Macrophages Orchestrate the Liver Tumor Microenvironment



Abstract
Simple Summary
Abstract
1. Introduction
2. Liver Macrophages
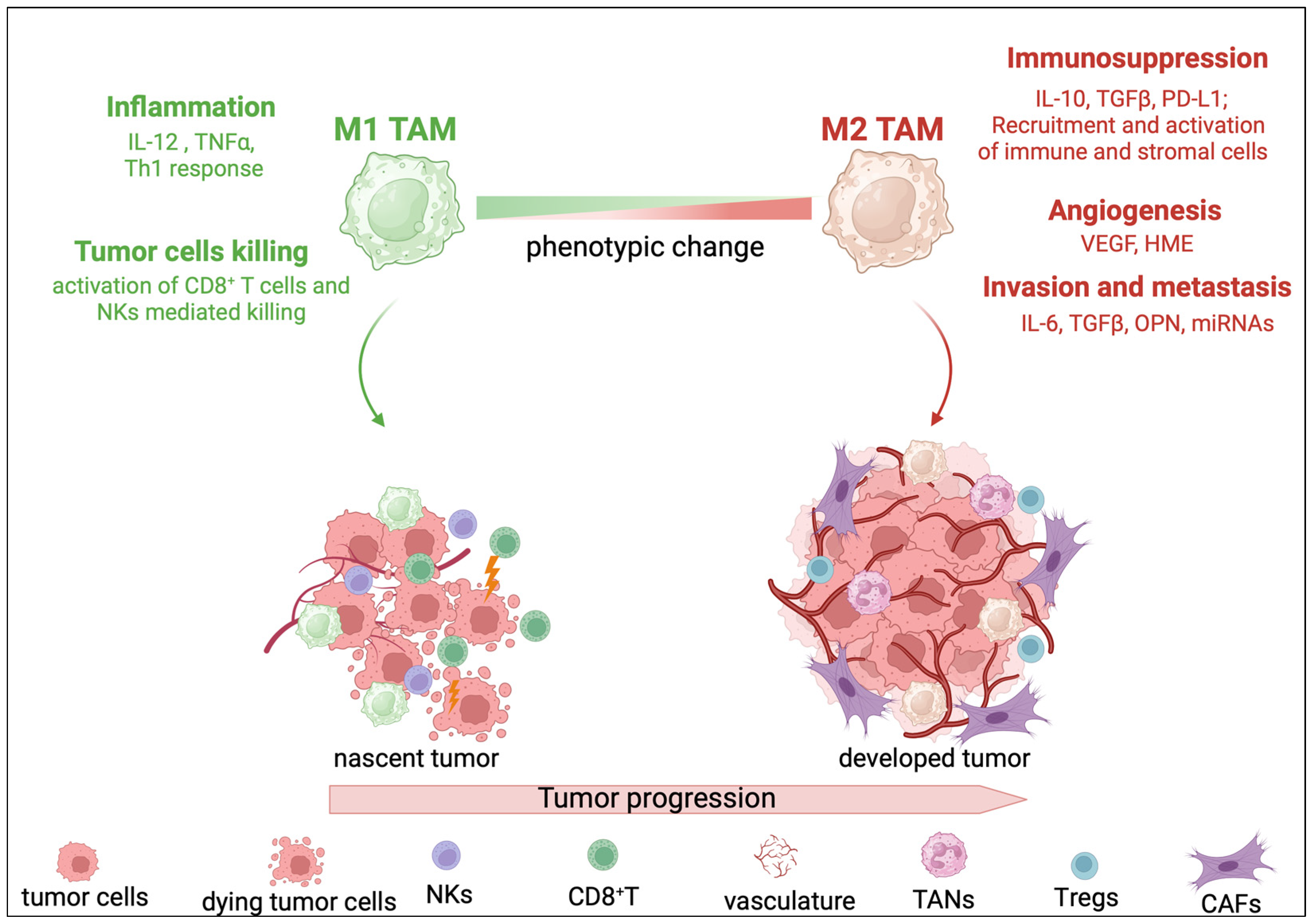
3. TAMs in Liver Cancer
4. TAM Modulation of the Liver TME
4.1. TAMs and Tumor Cells’ Cross-Talk
4.2. TAMs and Immune Cells’ Cross-Talk
4.3. TAMs and LSECs’ Cross-Talk
4.4. TAMs and CAFs’ Cross-Talk
5. Metabolic Regulation of TAMs
6. Microbiome Regulation of TAMs
7. The Epigenetic Regulation of TAMs
8. Targeting TAMs
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular Carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Banales, J.M.; Marin, J.J.G.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Cholangiocarcinoma 2020: The next Horizon in Mechanisms and Management. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 557–588. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ramadori, P.; Pfister, D.; Seehawer, M.; Zender, L.; Heikenwalder, M. The Immunological and Metabolic Landscape in Primary and Metastatic Liver Cancer. Nat. Rev. Cancer 2021, 21, 541–557. [Google Scholar] [CrossRef] [PubMed]
- Bridgewater, J.; Galle, P.R.; Khan, S.A.; Llovet, J.M.; Park, J.-W.; Patel, T.; Pawlik, T.M.; Gores, G.J. Guidelines for the Diagnosis and Management of Intrahepatic Cholangiocarcinoma. J. Hepatol. 2014, 60, 1268–1289. [Google Scholar] [CrossRef] [PubMed]
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular Carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef] [PubMed]
- Sarcognato, S.; Sacchi, D.; Fassan, M.; Fabris, L.; Cadamuro, M.; Zanus, G.; Cataldo, I.; Capelli, P.; Baciorri, F.; Cacciatore, M.; et al. Cholangiocarcinoma. Pathologica 2021, 113, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Roayaie, S.; Jibara, G.; Tabrizian, P.; Park, J.; Yang, J.; Yan, L.; Schwartz, M.; Han, G.; Izzo, F.; Chen, M.; et al. The Role of Hepatic Resection in the Treatment of Hepatocellular Cancer. Hepatology 2015, 62, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Shin, J.; Kim, D.Y.; Choi, G.H.; Kim, M.-J.; Choi, J.-Y. Postoperative Recurrence of Hepatocellular Carcinoma: The Importance of Distinguishing between Intrahepatic Metastasis and Multicentric Occurrence—Response. Clin. Cancer Res. 2019, 25, 5427. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Nault, J.-C.; Cheng, A.-L.; Sangro, B.; Llovet, J.M. Milestones in the Pathogenesis and Management of Primary Liver Cancer. J. Hepatol. 2020, 72, 209–214. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.-W.; Han, G.; Jassem, J.; et al. Lenvatinib versus Sorafenib in First-Line Treatment of Patients with Unresectable Hepatocellular Carcinoma: A Randomised Phase 3 Non-Inferiority Trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef] [PubMed]
- El-Diwany, R.; Pawlik, T.M.; Ejaz, A. Intrahepatic Cholangiocarcinoma. Surg. Oncol. Clin. N. Am. 2019, 28, 587–599. [Google Scholar] [CrossRef]
- Høgdall, D.; Lewinska, M.; Andersen, J.B. Desmoplastic Tumor Microenvironment and Immunotherapy in Cholangiocarcinoma. Trends Cancer 2018, 4, 239–255. [Google Scholar] [CrossRef]
- Banales, J.M.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Fouassier, L.; et al. Cholangiocarcinoma: Current Knowledge and Future Perspectives Consensus Statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef]
- Llovet, J.M.; Castet, F.; Heikenwalder, M.; Maini, M.K.; Mazzaferro, V.; Pinato, D.J.; Pikarsky, E.; Zhu, A.X.; Finn, R.S. Immunotherapies for Hepatocellular Carcinoma. Nat. Rev. Clin. Oncol. 2022, 19, 151–172. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental Regulation of Tumor Progression and Metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Pietras, K.; Östman, A. Hallmarks of Cancer: Interactions with the Tumor Stroma. Exp. Cell Res. 2010, 316, 1324–1331. [Google Scholar] [CrossRef]
- Atanasov, G.; Hau, H.-M.; Dietel, C.; Benzing, C.; Krenzien, F.; Brandl, A.; Wiltberger, G.; Matia, I.; Prager, I.; Schierle, K.; et al. Prognostic Significance of Macrophage Invasion in Hilar Cholangiocarcinoma. BMC Cancer 2015, 15, 790. [Google Scholar] [CrossRef] [PubMed]
- Horst, A.K.; Neumann, K.; Diehl, L.; Tiegs, G. Modulation of Liver Tolerance by Conventional and Nonconventional Antigen-Presenting Cells and Regulatory Immune Cells. Cell Mol. Immunol. 2016, 13, 277–292. [Google Scholar] [CrossRef]
- Protzer, U.; Maini, M.K.; Knolle, P.A. Living in the Liver: Hepatic Infections. Nat. Rev. Immunol. 2012, 12, 201–213. [Google Scholar] [CrossRef]
- Blériot, C.; Ginhoux, F. Understanding the Heterogeneity of Resident Liver Macrophages. Front. Immunol. 2019, 10, 2694. [Google Scholar] [CrossRef] [PubMed]
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; de Bruijn, M.F.; Geissmann, F.; et al. Tissue-Resident Macrophages Originate from Yolk-Sac-Derived Erythro-Myeloid Progenitors. Nature 2015, 518, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Hoeffel, G.; Chen, J.; Lavin, Y.; Low, D.; Almeida, F.F.; See, P.; Beaudin, A.E.; Lum, J.; Low, I.; Forsberg, E.C.; et al. C-Myb+ Erythro-Myeloid Progenitor-Derived Fetal Monocytes Give Rise to Adult Tissue-Resident Macrophages. Immunity 2015, 42, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Mass, E.; Ballesteros, I.; Farlik, M.; Halbritter, F.; Günther, P.; Crozet, L.; Jacome-Galarza, C.E.; Händler, K.; Klughammer, J.; Kobayashi, Y.; et al. Specification of Tissue-Resident Macrophages during Organogenesis. Science 2016, 353, aaf4238. [Google Scholar] [CrossRef] [PubMed]
- MacParland, S.A.; Liu, J.C.; Ma, X.-Z.; Innes, B.T.; Bartczak, A.M.; Gage, B.K.; Manuel, J.; Khuu, N.; Echeverri, J.; Linares, I.; et al. Single Cell RNA Sequencing of Human Liver Reveals Distinct Intrahepatic Macrophage Populations. Nat. Commun. 2018, 9, 4383. [Google Scholar] [CrossRef]
- Bain, C.C.; Hawley, C.A.; Garner, H.; Scott, C.L.; Schridde, A.; Steers, N.J.; Mack, M.; Joshi, A.; Guilliams, M.; Mowat, A.M.I.; et al. Long-Lived Self-Renewing Bone Marrow-Derived Macrophages Displace Embryo-Derived Cells to Inhabit Adult Serous Cavities. Nat. Commun. 2016, 7, ncomms11852. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-Resident Macrophages Self-Maintain Locally throughout Adult Life with Minimal Contribution from Circulating Monocytes. Immunity 2013, 38, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Yona, S.; Kim, K.-W.; Wolf, Y.; Mildner, A.; Varol, D.; Breker, M.; Strauss-Ayali, D.; Viukov, S.; Guilliams, M.; Misharin, A.; et al. Fate Mapping Reveals Origins and Dynamics of Monocytes and Tissue Macrophages under Homeostasis. Immunity 2013, 38, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Sakai, M.; Troutman, T.D.; Seidman, J.S.; Ouyang, Z.; Spann, N.J.; Abe, Y.; Ego, K.M.; Bruni, C.M.; Deng, Z.; Schlachetzki, J.C.M.; et al. Liver-Derived Signals Sequentially Reprogram Myeloid Enhancers to Initiate and Maintain Kupffer Cell Identity. Immunity 2019, 51, 655–670.e8. [Google Scholar] [CrossRef]
- Scott, C.L.; Zheng, F.; De Baetselier, P.; Martens, L.; Saeys, Y.; De Prijck, S.; Lippens, S.; Abels, C.; Schoonooghe, S.; Raes, G.; et al. Bone Marrow-Derived Monocytes Give Rise to Self-Renewing and Fully Differentiated Kupffer Cells. Nat. Commun. 2016, 7, 10321. [Google Scholar] [CrossRef]
- Borst, K.; Frenz, T.; Spanier, J.; Tegtmeyer, P.-K.; Chhatbar, C.; Skerra, J.; Ghita, L.; Namineni, S.; Lienenklaus, S.; Köster, M.; et al. Type I Interferon Receptor Signaling Delays Kupffer Cell Replenishment during Acute Fulminant Viral Hepatitis. J. Hepatol. 2018, 68, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Remmerie, A.; Martens, L.; Thoné, T.; Castoldi, A.; Seurinck, R.; Pavie, B.; Roels, J.; Vanneste, B.; De Prijck, S.; Vanhockerhout, M.; et al. Osteopontin Expression Identifies a Subset of Recruited Macrophages Distinct from Kupffer Cells in the Fatty Liver. Immunity 2020, 53, 641–657.e14. [Google Scholar] [CrossRef] [PubMed]
- Seidman, J.S.; Troutman, T.D.; Sakai, M.; Gola, A.; Spann, N.J.; Bennett, H.; Bruni, C.M.; Ouyang, Z.; Li, R.Z.; Sun, X.; et al. Niche-Specific Reprogramming of Epigenetic Landscapes Drives Myeloid Cell Diversity in Nonalcoholic Steatohepatitis. Immunity 2020, 52, 1057–1074.e7. [Google Scholar] [CrossRef] [PubMed]
- Bonnardel, J.; T’Jonck, W.; Gaublomme, D.; Browaeys, R.; Scott, C.L.; Martens, L.; Vanneste, B.; De Prijck, S.; Nedospasov, S.A.; Kremer, A.; et al. Stellate Cells, Hepatocytes, and Endothelial Cells Imprint the Kupffer Cell Identity on Monocytes Colonizing the Liver Macrophage Niche. Immunity 2019, 51, 638–654.e9. [Google Scholar] [CrossRef] [PubMed]
- Papachristoforou, E.; Ramachandran, P. Macrophages as Key Regulators of Liver Health and Disease. Int. Rev. Cell Mol. Biol. 2022, 368, 143–212. [Google Scholar] [PubMed]
- Guillot, A.; Tacke, F. Liver Macrophages: Old Dogmas and New Insights. Hepatol. Commun. 2019, 3, 730–743. [Google Scholar] [CrossRef] [PubMed]
- Koh, M.Y.; Gagea, M.; Sargis, T.; Lemos, R.; Grandjean, G.; Charbono, A.; Bekiaris, V.; Sedy, J.; Kiriakova, G.; Liu, X.; et al. A New HIF-1α/RANTES-driven Pathway to Hepatocellular Carcinoma Mediated by Germline Haploinsufficiency of SART1/HAF in Mice. Hepatology 2016, 63, 1576–1591. [Google Scholar] [CrossRef] [PubMed]
- Shim, Y.-R.; Jeong, W.-I. Recent Advances of Sterile Inflammation and Inter-Organ Cross-Talk in Alcoholic Liver Disease. Exp. Mol. Med. 2020, 52, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Shim, Y.; Seo, W.; Kim, M.; Choi, W.; Kim, H.; Kim, Y.E.; Yang, K.; Ryu, T.; Jeong, J.; et al. Mitochondrial Double-Stranded RNA in Exosome Promotes Interleukin-17 Production Through Toll-Like Receptor 3 in Alcohol-associated Liver Injury. Hepatology 2020, 72, 609–625. [Google Scholar] [CrossRef]
- Marra, F.; Tacke, F. Roles for Chemokines in Liver Disease. Gastroenterology 2014, 147, 577–594.e1. [Google Scholar] [CrossRef]
- Dambach, D.M.; Watson, L.M.; Gray, K.R.; Durham, S.K.; Laskin, D.L. Role of CCR2 in Macrophage Migration into the Liver during Acetaminophen-Induced Hepatotoxicity in the Mouse. Hepatology 2002, 35, 1093–1103. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Lambrecht, J.; Ju, C.; Tacke, F. Hepatic Macrophages in Liver Homeostasis and Diseases-Diversity, Plasticity and Therapeutic Opportunities. Cell Mol. Immunol. 2021, 18, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Ning, H.; Liu, M.; Lin, J.; Luo, S.; Zhu, W.; Xu, J.; Wu, W.-C.; Liang, J.; Shao, C.-K.; et al. Spleen Mediates a Distinct Hematopoietic Progenitor Response Supporting Tumor-Promoting Myelopoiesis. J. Clin. Investig. 2018, 128, 3425–3438. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Shi, Y.; Zhang, M.; Goswami, S.; Afridi, S.; Meng, L.; Ma, J.; Chen, Y.; Lin, Y.; Zhang, J.; et al. Global Immune Characterization of HBV/HCV-Related Hepatocellular Carcinoma Identifies Macrophage and T-Cell Subsets Associated with Disease Progression. Cell Discov. 2020, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Seow, J.J.W.; Dutertre, C.-A.; Pai, R.; Blériot, C.; Mishra, A.; Wong, R.M.M.; Singh, G.S.N.; Sudhagar, S.; Khalilnezhad, S.; et al. Onco-Fetal Reprogramming of Endothelial Cells Drives Immunosuppressive Macrophages in Hepatocellular Carcinoma. Cell 2020, 183, 377–394.e21. [Google Scholar] [CrossRef] [PubMed]
- Noy, R.; Pollard, J.W. Tumor-Associated Macrophages: From Mechanisms to Therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wu, L.; Zhong, Y.; Zhou, K.; Hou, Y.; Wang, Z.; Zhang, Z.; Xie, J.; Wang, C.; Chen, D.; et al. Single-Cell Landscape of the Ecosystem in Early-Relapse Hepatocellular Carcinoma. Cell 2021, 184, 404–421.e16. [Google Scholar] [CrossRef] [PubMed]
- Hasita, H.; Komohara, Y.; Okabe, H.; Masuda, T.; Ohnishi, K.; Lei, X.F.; Beppu, T.; Baba, H.; Takeya, M. Significance of Alternatively Activated Macrophages in Patients with Intrahepatic Cholangiocarcinoma. Cancer Sci. 2010, 101, 1913–1919. [Google Scholar] [CrossRef] [PubMed]
- MANTOVANI, A.; SOZZANI, S.; LOCATI, M.; ALLAVENA, P.; SICA, A. Macrophage Polarization: Tumor-Associated Macrophages as a Paradigm for Polarized M2 Mononuclear Phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Ruffell, B. Macrophages as Regulators of Tumour Immunity and Immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef]
- Gorrin-Rivas, M.J.; Arii, S.; Mori, A.; Takeda, Y.; Mizumoto, M.; Furutani, M.; Imamura, M. Implications of Human Macrophage Metalloelastase and Vascular Endothelial Growth Factor Gene Expression in Angiogenesis of Hepatocellular Carcinoma. Ann. Surg. 2000, 231, 67. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-S.; Heo, J.; Libbrecht, L.; Chu, I.-S.; Kaposi-Novak, P.; Calvisi, D.F.; Mikaelyan, A.; Roberts, L.R.; Demetris, A.J.; Sun, Z.; et al. A Novel Prognostic Subtype of Human Hepatocellular Carcinoma Derived from Hepatic Progenitor Cells. Nat. Med. 2006, 12, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Hoshida, Y.; Villanueva, A.; Kobayashi, M.; Peix, J.; Chiang, D.Y.; Camargo, A.; Gupta, S.; Moore, J.; Wrobel, M.J.; Lerner, J.; et al. Gene Expression in Fixed Tissues and Outcome in Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 1995–2004. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 2017, 66, 1300–1312. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; He, Y.; Luo, N.; Patel, S.J.; Han, Y.; Gao, R.; Modak, M.; Carotta, S.; Haslinger, C.; Kind, D.; et al. Landscape and Dynamics of Single Immune Cells in Hepatocellular Carcinoma. Cell 2019, 179, 829–845.e20. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.; Dobie, R.; Wilson-Kanamori, J.R.; Dora, E.F.; Henderson, B.E.P.; Luu, N.T.; Portman, J.R.; Matchett, K.P.; Brice, M.; Marwick, J.A.; et al. Resolving the Fibrotic Niche of Human Liver Cirrhosis at Single-Cell Level. Nature 2019, 575, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Massalha, H.; Bahar Halpern, K.; Abu-Gazala, S.; Jana, T.; Massasa, E.E.; Moor, A.E.; Buchauer, L.; Rozenberg, M.; Pikarsky, E.; Amit, I.; et al. A Single Cell Atlas of the Human Liver Tumor Microenvironment. Mol. Syst. Biol. 2020, 16, e9682. [Google Scholar] [CrossRef]
- Xu, L.; Yan, M.; Long, J.; Liu, M.; Yang, H.; Li, W. Identification of Macrophage Correlated Biomarkers to Predict the Prognosis in Patients with Intrahepatic Cholangiocarcinoma. Front. Oncol. 2022, 12, 967982. [Google Scholar] [CrossRef] [PubMed]
- Xiao, P.; Long, X.; Zhang, L.; Ye, Y.; Guo, J.; Liu, P.; Zhang, R.; Ning, J.; Yu, W.; Wei, F.; et al. Neurotensin/IL-8 Pathway Orchestrates Local Inflammatory Response and Tumor Invasion by Inducing M2 Polarization of Tumor-Associated Macrophages and Epithelial-Mesenchymal Transition of Hepatocellular Carcinoma Cells. Oncoimmunology 2018, 7, e1440166. [Google Scholar] [CrossRef]
- Zhou, T.; Zhou, Y.; Qian, M.; Fang, Y.; Ye, S.; Xin, W.; Yang, X.; Wu, H. Interleukin-6 Induced by YAP in Hepatocellular Carcinoma Cells Recruits Tumor-Associated Macrophages. J. Pharmacol. Sci. 2018, 138, 89–95. [Google Scholar] [CrossRef]
- He, Q.; Liu, M.; Huang, W.; Chen, X.; Zhang, B.; Zhang, T.; Wang, Y.; Liu, D.; Xie, M.; Ji, X.; et al. IL-1β-Induced Elevation of Solute Carrier Family 7 Member 11 Promotes Hepatocellular Carcinoma Metastasis Through Up-regulating Programmed Death Ligand 1 and Colony-Stimulating Factor 1. Hepatology 2021, 74, 3174–3193. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Yang, J.; Xu, D.; Gao, X.-M.; Zhang, Z.; Hsu, J.L.; Li, C.-W.; Lim, S.-O.; Sheng, Y.-Y.; Zhang, Y.; et al. Disruption of Tumour-Associated Macrophage Trafficking by the Osteopontin-Induced Colony-Stimulating Factor-1 Signalling Sensitises Hepatocellular Carcinoma to Anti-PD-L1 Blockade. Gut 2019, 68, 1653–1666. [Google Scholar] [CrossRef] [PubMed]
- Agirre-Lizaso, A.; Huici-Izagirre, M.; Urretabizkaia-Garmendia, J.; Rodrigues, P.M.; Banales, J.M.; Perugorria, M.J. Targeting the Heterogeneous Tumour-Associated Macrophages in Hepatocellular Carcinoma. Cancers 2023, 15, 4977. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pan, J.; Zheng, S.; Cai, D.; Luo, A.; Xia, Z.; Huang, J. Hepatocellular Carcinoma Cell-Derived Exosomal MiR-21-5p Induces Macrophage M2 Polarization by Targeting RhoB. Int. J. Mol. Sci. 2023, 24, 4593. [Google Scholar] [CrossRef] [PubMed]
- Zongqiang, H.; Jiapeng, C.; Yingpeng, Z.; Chuntao, Y.; Yiting, W.; Jiashun, Z.; Li, L. Exosomal MiR-452-5p Induce M2 Macrophage Polarization to Accelerate Hepatocellular Carcinoma Progression by Targeting TIMP3. J. Immunol. Res. 2022, 2022, 1032106. [Google Scholar] [CrossRef] [PubMed]
- Ning, J.; Ye, Y.; Bu, D.; Zhao, G.; Song, T.; Liu, P.; Yu, W.; Wang, H.; Li, H.; Ren, X.; et al. Imbalance of TGF-Β1/BMP-7 Pathways Induced by M2-Polarized Macrophages Promotes Hepatocellular Carcinoma Aggressiveness. Mol. Ther. 2021, 29, 2067–2087. [Google Scholar] [CrossRef]
- Luo, C.; Xin, H.; Zhou, Z.; Hu, Z.; Sun, R.; Yao, N.; Sun, Q.; Borjigin, U.; Wu, X.; Fan, J.; et al. Tumor-derived Exosomes Induce Immunosuppressive Macrophages to Foster Intrahepatic Cholangiocarcinoma Progression. Hepatology 2022, 76, 982–999. [Google Scholar] [CrossRef]
- Wang, K.C.; Chang, H.Y. Molecular Mechanisms of Long Noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef]
- Lecerf, C.; Peperstraete, E.; Le Bourhis, X.; Adriaenssens, E. Propagation and Maintenance of Cancer Stem Cells: A Major Influence of the Long Non-Coding RNA H19. Cells 2020, 9, 2613. [Google Scholar] [CrossRef]
- Yeung, O.W.H.; Lo, C.-M.; Ling, C.-C.; Qi, X.; Geng, W.; Li, C.-X.; Ng, K.T.P.; Forbes, S.J.; Guan, X.-Y.; Poon, R.T.P.; et al. Alternatively Activated (M2) Macrophages Promote Tumour Growth and Invasiveness in Hepatocellular Carcinoma. J. Hepatol. 2015, 62, 607–616. [Google Scholar] [CrossRef]
- Chen, Y.; Wen, H.; Zhou, C.; Su, Q.; Lin, Y.; Xie, Y.; Huang, Y.; Qiu, Q.; Lin, J.; Huang, X.; et al. TNF-α Derived from M2 Tumor-Associated Macrophages Promotes Epithelial-Mesenchymal Transition and Cancer Stemness through the Wnt/β-Catenin Pathway in SMMC-7721 Hepatocellular Carcinoma Cells. Exp. Cell Res. 2019, 378, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Li, X.; Chen, S.; Zeng, Q.; Zhao, Y.; Luo, F. Tumor-Associated Macrophage or Chemokine Ligand CCL17 Positively Regulates the Tumorigenesis of Hepatocellular Carcinoma. Med. Oncol. 2016, 33, 17. [Google Scholar] [CrossRef] [PubMed]
- Boulter, L.; Guest, R.V.; Kendall, T.J.; Wilson, D.H.; Wojtacha, D.; Robson, A.J.; Ridgway, R.A.; Samuel, K.; Van Rooijen, N.; Barry, S.T.; et al. WNT Signaling Drives Cholangiocarcinoma Growth and Can Be Pharmacologically Inhibited. J. Clin. Investig. 2015, 125, 1269–1285. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Luo, T.; Dong, P.; Zhang, N.; Chen, J.; Zhang, S.; Dong, L.; Janssen, H.L.A.; Zhang, S. M2-polarized Tumor-associated Macrophages Promote Epithelial-mesenchymal Transition via Activation of the AKT3/PRAS40 Signaling Pathway in Intrahepatic Cholangiocarcinoma. J. Cell Biochem. 2020, 121, 2828–2838. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, H.; Li, Z.; Chen, S.; Huang, X.; Zheng, Z.; Qian, X.; Zhang, L.; Long, G.; Xie, J.; et al. SHH/GLI2-TGF-Β1 Feedback Loop between Cancer Cells and Tumor-Associated Macrophages Maintains Epithelial-Mesenchymal Transition and Endoplasmic Reticulum Homeostasis in Cholangiocarcinoma. Pharmacol. Res. 2023, 187, 106564. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Lin, Z.; Liu, Y.; Jiang, Y.; Liu, K.; Tu, M.; Yao, N.; Qu, C.; Hong, J. Intrahepatic Cholangiocarcinoma Induced M2-Polarized Tumor-Associated Macrophages Facilitate Tumor Growth and Invasiveness. Cancer Cell Int. 2020, 20, 586. [Google Scholar] [CrossRef] [PubMed]
- Quaranta, V.; Schmid, M.C. Macrophage-Mediated Subversion of Anti-Tumour Immunity. Cells 2019, 8, 747. [Google Scholar] [CrossRef] [PubMed]
- Xiao, N.; Zhu, X.; Li, K.; Chen, Y.; Liu, X.; Xu, B.; Lei, M.; Xu, J.; Sun, H.-C. Blocking Siglec-10hi Tumor-Associated Macrophages Improves Anti-Tumor Immunity and Enhances Immunotherapy for Hepatocellular Carcinoma. Exp. Hematol. Oncol. 2021, 10, 36. [Google Scholar] [CrossRef]
- Ren, X.; Ju, Y.; Wang, C.; Wei, R.; Sun, H.; Zhang, Q. MARCKS on Tumor-Associated Macrophages Is Correlated with Immune Infiltrates and Poor Prognosis in Hepatocellular Carcinoma. Cancer Investig. 2021, 39, 756–768. [Google Scholar] [CrossRef]
- Borrego, F.; Ulbrecht, M.; Weiss, E.H.; Coligan, J.E.; Brooks, A.G. Recognition of Human Histocompatibility Leukocyte Antigen (HLA)-E Complexed with HLA Class I Signal Sequence–Derived Peptides by CD94/NKG2 Confers Protection from Natural Killer Cell–Mediated Lysis. J. Exp. Med. 1998, 187, 813–818. [Google Scholar] [CrossRef]
- Lu, C.; Rong, D.; Zhang, B.; Zheng, W.; Wang, X.; Chen, Z.; Tang, W. Current Perspectives on the Immunosuppressive Tumor Microenvironment in Hepatocellular Carcinoma: Challenges and Opportunities. Mol. Cancer 2019, 18, 130. [Google Scholar] [CrossRef] [PubMed]
- Kuang, D.-M.; Zhao, Q.; Peng, C.; Xu, J.; Zhang, J.-P.; Wu, C.; Zheng, L. Activated Monocytes in Peritumoral Stroma of Hepatocellular Carcinoma Foster Immune Privilege and Disease Progression through PD-L1. J. Exp. Med. 2009, 206, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zheng, B.; Goswami, S.; Meng, L.; Zhang, D.; Cao, C.; Li, T.; Zhu, F.; Ma, L.; Zhang, Z.; et al. PD1Hi CD8+ T Cells Correlate with Exhausted Signature and Poor Clinical Outcome in Hepatocellular Carcinoma. J. Immunother. Cancer 2019, 7, 331. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, Y.; Yan, Z.; Yang, H.; Sun, W.; Yao, Y.; Chen, Y.; Jiang, R. IL-6 Promotes PD-L1 Expression in Monocytes and Macrophages by Decreasing Protein Tyrosine Phosphatase Receptor Type O Expression in Human Hepatocellular Carcinoma. J. Immunother. Cancer 2020, 8, e000285. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Fan, L.; Yu, H.; Zhang, J.; He, Y.; Feng, D.; Wang, F.; Li, X.; Liu, Q.; Li, Y.; et al. Endoplasmic Reticulum Stress Causes Liver Cancer Cells to Release Exosomal MiR-23a-3p and Up-regulate Programmed Death Ligand 1 Expression in Macrophages. Hepatology 2019, 70, 241–258. [Google Scholar] [CrossRef] [PubMed]
- Walter, D.; Herrmann, E.; Schnitzbauer, A.A.; Zeuzem, S.; Hansmann, M.L.; Peveling-Oberhag, J.; Hartmann, S. <scp>PD</Scp> -L1 Expression in Extrahepatic Cholangiocarcinoma. Histopathology 2017, 71, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Loeuillard, E.; Yang, J.; Buckarma, E.; Wang, J.; Liu, Y.; Conboy, C.; Pavelko, K.D.; Li, Y.; O’Brien, D.; Wang, C.; et al. Targeting Tumor-Associated Macrophages and Granulocytic Myeloid-Derived Suppressor Cells Augments PD-1 Blockade in Cholangiocarcinoma. J. Clin. Investig. 2020, 130, 5380–5396. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Yang, L.; Yue, D.; Cao, L.; Li, L.; Wang, D.; Ping, Y.; Shen, Z.; Zheng, Y.; Wang, L.; et al. Macrophage-Derived CCL22 Promotes an Immunosuppressive Tumor Microenvironment via IL-8 in Malignant Pleural Effusion. Cancer Lett. 2019, 452, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Zhou, W.; Yin, S.; Zhou, Y.; Chen, T.; Qian, J.; Su, R.; Hong, L.; Lu, H.; Zhang, F.; et al. Blocking Triggering Receptor Expressed on Myeloid Cells-1-Positive Tumor-Associated Macrophages Induced by Hypoxia Reverses Immunosuppression and Anti-Programmed Cell Death Ligand 1 Resistance in Liver Cancer. Hepatology 2019, 70, 198–214. [Google Scholar] [CrossRef]
- Zhou, J.; Ding, T.; Pan, W.; Zhu, L.; Li, L.; Zheng, L. Increased Intratumoral Regulatory T Cells Are Related to Intratumoral Macrophages and Poor Prognosis in Hepatocellular Carcinoma Patients. Int. J. Cancer 2009, 125, 1640–1648. [Google Scholar] [CrossRef]
- Sica, A.; Bronte, V. Altered Macrophage Differentiation and Immune Dysfunction in Tumor Development. J. Clin. Investig. 2007, 117, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Kuang, D.-M.; Pan, W.-D.; Wan, Y.-L.; Lao, X.-M.; Wang, D.; Li, X.-F.; Zheng, L. Monocyte/Macrophage-Elicited Natural Killer Cell Dysfunction in Hepatocellular Carcinoma Is Mediated by CD48/2B4 Interactions. Hepatology 2013, 57, 1107–1116. [Google Scholar] [CrossRef]
- Duan, M.; Goswami, S.; Shi, J.-Y.; Wu, L.-J.; Wang, X.-Y.; Ma, J.-Q.; Zhang, Z.; Shi, Y.; Ma, L.-J.; Zhang, S.; et al. Activated and Exhausted MAIT Cells Foster Disease Progression and Indicate Poor Outcome in Hepatocellular Carcinoma. Clin. Cancer Res. 2019, 25, 3304–3316. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, C.L.; Filipovic, I.; Cornillet, M.; O’Rourke, C.J.; Berglin, L.; Jansson, H.; Sun, D.; Strauss, O.; Hertwig, L.; Johansson, H.; et al. Mucosal-associated Invariant T-cell Tumor Infiltration Predicts Long-term Survival in Cholangiocarcinoma. Hepatology 2022, 75, 1154–1168. [Google Scholar] [CrossRef] [PubMed]
- Ruf, B.; Bruhns, M.; Babaei, S.; Kedei, N.; Ma, L.; Revsine, M.; Benmebarek, M.-R.; Ma, C.; Heinrich, B.; Subramanyam, V.; et al. Tumor-Associated Macrophages Trigger MAIT Cell Dysfunction at the HCC Invasive Margin. Cell 2023, 186, 3686–3705.e32. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Wang, P.; Sun, R.; Li, J.; Hu, Z.; Xin, H.; Luo, C.; Zhou, J.; Fan, J.; Zhou, S. Tumor-Associated Neutrophils and Macrophages Interaction Contributes to Intrahepatic Cholangiocarcinoma Progression by Activating STAT3. J. Immunother. Cancer 2021, 9, e001946. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.-P.; Jiang, Z.-Z.; Guo, H.-F.; Zhou, M.-M.; Huang, Y.-F.; Ning, W.-R.; Huang, J.-H.; Zheng, L.; Wu, Y. Glycolytic Activation of Monocytes Regulates the Accumulation and Function of Neutrophils in Human Hepatocellular Carcinoma. J. Hepatol. 2020, 73, 906–917. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.-L.; Zhou, Z.-J.; Hu, Z.-Q.; Huang, X.-W.; Wang, Z.; Chen, E.-B.; Fan, J.; Cao, Y.; Dai, Z.; Zhou, J. Tumor-Associated Neutrophils Recruit Macrophages and T-Regulatory Cells to Promote Progression of Hepatocellular Carcinoma and Resistance to Sorafenib. Gastroenterology 2016, 150, 1646–1658.e17. [Google Scholar] [CrossRef] [PubMed]
- Limmer, A.; Ohl, J.; Kurts, C.; Ljunggren, H.-G.; Reiss, Y.; Groettrup, M.; Momburg, F.; Arnold, B.; Knolle, P.A. Efficient Presentation of Exogenous Antigen by Liver Endothelial Cells to CD8+ T Cells Results in Antigen-Specific T-Cell Tolerance. Nat. Med. 2000, 6, 1348–1354. [Google Scholar] [CrossRef]
- Yoong, K.F.; McNab, G.; Hübscher, S.G.; Adams, D.H. Vascular Adhesion Protein-1 and ICAM-1 Support the Adhesion of Tumor-Infiltrating Lymphocytes to Tumor Endothelium in Human Hepatocellular Carcinoma. J. Immunol. 1998, 160, 3978–3988. [Google Scholar] [CrossRef]
- Wilkinson, A.L.; Qurashi, M.; Shetty, S. The Role of Sinusoidal Endothelial Cells in the Axis of Inflammation and Cancer Within the Liver. Front. Physiol. 2020, 11, 990. [Google Scholar] [CrossRef] [PubMed]
- Rantakari, P.; Jäppinen, N.; Lokka, E.; Mokkala, E.; Gerke, H.; Peuhu, E.; Ivaska, J.; Elima, K.; Auvinen, K.; Salmi, M. Fetal Liver Endothelium Regulates the Seeding of Tissue-Resident Macrophages. Nature 2016, 538, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, T.; Kanto, T.; Kuroda, S.; Yoshio, S.; Higashitani, K.; Kakita, N.; Miyazaki, M.; Sakakibara, M.; Hiramatsu, N.; Kasahara, A.; et al. TIE2-Expressing Monocytes as a Diagnostic Marker for Hepatocellular Carcinoma Correlates with Angiogenesis. Hepatology 2013, 57, 1416–1425. [Google Scholar] [CrossRef] [PubMed]
- Subimerb, C.; Pinlaor, S.; Lulitanond, V.; Khuntikeo, N.; Okada, S.; McGrath, M.S.; Wongkham, S. Circulating CD14+CD16+ Monocyte Levels Predict Tissue Invasive Character of Cholangiocarcinoma. Clin. Exp. Immunol. 2010, 161, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Bartneck, M.; Schrammen, P.L.; Möckel, D.; Govaere, O.; Liepelt, A.; Krenkel, O.; Ergen, C.; McCain, M.V.; Eulberg, D.; Luedde, T.; et al. The CCR2+ Macrophage Subset Promotes Pathogenic Angiogenesis for Tumor Vascularization in Fibrotic Livers. Cell Mol. Gastroenterol. Hepatol. 2019, 7, 371–390. [Google Scholar] [CrossRef] [PubMed]
- Zang, M.; Li, Y.; He, H.; Ding, H.; Chen, K.; Du, J.; Chen, T.; Wu, Z.; Liu, H.; Wang, D.; et al. IL-23 Production of Liver Inflammatory Macrophages to Damaged Hepatocytes Promotes Hepatocellular Carcinoma Development after Chronic Hepatitis B Virus Infection. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2018, 1864, 3759–3770. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The Biology and Function of Fibroblasts in Cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Chuaysri, C.; Thuwajit, P.; Paupairoj, A.; Chau-In, S.; Suthiphongchai, T.; Thuwajit, C. Alpha-Smooth Muscle Actin-Positive Fibroblasts Promote Biliary Cell Proliferation and Correlate with Poor Survival in Cholangiocarcinoma. Oncol. Rep. 2009, 21, 957–969. [Google Scholar] [CrossRef]
- Galbo, P.M.; Zang, X.; Zheng, D. Molecular Features of Cancer-Associated Fibroblast Subtypes and Their Implication on Cancer Pathogenesis, Prognosis, and Immunotherapy Resistance. Clin. Cancer Res. 2021, 27, 2636–2647. [Google Scholar] [CrossRef]
- Yin, Z.; Dong, C.; Jiang, K.; Xu, Z.; Li, R.; Guo, K.; Shao, S.; Wang, L. Heterogeneity of Cancer-Associated Fibroblasts and Roles in the Progression, Prognosis, and Therapy of Hepatocellular Carcinoma. J. Hematol. Oncol. 2019, 12, 101. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of Hepatic Stellate Cell Activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Lin, Y.; Shi, Y.; Li, B.; Liu, W.; Yin, W.; Dang, Y.; Chu, Y.; Fan, J.; He, R. FAP Promotes Immunosuppression by Cancer-Associated Fibroblasts in the Tumor Microenvironment via STAT3–CCL2 Signaling. Cancer Res. 2016, 76, 4124–4135. [Google Scholar] [CrossRef]
- Deng, Y.; Cheng, J.; Fu, B.; Liu, W.; Chen, G.; Zhang, Q.; Yang, Y. Hepatic Carcinoma-Associated Fibroblasts Enhance Immune Suppression by Facilitating the Generation of Myeloid-Derived Suppressor Cells. Oncogene 2017, 36, 1090–1101. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Wei, Y.; Han, D.; Li, Y.; Shi, S.; Jiao, D.; Wu, J.; Zhang, Q.; Shi, C.; Yang, L.; et al. Interaction with CD68 and Regulation of GAS6 Expression by Endosialin in Fibroblasts Drives Recruitment and Polarization of Macrophages in Hepatocellular Carcinoma. Cancer Res. 2020, 80, 3892–3905. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Morine, Y.; Tokuda, K.; Yamada, S.; Saito, Y.; Nishi, M.; Ikemoto, T.; Shimada, M. Cancer-associated Fibroblast-induced M2-polarized Macrophages Promote Hepatocellular Carcinoma Progression via the Plasminogen Activator Inhibitor-1 Pathway. Int. J. Oncol. 2021, 59, 59. [Google Scholar] [CrossRef]
- Liu, Y.; Xun, Z.; Ma, K.; Liang, S.; Li, X.; Zhou, S.; Sun, L.; Liu, Y.; Du, Y.; Guo, X.; et al. Identification of a Tumour Immune Barrier in the HCC Microenvironment That Determines the Efficacy of Immunotherapy. J. Hepatol. 2023, 78, 770–782. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mantovani, A. Orchestration of Metabolism by Macrophages. Cell Metab. 2012, 15, 432–437. [Google Scholar] [CrossRef]
- Hayes, C.; Donohoe, C.L.; Davern, M.; Donlon, N.E. The Oncogenic and Clinical Implications of Lactate Induced Immunosuppression in the Tumour Microenvironment. Cancer Lett. 2021, 500, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional Polarization of Tumour-Associated Macrophages by Tumour-Derived Lactic Acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- de-Brito, N.M.; Duncan-Moretti, J.; da-Costa, H.C.; Saldanha-Gama, R.; Paula-Neto, H.A.; Dorighello, G.G.; Simões, R.L.; Barja-Fidalgo, C. Aerobic Glycolysis Is a Metabolic Requirement to Maintain the M2-like Polarization of Tumor-Associated Macrophages. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2020, 1867, 118604. [Google Scholar] [CrossRef] [PubMed]
- Ning, W.-R.; Jiang, D.; Liu, X.-C.; Huang, Y.-F.; Peng, Z.-P.; Jiang, Z.-Z.; Kang, T.; Zhuang, S.-M.; Wu, Y.; Zheng, L. Carbonic Anhydrase XII Mediates the Survival and Prometastatic Functions of Macrophages in Human Hepatocellular Carcinoma. J. Clin. Investig. 2022, 132, e153110. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer Metabolism: Fatty Acid Oxidation in the Limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, Z.; Wen, H.; Guo, Y.; Xu, F.; Zhu, Q.; Yuan, W.; Luo, R.; Lu, C.; Liu, R.; et al. Immunosuppressive TREM2(+) Macrophages Are Associated with Undesirable Prognosis and Responses to Anti-PD-1 Immunotherapy in Non-Small Cell Lung Cancer. Cancer Immunol. Immunother. 2022, 71, 2511–2522. [Google Scholar] [CrossRef]
- Sathe, A.; Mason, K.; Grimes, S.M.; Zhou, Z.; Lau, B.T.; Bai, X.; Su, A.; Tan, X.; Lee, H.; Suarez, C.J.; et al. Colorectal Cancer Metastases in the Liver Establish Immunosuppressive Spatial Networking between Tumor-Associated SPP1 + Macrophages and Fibroblasts. Clin. Cancer Res. 2023, 29, 244–260. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, H.; Zhao, Z.-B.; Zhu, W.; Feng, P.-P.; Zhu, X.-W.; Gong, J.-P. SIRT4 Silencing in Tumor-Associated Macrophages Promotes HCC Development via PPARδ Signalling-Mediated Alternative Activation of Macrophages. J. Exp. Clin. Cancer Res. 2019, 38, 469. [Google Scholar] [CrossRef]
- Choi, J.; Stradmann-Bellinghausen, B.; Yakubov, E.; Savaskan, N.E.; Régnier-Vigouroux, A. Glioblastoma Cells Induce Differential Glutamatergic Gene Expressions in Human Tumor-Associated Microglia/Macrophages and Monocyte-Derived Macrophages. Cancer Biol. Ther. 2015, 16, 1205–1213. [Google Scholar] [CrossRef]
- Huang, J.; Wu, Q.; Geller, D.A.; Yan, Y. Macrophage Metabolism, Phenotype, Function, and Therapy in Hepatocellular Carcinoma (HCC). J. Transl. Med. 2023, 21, 815. [Google Scholar] [CrossRef]
- Li, S.; Yu, J.; Huber, A.; Kryczek, I.; Wang, Z.; Jiang, L.; Li, X.; Du, W.; Li, G.; Wei, S.; et al. Metabolism Drives Macrophage Heterogeneity in the Tumor Microenvironment. Cell Rep. 2022, 39, 110609. [Google Scholar] [CrossRef]
- Kuchuk, O.; Tuccitto, A.; Citterio, D.; Huber, V.; Camisaschi, C.; Milione, M.; Vergani, B.; Villa, A.; Alison, M.R.; Carradori, S.; et al. PH Regulators to Target the Tumor Immune Microenvironment in Human Hepatocellular Carcinoma. Oncoimmunology 2018, 7, e1445452. [Google Scholar] [CrossRef]
- Wang, R.; Tang, R.; Li, B.; Ma, X.; Schnabl, B.; Tilg, H. Gut Microbiome, Liver Immunology, and Liver Diseases. Cell Mol. Immunol. 2021, 18, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H. Immune Regulation by Microbiome Metabolites. Immunology 2018, 154, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Dapito, D.H.; Mencin, A.; Gwak, G.-Y.; Pradere, J.-P.; Jang, M.-K.; Mederacke, I.; Caviglia, J.M.; Khiabanian, H.; Adeyemi, A.; Bataller, R.; et al. Promotion of Hepatocellular Carcinoma by the Intestinal Microbiota and TLR4. Cancer Cell 2012, 21, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Vespasiani-Gentilucci, U.; Carotti, S.; Perrone, G.; Mazzarelli, C.; Galati, G.; Onetti-Muda, A.; Picardi, A.; Morini, S. Hepatic Toll-like Receptor 4 Expression Is Associated with Portal Inflammation and Fibrosis in Patients with NAFLD. Liver Int. 2015, 35, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.; Ding, Y.; Saedi, N.; Choi, M.; Sridharan, G.V.; Sherr, D.H.; Yarmush, M.L.; Alaniz, R.C.; Jayaraman, A.; Lee, K. Gut Microbiota-Derived Tryptophan Metabolites Modulate Inflammatory Response in Hepatocytes and Macrophages. Cell Rep. 2018, 23, 1099–1111. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Li, H.; Hu, J.; Zheng, J.; Zhou, J.; Botchlett, R.; Matthews, D.; Zeng, T.; Chen, L.; Xiao, X.; et al. Indole Alleviates Diet-Induced Hepatic Steatosis and Inflammation in a Manner Involving Myeloid Cell 6-Phosphofructo-2-Kinase/Fructose-2,6-Biphosphatase 3. Hepatology 2020, 72, 1191–1203. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, A.; Nakatani, A.; Hasegawa, S.; Irie, J.; Ozawa, K.; Tsujimoto, G.; Suganami, T.; Itoh, H.; Kimura, I. The Short Chain Fatty Acid Receptor GPR43 Regulates Inflammatory Signals in Adipose Tissue M2-Type Macrophages. PLoS ONE 2017, 12, e0179696. [Google Scholar] [CrossRef] [PubMed]
- Ohira, H.; Fujioka, Y.; Katagiri, C.; Mamoto, R.; Aoyama-Ishikawa, M.; Amako, K.; Izumi, Y.; Nishiumi, S.; Yoshida, M.; Usami, M.; et al. Butyrate Attenuates Inflammation and Lipolysis Generated by the Interaction of Adipocytes and Macrophages. J. Atheroscler. Thromb. 2013, 20, 425–442. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Chen, J.; Qiao, Y. Epigenetic Modifications in Tumor-Associated Macrophages: A New Perspective for an Old Foe. Front. Immunol. 2022, 13, 836223. [Google Scholar] [CrossRef]
- Wang, F.; Malnassy, G.; Qiu, W. The Epigenetic Regulation of Microenvironment in Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 653037. [Google Scholar] [CrossRef]
- de Groot, A.E.; Pienta, K.J. Epigenetic Control of Macrophage Polarization: Implications for Targeting Tumor-Associated Macrophages. Oncotarget 2018, 9, 20908–20927. [Google Scholar] [CrossRef] [PubMed]
- Cui, B.; Fan, X.; Zhou, D.; He, L.; Li, Y.; Li, D.; Lin, H. CSF1R Methylation Is a Key Regulatory Mechanism of Tumor-associated Macrophages in Hepatocellular Carcinoma. Oncol. Lett. 2020, 20, 1835–1845. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Yang, Y.; Li, C. SIRT1 Inhibits Hepatocellular Carcinoma Metastasis by Promoting M1 Macrophage Polarization via NF-ΚB Pathway. Onco Targets Ther. 2019, 12, 2519–2529. [Google Scholar] [CrossRef]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic Regulation of Gene Expression by Histone Lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef]
- Jin, M.; Cao, W.; Chen, B.; Xiong, M.; Cao, G. Tumor-Derived Lactate Creates a Favorable Niche for Tumor via Supplying Energy Source for Tumor and Modulating the Tumor Microenvironment. Front. Cell Dev. Biol. 2022, 10, 808859. [Google Scholar] [CrossRef]
- Ye, Y.; Xu, Y.; Lai, Y.; He, W.; Li, Y.; Wang, R.; Luo, X.; Chen, R.; Chen, T. Long Non-coding RNA Cox-2 Prevents Immune Evasion and Metastasis of Hepatocellular Carcinoma by Altering M1/M2 Macrophage Polarization. J. Cell Biochem. 2018, 119, 2951–2963. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Sun, P.; Zhang, C.; Li, Z.; Cui, K.; Zhou, W. MiR-98 Modulates Macrophage Polarization and Suppresses the Effects of Tumor-Associated Macrophages on Promoting Invasion and Epithelial–Mesenchymal Transition of Hepatocellular Carcinoma. Cancer Cell Int. 2018, 18, 95. [Google Scholar] [CrossRef]
- Zhou, S.; Hu, Z.; Zhou, Z.; Dai, Z.; Wang, Z.; Cao, Y.; Fan, J.; Huang, X.; Zhou, J. MiR-28-5p-IL-34-macrophage Feedback Loop Modulates Hepatocellular Carcinoma Metastasis. Hepatology 2016, 63, 1560–1575. [Google Scholar] [CrossRef]
- Zhao, J.; Li, H.; Zhao, S.; Wang, E.; Zhu, J.; Feng, D.; Zhu, Y.; Dou, W.; Fan, Q.; Hu, J.; et al. Epigenetic Silencing of MiR-144/451a Cluster Contributes to HCC Progression via Paracrine HGF/MIF-Mediated TAM Remodeling. Mol. Cancer 2021, 20, 46. [Google Scholar] [CrossRef]
- Cassetta, L.; Pollard, J.W. Targeting Macrophages: Therapeutic Approaches in Cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef]
- Li, X.; Yao, W.; Yuan, Y.; Chen, P.; Li, B.; Li, J.; Chu, R.; Song, H.; Xie, D.; Jiang, X.; et al. Targeting of Tumour-Infiltrating Macrophages via CCL2/CCR2 Signalling as a Therapeutic Strategy against Hepatocellular Carcinoma. Gut 2017, 66, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ramjiawan, R.R.; Reiberger, T.; Ng, M.R.; Hato, T.; Huang, Y.; Ochiai, H.; Kitahara, S.; Unan, E.C.; Reddy, T.P.; et al. CXCR4 Inhibition in Tumor Microenvironment Facilitates Anti-programmed Death Receptor-1 Immunotherapy in Sorafenib-treated Hepatocellular Carcinoma in Mice. Hepatology 2015, 61, 1591–1602. [Google Scholar] [CrossRef] [PubMed]
- Song, J.-S.; Chang, C.-C.; Wu, C.-H.; Dinh, T.K.; Jan, J.-J.; Huang, K.-W.; Chou, M.-C.; Shiue, T.-Y.; Yeh, K.-C.; Ke, Y.-Y.; et al. A Highly Selective and Potent CXCR4 Antagonist for Hepatocellular Carcinoma Treatment. Proc. Natl. Acad. Sci. USA 2021, 118, e2015433118. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zheng, D.-X.; Yu, X.-J.; Sun, H.-W.; Xu, Y.-T.; Zhang, Y.-J.; Xu, J. Macrophages Induce CD47 Upregulation via IL-6 and Correlate with Poor Survival in Hepatocellular Carcinoma Patients. Oncoimmunology 2019, 8, e1652540. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kγ Is a Molecular Switch That Controls Immune Suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, A.; Sarker, D.; Reebye, V.; Jarvis, S.; Sodergren, M.H.; Kossenkov, A.; Sanseviero, E.; Raulf, N.; Vasara, J.; Andrikakou, P.; et al. Upregulation of C/EBPα Inhibits Suppressive Activity of Myeloid Cells and Potentiates Antitumor Response in Mice and Patients with Cancer. Clin. Cancer Res. 2021, 27, 5961–5978. [Google Scholar] [CrossRef]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human Chimeric Antigen Receptor Macrophages for Cancer Immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| TAM Target | TAM Targeting Agent | Combination Therapy | Liver Cancer Type | ClinicalTrial.Gov Reference |
|---|---|---|---|---|
| TAM depletion | ||||
| CSF-1/CSF-1R | anti-CSF-1R mAb (Cabiralizumab) | anti-PD-1 mAb (Nivolumab) | HCC | NCT04050462 |
| multi-protein kinase inhibitor (Regorafenib) | anti-PD-1 mAb (Nivolumab) | HCC | NCT04170556 | |
| anti-CSF-1R mAb (SNDX-6352) | anti-PD-L1 mAb (Durvalumab) | iCCA | NCT04301778 | |
| Inhibition of TAM recruitment | ||||
| CCR2/CCR5 | CCR2/CCR5 inhibitor (BMS-813160) | anti-PD-1 mAb (Nivolumab) | HCC | NCT04123379 |
| TAM reprogramming | ||||
| CD47/SIRPα | anti-hu SIRPα Ab | N/A | HCC | NCT02868255 |
| STAT6 | exoASO-STAT6 (CDK-004) | N/A | HCC | NCT05375604 |
| PI3Kγ | PI3Kγ inhibitor (SF1126) | anti-PD-1 mAb (Nivolumab) | HCC | NCT03059147 |
| TLR7/TLR8 | TLR7 agonist (RO7119929) | N/A | HCC and biliary tract cancer | NCT04338685 |
| C/EBPα | Small activating RNA (MTL-CEBA) | Kinase inhibitor (Sorafenib) | HCC | NCT02716012 |
| Chemotherapy and VEGF-A inhibitor | HCC | NCT05097911 | ||
| anti-PD-1 mAb (Pembrolizumab) | HCC and biliary tract cancer | NCT04105335 | ||
| HER2 | CAR-M | N/A | HCC and biliary tract cancer | NCT04660929 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quaranta, V.; Ballarò, C.; Giannelli, G. Macrophages Orchestrate the Liver Tumor Microenvironment. Cancers 2024, 16, 1772. https://doi.org/10.3390/cancers16091772
Quaranta V, Ballarò C, Giannelli G. Macrophages Orchestrate the Liver Tumor Microenvironment. Cancers. 2024; 16(9):1772. https://doi.org/10.3390/cancers16091772
Chicago/Turabian StyleQuaranta, Valeria, Costanza Ballarò, and Gianluigi Giannelli. 2024. "Macrophages Orchestrate the Liver Tumor Microenvironment" Cancers 16, no. 9: 1772. https://doi.org/10.3390/cancers16091772
APA StyleQuaranta, V., Ballarò, C., & Giannelli, G. (2024). Macrophages Orchestrate the Liver Tumor Microenvironment. Cancers, 16(9), 1772. https://doi.org/10.3390/cancers16091772