
The RAGE Inhibitor TTP488 (Azeliragon) Demonstrates Anti-Tumor Activity and Enhances the Efficacy of Radiation Therapy in Pancreatic Cancer Cell Lines
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Cell Proliferation
2.3. Tumor Model
2.4. Clonogenic Assay
2.5. Western Blot
2.6. Flow Cytometric Analysis
2.7. Statistical Analysis
3. Results
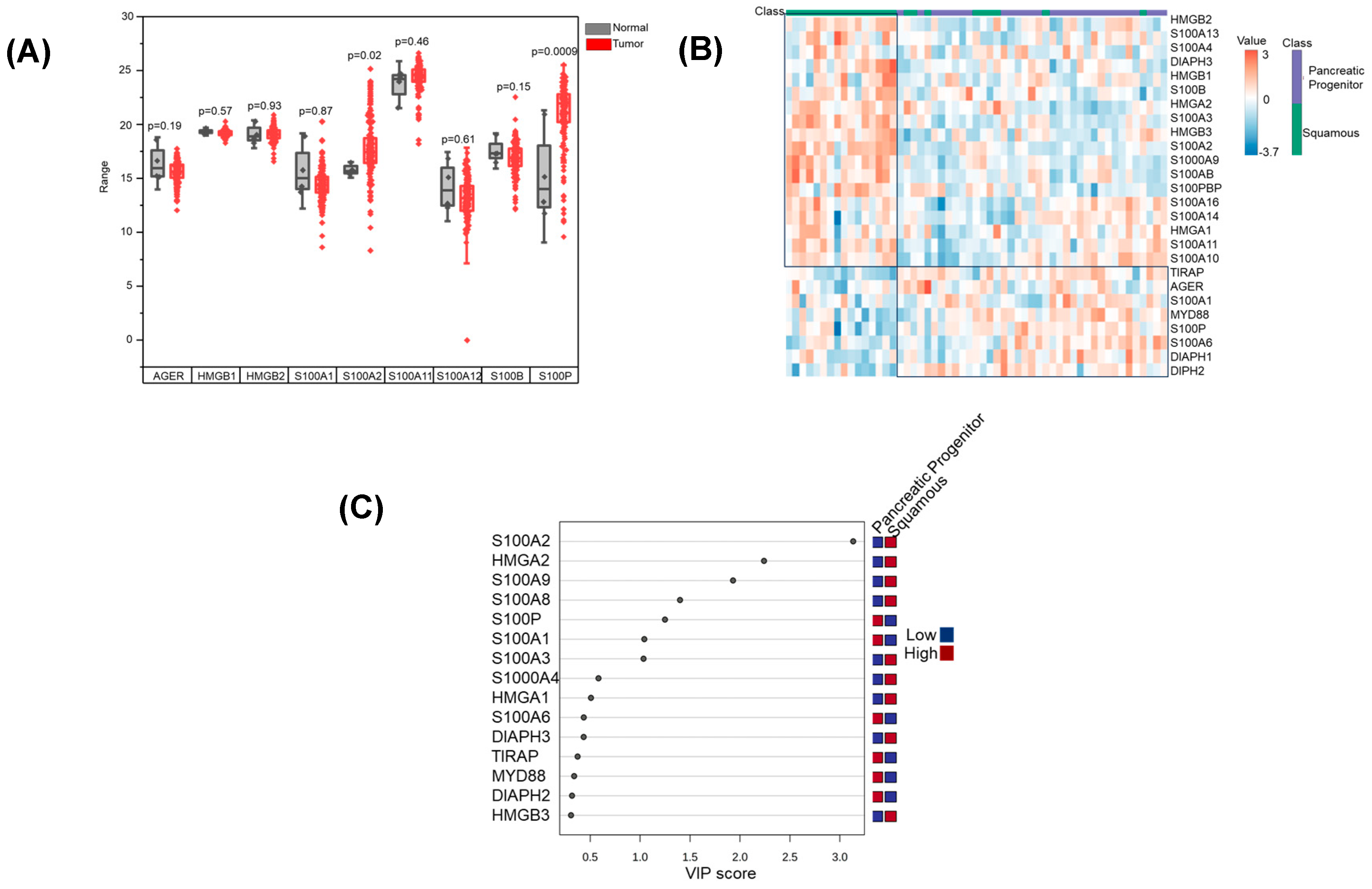
3.1. RAGE Pathway Activation in Pancreatic Cancer
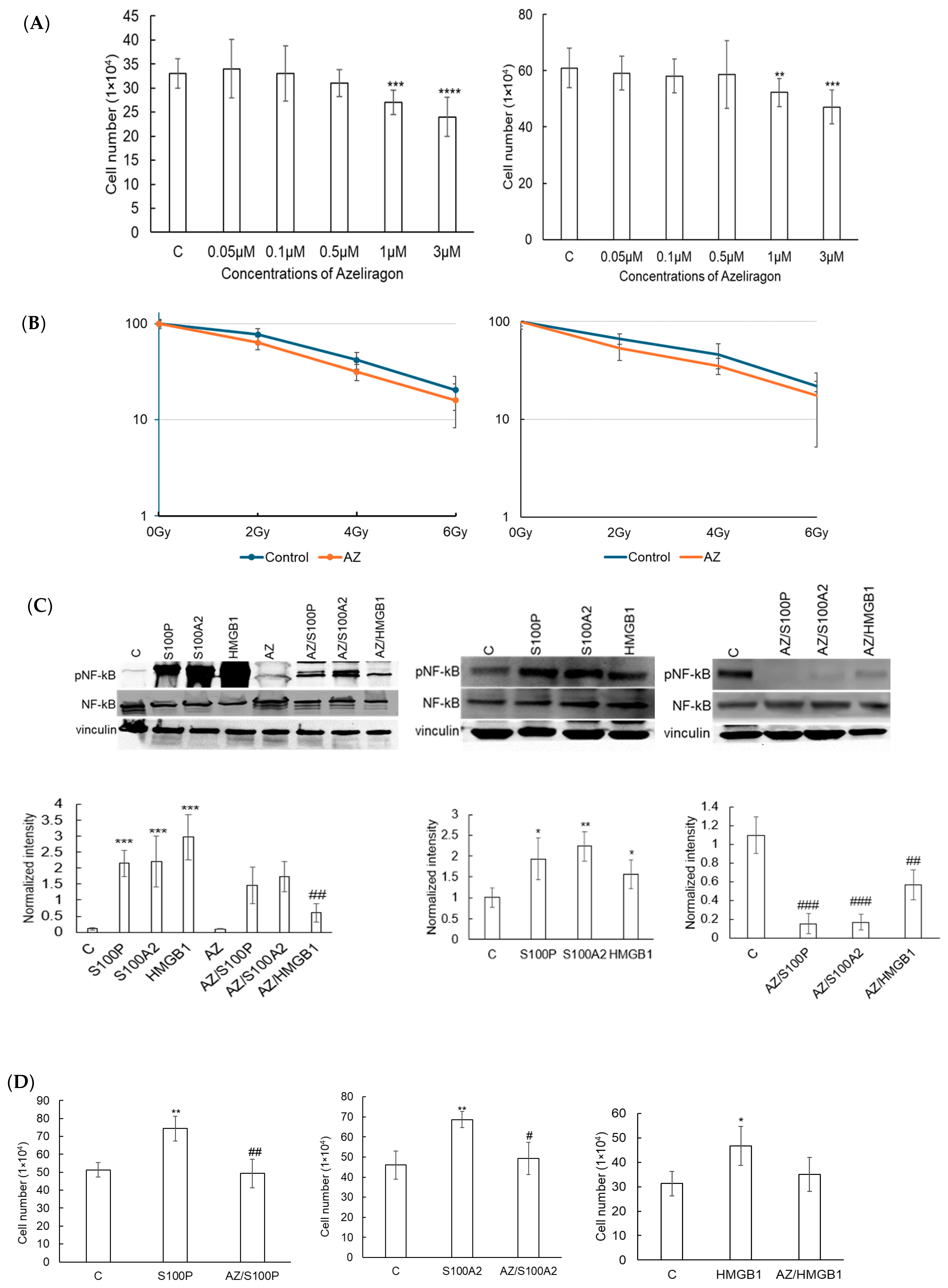
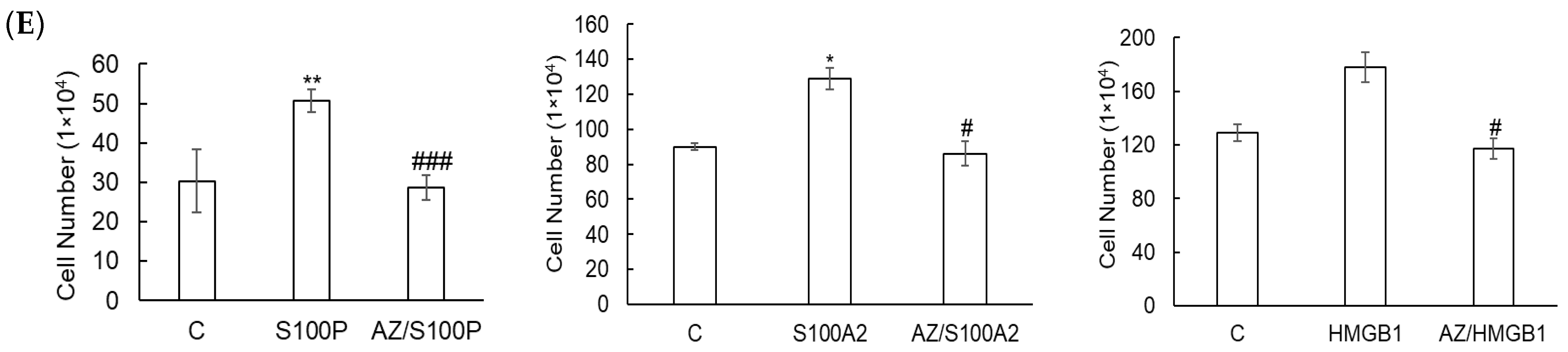
3.2. Azeliragon Inhibits RAGE Pathway Activation in Pancreatic Cancer Cell Lines
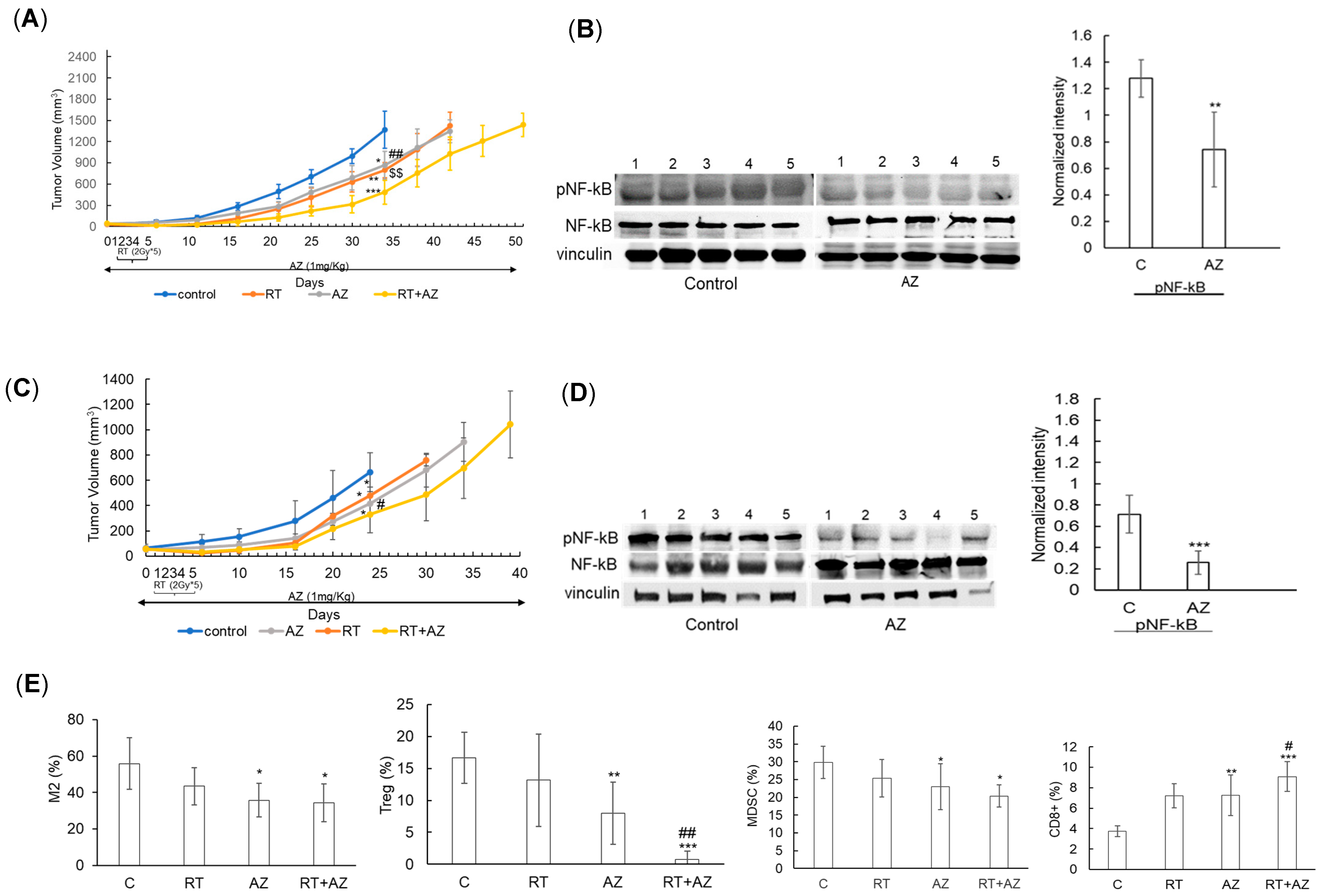
3.3. Azeliragon Demonstrates Anti-Tumor Activity and Modulates the Immune Microenvironment in Pancreatic Cancer Cell Lines In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Quiñonero, F.; Mesas, C.; Doello, K.; Cabeza, L.; Perazzoli, G.; Jimenez-Luna, C.; Rama, A.R.; Melguizo, C.; Prados, J. The Challenge of Drug Resistance in Pancreatic Ductal Adenocarcinoma: A Current Overview. Cancer Biol. Med. 2019, 16, 688–699. [Google Scholar] [CrossRef] [PubMed]
- Espona-Fiedler, M.; Patthey, C.; Lindblad, S.; Sarró, I.; Öhlund, D. Overcoming Therapy Resistance in Pancreatic Cancer: New Insights and Future Directions. Biochem. Pharmacol. 2024, 229, 116492. [Google Scholar] [CrossRef]
- Li, Y.; Xiang, S.; Pan, W.; Wang, J.; Zhan, H.; Liu, S. Targeting Tumor Immunosuppressive Microenvironment for Pancreatic Cancer Immunotherapy: Current Research and Future Perspective. Front. Oncol. 2023, 13, 1166860. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.M.; Hofmann, M.; Taguchi, A.; Yan, S.D.; Stern, D.M. RAGE: A Multiligand Receptor Contributing to the Cellular Response in Diabetic Vasculopathy and Inflammation. Semin. Thromb. Hemost. 2000, 26, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, C.D.; Fuentes, M.K.; Huang, E.H.; Arumugam, T. RAGE and RAGE Ligands in Cancer. Curr. Mol. Med. 2007, 7, 777–789. [Google Scholar] [CrossRef]
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding RAGE, the Receptor for Advanced Glycation End Products. J. Mol. Med. 2005, 83, 876–886. [Google Scholar] [CrossRef] [PubMed]
- Sparvero, L.J.; Asafu-Adjei, D.; Kang, R.; Tang, D.; Amin, N.; Im, J.; Rutledge, R.; Lin, B.; Amoscato, A.A.; Zeh, H.J.; et al. RAGE (Receptor for Advanced Glycation Endproducts), RAGE Ligands, and Their Role in Cancer and Inflammation. J. Transl. Med. 2009, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Chavakis, T.; Bierhaus, A.; Al-Fakhri, N.; Schneider, D.; Witte, S.; Linn, T.; Nagashima, M.; Morser, J.; Arnold, B.; Preissner, K.T.; et al. The Pattern Recognition Receptor (RAGE) Is a Counterreceptor for Leukocyte Integrins: A Novel Pathway for Inflammatory Cell Recruitment. J. Exp. Med. 2003, 198, 1507–1515. [Google Scholar] [CrossRef]
- Foell, D.; Wittkowski, H.; Vogl, T.; Roth, J. S100 Proteins Expressed in Phagocytes: A Novel Group of Damage-Associated Molecular Pattern Molecules. J. Leukoc. Biol. 2007, 81, 28–37. [Google Scholar] [CrossRef]
- Hsieh, H.-L.; Schäfer, B.W.; Sasaki, N.; Heizmann, C.W. Expression Analysis of S100 Proteins and RAGE in Human Tumors Using Tissue Microarrays. Biochem. Biophys. Res. Commun. 2003, 307, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, H.; Nakaigawa, N.; Miyoshi, Y.; Fujinami, K.; Kubota, Y.; Uemura, H. Receptor for Advanced Glycation End Products (RAGE) and Its Ligand, Amphoterin Are Overexpressed and Associated with Prostate Cancer Development. Prostate 2005, 64, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Kuniyasu, H.; Chihara, Y.; Takahashi, T. Co-Expression of Receptor for Advanced Glycation End Products and the Ligand Amphoterin Associates Closely with Metastasis of Colorectal Cancer. Oncol. Rep. 2003, 10, 445–448. [Google Scholar] [CrossRef] [PubMed]
- Faruqui, T.; Khan, M.S.; Akhter, Y.; Khan, S.; Rafi, Z.; Saeed, M.; Han, I.; Choi, E.-H.; Yadav, D.K. RAGE Inhibitors for Targeted Therapy of Cancer: A Comprehensive Review. Int. J. Mol. Sci. 2022, 24, 266. [Google Scholar] [CrossRef] [PubMed]
- DiNorcia, J.; Lee, M.K.; Moroziewicz, D.N.; Winner, M.; Suman, P.; Bao, F.; Remotti, H.E.; Zou, Y.S.; Yan, S.F.; Qiu, W.; et al. RAGE Gene Deletion Inhibits the Development and Progression of Ductal Neoplasia and Prolongs Survival in a Murine Model of Pancreatic Cancer. J. Gastrointest. Surg. 2012, 16, 104–112; discussion 112. [Google Scholar] [CrossRef]
- Kang, R.; Loux, T.; Tang, D.; Schapiro, N.E.; Vernon, P.; Livesey, K.M.; Krasinskas, A.; Lotze, M.T.; Zeh, H.J. The Expression of the Receptor for Advanced Glycation Endproducts (RAGE) Is Permissive for Early Pancreatic Neoplasia. Proc. Natl. Acad. Sci. USA 2012, 109, 7031–7036. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Tang, D.; Schapiro, N.E.; Loux, T.; Livesey, K.M.; Billiar, T.R.; Wang, H.; Van Houten, B.; Lotze, M.T.; Zeh, H.J. The HMGB1/RAGE Inflammatory Pathway Promotes Pancreatic Tumor Growth by Regulating Mitochondrial Bioenergetics. Oncogene 2014, 33, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Tang, D.; Livesey, K.M.; Schapiro, N.E.; Lotze, M.T.; Zeh, H.J. The Receptor for Advanced Glycation End-Products (RAGE) Protects Pancreatic Tumor Cells against Oxidative Injury. Antioxid. Redox Signal. 2011, 15, 2175–2184. [Google Scholar] [CrossRef] [PubMed]
- Swami, P.; Thiyagarajan, S.; Vidger, A.; Indurthi, V.S.K.; Vetter, S.W.; Leclerc, E. RAGE Up-Regulation Differently Affects Cell Proliferation and Migration in Pancreatic Cancer Cells. Int. J. Mol. Sci. 2020, 21, 7723. [Google Scholar] [CrossRef]
- Arumugam, T.; Simeone, D.M.; Van Golen, K.; Logsdon, C.D. S100P Promotes Pancreatic Cancer Growth, Survival, and Invasion. Clin. Cancer Res. 2005, 11, 5356–5364. [Google Scholar] [CrossRef]
- Arumugam, T.; Ramachandran, V.; Gomez, S.B.; Schmidt, A.M.; Logsdon, C.D. S100P-Derived RAGE Antagonistic Peptide Reduces Tumor Growth and Metastasis. Clin. Cancer Res. 2012, 18, 4356–4364. [Google Scholar] [CrossRef] [PubMed]
- Swami, P.; O’Connell, K.A.; Thiyagarajan, S.; Crawford, A.; Patil, P.; Radhakrishnan, P.; Shin, S.; Caffrey, T.C.; Grunkemeyer, J.; Neville, T.; et al. Inhibition of the Receptor for Advanced Glycation End Products Enhances the Cytotoxic Effect of Gemcitabine in Murine Pancreatic Tumors. Biomolecules 2021, 11, 526. [Google Scholar] [CrossRef] [PubMed]
- Lander, H.M.; Tauras, J.M.; Ogiste, J.S.; Hori, O.; Moss, R.A.; Schmidt, A.M. Activation of the Receptor for Advanced Glycation End Products Triggers a P21(Ras)-Dependent Mitogen-Activated Protein Kinase Pathway Regulated by Oxidant Stress. J. Biol. Chem. 1997, 272, 17810–17814. [Google Scholar] [CrossRef] [PubMed]
- Huttunen, H.J.; Fages, C.; Kuja-Panula, J.; Ridley, A.J.; Rauvala, H. Receptor for Advanced Glycation End Products-Binding COOH-Terminal Motif of Amphoterin Inhibits Invasive Migration and Metastasis. Cancer Res. 2002, 62, 4805–4811. [Google Scholar] [PubMed]
- Kang, R.; Tang, D.; Schapiro, N.E.; Livesey, K.M.; Farkas, A.; Loughran, P.; Bierhaus, A.; Lotze, M.T.; Zeh, H.J. The Receptor for Advanced Glycation End Products (RAGE) Sustains Autophagy and Limits Apoptosis, Promoting Pancreatic Tumor Cell Survival. Cell Death Differ. 2010, 17, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, A.; Blood, D.C.; del Toro, G.; Canet, A.; Lee, D.C.; Qu, W.; Tanji, N.; Lu, Y.; Lalla, E.; Fu, C.; et al. Blockade of RAGE-Amphoterin Signalling Suppresses Tumour Growth and Metastases. Nature 2000, 405, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Bierhaus, A.; Schiekofer, S.; Schwaninger, M.; Andrassy, M.; Humpert, P.M.; Chen, J.; Hong, M.; Luther, T.; Henle, T.; Klöting, I.; et al. Diabetes-Associated Sustained Activation of the Transcription Factor Nuclear Factor-KappaB. Diabetes 2001, 50, 2792–2808. [Google Scholar] [CrossRef]
- Huttunen, H.J.; Fages, C.; Rauvala, H. Receptor for Advanced Glycation End Products (RAGE)-Mediated Neurite Outgrowth and Activation of NF-KappaB Require the Cytoplasmic Domain of the Receptor but Different Downstream Signaling Pathways. J. Biol. Chem. 1999, 274, 19919–19924. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Abbruzzese, J.L.; Evans, D.B.; Larry, L.; Cleary, K.R.; Chiao, P.J. The Nuclear Factor-Kappa B RelA Transcription Factor Is Constitutively Activated in Human Pancreatic Adenocarcinoma Cells. Clin. Cancer Res. 1999, 5, 119–127. [Google Scholar] [PubMed]
- Singh, V.; Gupta, D.; Arora, R. NF-KB as a Key Player in Regulation of Cellular Radiation Responses and Identification of Radiation Countermeasures. Discoveries 2015, 3, e35. [Google Scholar] [CrossRef] [PubMed]
- Camara, R.; Ogbeni, D.; Gerstmann, L.; Ostovar, M.; Hurer, E.; Scott, M.; Mahmoud, N.G.; Radon, T.; Crnogorac-Jurcevic, T.; Patel, P.; et al. Discovery of Novel Small Molecule Inhibitors of S100P with In Vitro Anti-Metastatic Effects on Pancreatic Cancer Cells. Eur. J. Med. Chem. 2020, 203, 112621. [Google Scholar] [CrossRef]
- Arumugam, T.; Ramachandran, V.; Logsdon, C.D. Effect of Cromolyn on S100P Interactions with RAGE and Pancreatic Cancer Growth and Invasion in Mouse Models. J. Natl. Cancer Inst. 2006, 98, 1806–1818. [Google Scholar] [CrossRef]
- National Center for Biotechnology PubChem Compound Summary for CID 11180124, Azeliragon. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Azeliragon (accessed on 19 December 2024).
- Burstein, A.H.; Sabbagh, M.; Andrews, R.; Valcarce, C.; Dunn, I.; Altstiel, L. Development of Azeliragon, an Oral Small Molecule Antagonist of the Receptor for Advanced Glycation Endproducts, for the Potential Slowing of Loss of Cognition in Mild Alzheimer’s Disease. J. Prev. Alzheimers Dis. 2018, 5, 149–154. [Google Scholar] [CrossRef]
- Magna, M.; Hwang, G.H.; McIntosh, A.; Drews-Elger, K.; Takabatake, M.; Ikeda, A.; Mera, B.J.; Kwak, T.; Miller, P.; Lippman, M.E.; et al. RAGE Inhibitor TTP488 (Azeliragon) Suppresses Metastasis in Triple-Negative Breast Cancer. NPJ Breast Cancer 2023, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Azeliragon and Chemoradiotherapy in Newly Diagnosed Glioblastoma. Available online: https://clinicaltrials.gov/study/NCT05635734?intr=azeliragon&rank=3 (accessed on 19 December 2024).
- ClinicalTrials.gov. Study of Effect of Azeliragon in Patients Refractory to First-Line Metastatic Pancreatic Cancer. Available online: https://clinicaltrials.gov/study/NCT05766748?intr=azeliragon&rank=2 (accessed on 19 December 2024).
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic Analyses Identify Molecular Subtypes of Pancreatic Cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Gu, A.; Li, J.; Qiu, S.; Hao, S.; Yue, Z.-Y.; Zhai, S.; Li, M.-Y.; Liu, Y. Pancreatic Cancer Environment: From Patient-Derived Models to Single-Cell Omics. Mol. Omics 2024, 20, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, C.; Riehl, A.; Durchdewald, M.; Németh, J.; Fürstenberger, G.; Müller-Decker, K.; Enk, A.; Arnold, B.; Bierhaus, A.; Nawroth, P.P.; et al. RAGE Signaling Sustains Inflammation and Promotes Tumor Development. J. Exp. Med. 2008, 205, 275–285. [Google Scholar] [CrossRef]
- Lawrence, T. The Nuclear Factor NF-KappaB Pathway in Inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [PubMed]
- Mancino, A.; Lawrence, T. Nuclear Factor-KappaB and Tumor-Associated Macrophages. Clin. Cancer Res. 2010, 16, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-ΚB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, K.; Saung, M.T.; Jing, H.; Herbst, B.; Zarecki, M.; Muth, S.; Wu, A.; Bigelow, E.; Chen, L.; Li, K.; et al. Interrogating the Immune-Modulating Roles of Radiation Therapy for a Rational Combination with Immune-Checkpoint Inhibitors in Treating Pancreatic Cancer. J. Immunother. Cancer 2020, 8, e000351. [Google Scholar] [CrossRef]
- Demaria, S.; Bhardwaj, N.; McBride, W.H.; Formenti, S.C. Combining Radiotherapy and Immunotherapy: A Revived Partnership. Int. J. Radiat. Oncol. Biol. Phys. 2005, 63, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-C.; Chen, K.-C.; Chang, G.-C.; Lin, H.; Wu, C.-C.; Kao, W.-H.; Teng, C.-L.J.; Hsu, S.-L.; Yang, T.-Y. RAGE Acts as an Oncogenic Role and Promotes the Metastasis of Human Lung Cancer. Cell Death Dis. 2020, 11, 265. [Google Scholar] [CrossRef] [PubMed]
- Galasko, D.; Bell, J.; Mancuso, J.Y.; Kupiec, J.W.; Sabbagh, M.N.; van Dyck, C.; Thomas, R.G.; Aisen, P.S. Alzheimer’s Disease Cooperative Study Clinical Trial of an Inhibitor of RAGE-Aβ Interactions in Alzheimer Disease. Neurology 2014, 82, 1536–1542. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alka, K.; Oyeniyi, J.F.; Mohammad, G.; Zhao, Y.; Marcus, S.; Chinnaiyan, P. The RAGE Inhibitor TTP488 (Azeliragon) Demonstrates Anti-Tumor Activity and Enhances the Efficacy of Radiation Therapy in Pancreatic Cancer Cell Lines. Cancers 2025, 17, 17. https://doi.org/10.3390/cancers17010017
Alka K, Oyeniyi JF, Mohammad G, Zhao Y, Marcus S, Chinnaiyan P. The RAGE Inhibitor TTP488 (Azeliragon) Demonstrates Anti-Tumor Activity and Enhances the Efficacy of Radiation Therapy in Pancreatic Cancer Cell Lines. Cancers. 2025; 17(1):17. https://doi.org/10.3390/cancers17010017
Chicago/Turabian StyleAlka, Kumari, Jacob F. Oyeniyi, Ghulam Mohammad, Yi Zhao, Stephen Marcus, and Prakash Chinnaiyan. 2025. "The RAGE Inhibitor TTP488 (Azeliragon) Demonstrates Anti-Tumor Activity and Enhances the Efficacy of Radiation Therapy in Pancreatic Cancer Cell Lines" Cancers 17, no. 1: 17. https://doi.org/10.3390/cancers17010017
APA StyleAlka, K., Oyeniyi, J. F., Mohammad, G., Zhao, Y., Marcus, S., & Chinnaiyan, P. (2025). The RAGE Inhibitor TTP488 (Azeliragon) Demonstrates Anti-Tumor Activity and Enhances the Efficacy of Radiation Therapy in Pancreatic Cancer Cell Lines. Cancers, 17(1), 17. https://doi.org/10.3390/cancers17010017