.; Desel, C.; Lorenzen, I.; et al. ADAM17-overexpressing breast cancer cells selectively targeted by antibody-toxin conjugates. Cancer Immunol. Immunother. 2013, 62, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Trad, A.; Baumgart, A.; Hüske, L.; Lorenzen, I.; Chalaris, A.; Grötzinger, J.; Dechow, T.; Scheller, J.; Rose-John, S. A novel bispecific single-chain antibody for ADAM17 and CD3 induces T-cell-mediated lysis of prostate cancer cells. Biochem. J. 2012, 445, 135–144. [Google Scholar] [CrossRef]
- Mierke, C.T. The versatile roles of ADAM8 in cancer cell migration, mechanics, and extracellular matrix remodeling. Front. Cell Dev. Biol. 2023, 11, 1130823. [Google Scholar] [CrossRef]
- Mineva, N.D.; Pianetti, S.; Das, S.G.; Srinivasan, S.; Billiald, N.M.; Sonenshein, G.E. A Novel Class of Human ADAM8 Inhibitory Antibodies for Treatment of Triple-Negative Breast Cancer. Pharmaceutics 2024, 16, 536. [Google Scholar] [CrossRef]
- Lei, D.; Zhang, F.; Yao, D.; Xiong, N.; Jiang, X.; Zhao, H. Galangin increases ERK1/2 phosphorylation to decrease ADAM9 expression and prevents invasion in A172 glioma cells. Mol. Med. Rep. 2018, 17, 667–673. [Google Scholar] [CrossRef]
- Moss, M.L.; Rasmussen, F.H. Fluorescent substrates for the proteinases ADAM17, ADAM10, ADAM8, and ADAM12 useful for high-throughput inhibitor screening. Anal. Biochem. 2007, 366, 144–148. [Google Scholar] [CrossRef]
- Zhong, S.; Khalil, R.A. A Disintegrin and Metalloproteinase (ADAM) and ADAM with thrombospondin motifs (ADAMTS) family in vascular biology and disease. Biochem. Pharmacol. 2019, 164, 188–204. [Google Scholar] [CrossRef]
- Madoux, F.; Dreymuller, D.; Pettiloud, J.-P.; Santos, R.; Becker-Pauly, C.; Ludwig, A.; Fields, G.B.; Bannister, T.; Spicer, T.P.; Cudic, M.; et al. Discovery of an enzyme and substrate selective inhibitor of ADAM10 using an exosite-binding glycosylated substrate. Sci. Rep. 2016, 6, 11. [Google Scholar] [CrossRef]
- Diez, J.; Selsted, M.E.; Bannister, T.D.; Minond, D. An ADAM10 Exosite Inhibitor Is Efficacious in an In Vivo Collagen-Induced Arthritis Model. Pharmaceuticals 2024, 17, 87. [Google Scholar] [CrossRef] [PubMed]
- Pece, R.; Tavella, S.; Costa, D.; Varesano, S.; Camodeca, C.; Cuffaro, D.; Nuti, E.; Rossello, A.; Alfano, M.; D’Arrigo, C.; et al. Inhibitors of ADAM10 reduce Hodgkin lymphoma cell growth in 3D microenvironments and enhance brentuximab-vedotin effect. Haematologica 2022, 107, 909–920. [Google Scholar] [CrossRef] [PubMed]
- Saha, N.; Xu, K.; Zhu, Z.; Robev, D.; Kalidindi, T.; Xu, Y.; Himanen, J.; de Stanchina, E.; Pillarsetty, N.V.K.; Dimitrov, D.S.; et al. Inhibitory monoclonal antibody targeting ADAM17 expressed on cancer cells. Transl. Oncol. 2022, 15, 101265. [Google Scholar] [CrossRef] [PubMed]
- Atapattu, L.; Saha, N.; Llerena, C.; Vail, M.E.; Scott, A.M.; Nikolov, D.B.; Lackmann, M.; Janes, P.W. Antibodies binding the ADAM10 substrate recognition domain inhibit Eph function. J. Cell Sci. 2012, 125, 6084–6093. [Google Scholar] [CrossRef]
- Saha, N.; Baek, D.-S.; Mendoza, R.P.; Robev, D.; Xu, Y.; Goldgur, Y.; De La Cruz, M.J.; de Stanchina, E.; Janes, P.W.; Xu, K.; et al. Fully human monoclonal antibody targeting activated ADAM10 on colorectal cancer cells. Biomed. Pharmacother. 2023, 161, 114494. [Google Scholar] [CrossRef]
- Yan, H.; Vail, M.E.; Hii, L.; Guo, N.; McMurrick, P.J.; Oliva, K.; Wilkins, S.; Saha, N.; Nikolov, D.B.; Lee, F.T.; et al. Preferential Antibody and Drug Conjugate Targeting of the ADAM10 Metalloprotease in Tumours. Cancers 2022, 14, 3171. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| ADAM | Cancer Type(s) | Key Functions in Cancer | Substrates | Prognosis | References |
|---|---|---|---|---|---|
| ADAM8 | Pancreatic, Breast, Lung, Prostate, and GBM | Tumour growth, invasion, and metastasis | CD23, TNF-R1, IL-1R2 collagen I, fibronectin, periostin, KL1, TNFα, APP, CHL1, and PD-L1 | Poor prognosis | [58,59,60,61,62,63,64,65,66,67] |
| ADAM9 | Pancreatic, Renal, Breast, and Oral | Tumour cell invasion and vascularization | HB-EGF and the interleukin-11 receptor | Poor prognosis | [68,69,70,71,72,73,74,75,76] |
| ADAM12 | Colon, Breast, and Liver | Migration, invasion, angiogenesis, and stem cell phenotype | EGFR | Poor prognosis and therapy resistance | [77,78,79,80,81,82,83,84] |
| ADAM10 | Breast, Osteosarcoma, Pancreatic, and Lung | Cell migration, invasion, metastasis, fibrosis, and immune evasion | Notch, Eph/ephrins, TNF, CX3CL, CXCL16, L1-CAM, N-cadherin, and E-cadherin | Poor prognosis, with overexpression linked to metastasis | [11,42,46,85,86,87,88,89,90,91,92,93,94,95,96,97] |
| ADAM17 | Pancreatic, Breast, Prostate, Colon, Lung, and especially K-Ras mutant tumours | Proliferation, invasion, and angiogenesis via EGFR activation | TNF-α, NRG1, Amphiregulin, Epiregulin, Heparin-binding EGF, and TGF-α | Poor prognosis linked to therapy resistance | [98,99,100,101,102,103,104,105,106,107,108] |
| Target | Drug | Type | Mechanism of Action (MOA) | Clinical Status | Clinicaltrials.gov Identifier, Reference |
|---|---|---|---|---|---|
| ADAM8 | BK-1361 | Small-MW inhibitor | Suppresses multimerization of ADAM8 | Preclinical | [124] |
| ADAM8 | Propofol | Small-MW inhibitor | Inhibition of specificity protein 1 (SP1), a key regulator of ADAM8 gene expression | Preclinical | [125] |
| ADAM9 (indirect) | Sorafenib and Regorafenib | Tyrosine kinase inhibitor | Reduces ADAM9/10 expression and MICA shedding, boosting natural killer cell activity | Preclinical | [126,127,128] |
| ADAM9 | Fisetin | Natural flavonoid | Increasing ERK1/2 activation through phosphorylation | Preclinical | [129,130] |
| ADAM9 | IMGC936 | Antibody–drug conjugate | Delivers a cytotoxic DM21-maytansinoid payload to tumour cells, inducing cell death | Phase 1/2 | [131] NCT04622774 |
| ADAM12 | TIMP3 | Small-MW inhibitor | Blocking the substrate-binding site in the cysteine-rich domain | Preclinical | [44] |
| ADAM12 | KB-R7785 | Small-MW inhibitor | Binds the ADAM12 active site, blocks the release of HB-EGF, and activates EGFR | Preclinical | [132] |
| ADAM10, ADAM17 | GI254023X | Small-MW inhibitor | Prevents the shedding of the IL-6 receptor | Preclinical | [4,9,117,123,133,134,135] |
| ADAM10 | INCB8765 | Small-MW inhibitor | Blocks EGF ligand processing (not selective) | Preclinical | [136,137] |
| ADAM10, ADAM17 | INCB3619 | Small-MW inhibitor | Prevents the secretion of the HER3 ligand neuregulin | Preclinical | [137] |
| ADAM10, ADAM17 | INCB7839 | Small-MW inhibitor | EGFR/HER2 inhibitor | Phase 1/2 | [138] NCT02141451 NCT04295759 |
| ADAM10 | 8C7 | Antibody and ADCs | Targets active ADAM10 and inhibits the cleavage of membrane-bound ligands, Eph, and notch signalling | Preclinical | [20,46] |
| ADAM17 | D1(A12) | Antibody | Attaches to both the M and D+C regions and blocks the release of various ADAM17 substrates | Preclinical | [139,140] |
| ADAM17 | MEDI3622 | Antibody | Binds the ADAM17 MP domain and inhibits amphiregulin shedding | Preclinical | [141,142,143,144] |
| ADAM17 | A300E | Antibody | Binds the ADAM17 C domain; bispecific anti-CD3 promotes anticancer T cell activity | Preclinical | [145,146] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arora, S.; Scott, A.M.; Janes, P.W. ADAM Proteases in Cancer: Biological Roles, Therapeutic Challenges, and Emerging Opportunities. Cancers 2025, 17, 1703. https://doi.org/10.3390/cancers17101703
Arora S, Scott AM, Janes PW. ADAM Proteases in Cancer: Biological Roles, Therapeutic Challenges, and Emerging Opportunities. Cancers. 2025; 17(10):1703. https://doi.org/10.3390/cancers17101703
Chicago/Turabian StyleArora, Sakshi, Andrew M. Scott, and Peter W. Janes. 2025. "ADAM Proteases in Cancer: Biological Roles, Therapeutic Challenges, and Emerging Opportunities" Cancers 17, no. 10: 1703. https://doi.org/10.3390/cancers17101703
APA StyleArora, S., Scott, A. M., & Janes, P. W. (2025). ADAM Proteases in Cancer: Biological Roles, Therapeutic Challenges, and Emerging Opportunities. Cancers, 17(10), 1703. https://doi.org/10.3390/cancers17101703