
Deciphering the Role of BCAR3 in Cancer Progression: Gene Regulation, Signal Transduction, and Therapeutic Implications

Abstract
Simple Summary
Abstract
1. Introduction
2. BCAR3 Gene Expression, mRNA Regulation, and Post-Translational Protein Structure
3. The Intracellular Function of BCAR3
3.1. The Role of BCAR3 in Integrin Signaling and Cancer Cell Migration
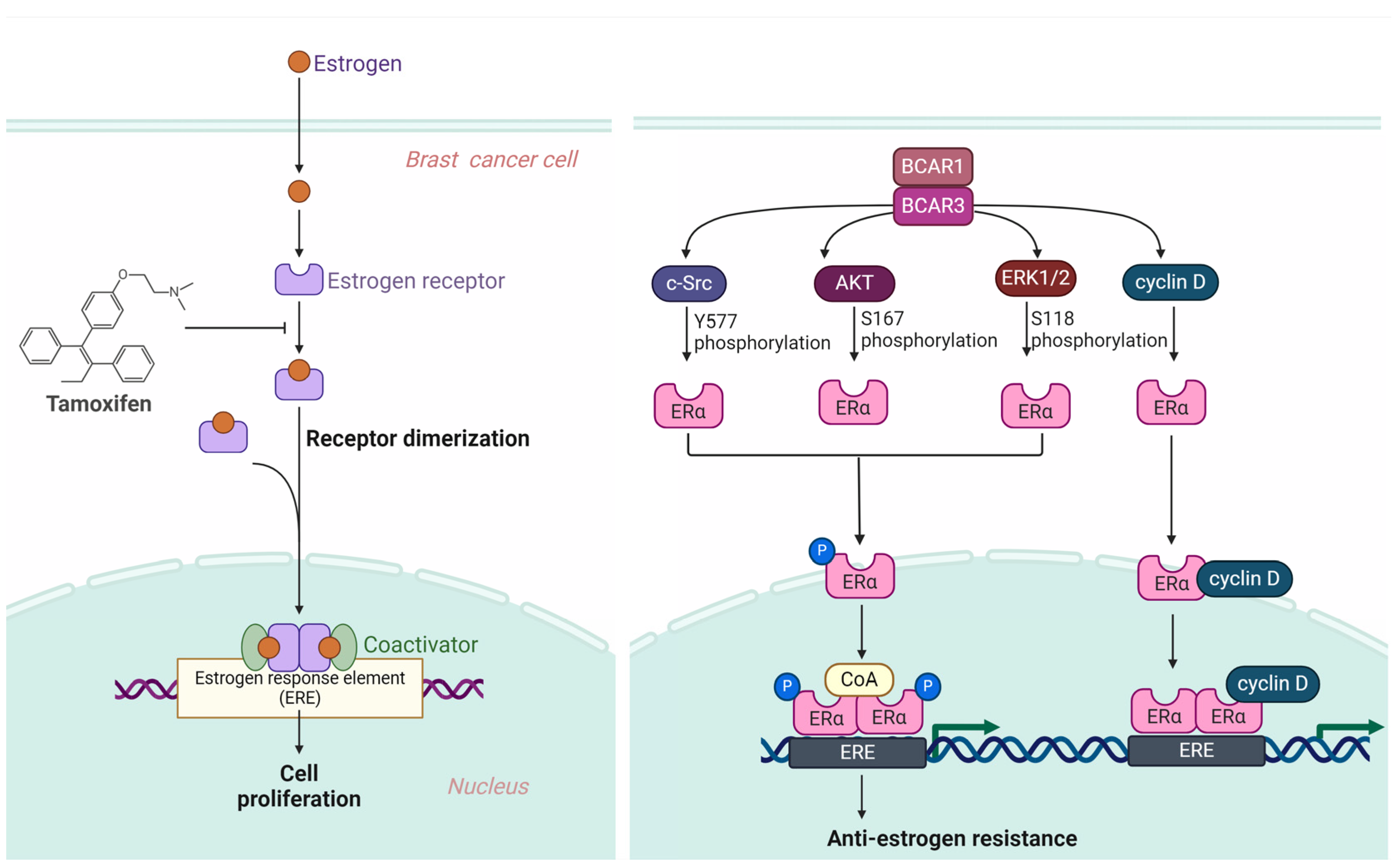
3.2. BCAR3 Triggers Anti-Estrogen Resistance
3.3. Cell Cycle Regulation by BCAR3
3.4. BCAR3 and Immune Regulation in Cancer Therapy
4. Future Research Directions for BCAR3
4.1. Transcription Factor Binding Studies
4.2. Isoform-Specific Expression Profiling
4.3. Therapeutic Targeting
4.4. Exploring the Role of BCAR3 in Immune Cell Function within the Tumor Microenvironment
4.5. Identifying Roles in Cancer Stem Cells
5. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lu, Y.; Brush, J.; Stewart, T.A. NSP1 defines a novel family of adaptor proteins linking integrin and tyrosine kinase receptors to the c-Jun N-terminal kinase/stress-activated protein kinase signaling pathway. J. Biol. Chem. 1999, 274, 10047–10052. [Google Scholar] [CrossRef] [PubMed]
- Steenkiste, E.M.; Berndt, J.D.; Pilling, C.; Simpkins, C.; Cooper, J.A. A Cas-BCAR3 co-regulatory circuit controls lamellipodia dynamics. eLife 2021, 10, e67078. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Cheng, S.Y.; Chen, M.; Lim, C.J.; Pallen, C.J. Protein tyrosine phosphatase α phosphotyrosyl-789 binds BCAR3 to position Cas for activation at integrin-mediated focal adhesions. Mol. Cell. Biol. 2012, 32, 3776–3789. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cai, D.; Clayton, L.K.; Smolyar, A.; Lerner, A. AND-34, a novel p130Cas-binding thymic stromal cell protein regulated by adhesion and inflammatory cytokines. J. Immunol. 1999, 163, 2104–2112. [Google Scholar] [CrossRef] [PubMed]
- van Agthoven, T.; van Agthoven, T.L.; Dekker, A.; van der Spek, P.J.; Vreede, L.; Dorssers, L.C. Identification of BCAR3 by a random search for genes involved in antiestrogen resistance of human breast cancer cells. Embo J. 1998, 17, 2799–2808. [Google Scholar] [CrossRef]
- Vervoort, V.S.; Roselli, S.; Oshima, R.G.; Pasquale, E.B. Splice variants and expression patterns of SHEP1, BCAR3 and NSP1, a gene family involved in integrin and receptor tyrosine kinase signaling. Gene 2007, 391, 161–170. [Google Scholar] [CrossRef]
- Arras, J.; Thomas, K.S.; Myers, P.J.; Cross, A.M.; Osei, A.D.; Vazquez, G.E.; Atkins, K.A.; Conaway, M.R.; Jones, M.K.; Lazzara, M.J.; et al. Breast Cancer Antiestrogen Resistance 3 (BCAR3) promotes tumor growth and progression in triple-negative breast cancer. Am. J. Cancer. Res. 2021, 11, 4768–4787. [Google Scholar] [PubMed]
- Schrecengost, R.S.; Riggins, R.B.; Thomas, K.S.; Guerrero, M.S.; Bouton, A.H. Breast cancer antiestrogen resistance-3 expression regulates breast cancer cell migration through promotion of p130Cas membrane localization and membrane ruffling. Cancer Res. 2007, 67, 6174–6182. [Google Scholar] [CrossRef] [PubMed]
- Pavanelli, A.C.; Mangone, F.R.; Yoganathan, P.; Bessa, S.A.; Nonogaki, S.; de Toledo Osório, C.A.B.; de Andrade, V.P.; Soares, I.C.; de Mello, E.S.; Mulligan, L.M.; et al. Comprehensive immunohistochemical analysis of RET, BCAR1, and BCAR3 expression in patients with Luminal A and B breast cancer subtypes. Breast Cancer Res. Treat. 2022, 192, 43–52. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, Y.; Wang, Y.; Wang, C.; Shuai, Y.; Luo, J.; Liu, R. BCAR3 promotes head and neck cancer growth and is associated with poor prognosis. Cell Death Discov. 2021, 7, 316. [Google Scholar] [CrossRef]
- Zhang, W.; Lin, Y.; Liu, X.; He, X.; Zhang, Y.; Fu, W.; Yang, Z.; Yang, P.; Wang, J.; Hu, K.; et al. Prediction and prognostic significance of BCAR3 expression in patients with multiple myeloma. J. Transl. Med. 2018, 16, 363. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Mi, X.; Liu, N.; Li, X.; Li, X.N.; Yang, Y.; Lu, X.; Fang, Y.; Jin, N.Y. MiR-199a/b-3p inhibits colorectal cancer cell proliferation, migration and invasion through targeting PAK4 and BCAR3. Eur. J. Med. Res. 2022, 27, 121. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Liu, J.; Wang, H.; Chen, P.; Wang, D. MicroRNA-126-5p downregulates BCAR3 expression to promote cell migration and invasion in endometriosis. Mol. Cell Endocrinol. 2019, 494, 110486. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Diebel, K.W.; Holy, J.; Skildum, A.; Odean, E.; Hicks, D.A.; Schotl, B.; Abrahante, J.E.; Spillman, M.A.; Bemis, L.T. A tRNA fragment, tRF5-Glu, regulates BCAR3 expression and proliferation in ovarian cancer cells. Oncotarget 2017, 8, 95377–95391. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Iyer, A.; Felekkis, K.N.; Near, R.I.; Luo, Z.; Chernoff, J.; Albanese, C.; Pestell, R.G.; Lerner, A. AND-34/BCAR3, a GDP exchange factor whose overexpression confers antiestrogen resistance, activates Rac, PAK1, and the cyclin D1 promoter. Cancer Res. 2003, 63, 6802–6808. [Google Scholar] [PubMed]
- Schuh, N.R.; Guerrero, M.S.; Schrecengost, R.S.; Bouton, A.H. BCAR3 regulates Src/p130 Cas association, Src kinase activity, and breast cancer adhesion signaling. J. Biol. Chem. 2010, 285, 2309–2317. [Google Scholar] [CrossRef] [PubMed]
- Dodelet, V.C.; Pazzagli, C.; Zisch, A.H.; Hauser, C.A.; Pasquale, E.B. A novel signaling intermediate, SHEP1, directly couples Eph receptors to R-Ras and Rap1A. J. Biol. Chem. 1999, 274, 31941–31946. [Google Scholar] [CrossRef] [PubMed]
- Machida, K.; Mayer, B.J. The SH2 domain: Versatile signaling module and pharmaceutical target. Biochim. Biophys. Acta 2005, 1747, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Schaffhausen, B. SH2 domain structure and function. Biochim. Biophys. Acta 1995, 1242, 61–75. [Google Scholar] [CrossRef]
- Gotoh, T.; Cai, D.; Tian, X.; Feig, L.A.; Lerner, A. p130Cas regulates the activity of AND-34, a novel Ral, Rap1, and R-Ras guanine nucleotide exchange factor. J. Biol. Chem. 2000, 275, 30118–30123. [Google Scholar] [CrossRef]
- Wallez, Y.; Riedl, S.J.; Pasquale, E.B. Association of the breast cancer antiestrogen resistance protein 1 (BCAR1) and BCAR3 scaffolding proteins in cell signaling and antiestrogen resistance. J. Biol. Chem. 2014, 289, 10431–10444. [Google Scholar] [CrossRef] [PubMed]
- Camacho Leal Mdel, P.; Sciortino, M.; Tornillo, G.; Colombo, S.; Defilippi, P.; Cabodi, S. p130Cas/BCAR1 scaffold protein in tissue homeostasis and pathogenesis. Gene 2015, 562, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Centonze, G.; Natalini, D.; Salemme, V.; Costamagna, A.; Cabodi, S.; Defilippi, P. p130Cas/BCAR1 and p140Cap/SRCIN1 Adaptors: The Yin Yang in Breast Cancer? Front. Cell Dev. Biol. 2021, 9, 729093. [Google Scholar] [CrossRef] [PubMed]
- Felekkis, K.N.; Narsimhan, R.P.; Near, R.; Castro, A.F.; Zheng, Y.; Quilliam, L.A.; Lerner, A. AND-34 activates phosphatidylinositol 3-kinase and induces anti-estrogen resistance in a SH2 and GDP exchange factor-like domain-dependent manner. Mol. Cancer Res. 2005, 3, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Makkinje, A.; Vanden Borre, P.; Near, R.I.; Patel, P.S.; Lerner, A. Breast cancer anti-estrogen resistance 3 (BCAR3) protein augments binding of the c-Src SH3 domain to Crk-associated substrate (p130cas). J. Biol. Chem. 2012, 287, 27703–27714. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Canaff, L.; Rajadurai, C.V.; Fils-Aimé, N.; Tian, J.; Dai, M.; Korah, J.; Villatoro, M.; Park, M.; Ali, S.; et al. Breast cancer anti-estrogen resistance 3 inhibits transforming growth factor β/Smad signaling and associates with favorable breast cancer disease outcomes. Breast Cancer Res. 2014, 16, 476. [Google Scholar] [CrossRef] [PubMed]
- Cross, A.M.; Wilson, A.L.; Guerrero, M.S.; Thomas, K.S.; Bachir, A.I.; Kubow, K.E.; Horwitz, A.R.; Bouton, A.H. Breast cancer antiestrogen resistance 3-p130(Cas) interactions promote adhesion disassembly and invasion in breast cancer cells. Oncogene 2016, 35, 5850–5859. [Google Scholar] [CrossRef] [PubMed]
- Nojima, Y.; Morino, N.; Mimura, T.; Hamasaki, K.; Furuya, H.; Sakai, R.; Sato, T.; Tachibana, K.; Morimoto, C.; Yazaki, Y.; et al. Integrin-mediated cell adhesion promotes tyrosine phosphorylation of p130Cas, a Src homology 3-containing molecule having multiple Src homology 2-binding motifs. J. Biol. Chem. 1995, 270, 15398–15402. [Google Scholar] [CrossRef] [PubMed]
- Miranti, C.K.; Brugge, J.S. Sensing the environment: A historical perspective on integrin signal transduction. Nat. Cell Biol. 2002, 4, E83–E90. [Google Scholar] [CrossRef]
- Critchley, D.R. Focal adhesions—The cytoskeletal connection. Curr. Opin. Cell Biol. 2000, 12, 133–139. [Google Scholar] [CrossRef]
- Guan, J.L. Role of focal adhesion kinase in integrin signaling. Int. J. Biochem. Cell Biol. 1997, 29, 1085–1096. [Google Scholar] [CrossRef] [PubMed]
- Toutant, M.; Costa, A.; Studler, J.M.; Kadaré, G.; Carnaud, M.; Girault, J.A. Alternative splicing controls the mechanisms of FAK autophosphorylation. Mol. Cell Biol. 2002, 22, 7731–7743. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Harrison, S.C.; Eck, M.J. Three-dimensional structure of the tyrosine kinase c-Src. Nature 1997, 385, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Young, M.A.; Gonfloni, S.; Superti-Furga, G.; Roux, B.; Kuriyan, J. Dynamic coupling between the SH2 and SH3 domains of c-Src and Hck underlies their inactivation by C-terminal tyrosine phosphorylation. Cell 2001, 105, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Song, H.E.; Lee, Y.; Kim, E.; Cho, C.Y.; Jung, O.; Lee, D.; Lee, E.G.; Nam, S.H.; Kang, M.; Macalino, S.J.Y.; et al. N-terminus-independent activation of c-Src via binding to a tetraspan(in) TM4SF5 in hepatocellular carcinoma is abolished by the TM4SF5 C-terminal peptide application. Theranostics 2021, 11, 8092–8111. [Google Scholar] [CrossRef] [PubMed]
- Cary, L.A.; Han, D.C.; Polte, T.R.; Hanks, S.K.; Guan, J.L. Identification of p130Cas as a mediator of focal adhesion kinase-promoted cell migration. J. Cell Biol. 1998, 140, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Matsui, H.; Harada, I.; Sawada, Y. Src, p130Cas, and Mechanotransduction in Cancer Cells. Genes Cancer 2012, 3, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Burnham, M.R.; Bruce-Staskal, P.J.; Harte, M.T.; Weidow, C.L.; Ma, A.; Weed, S.A.; Bouton, A.H. Regulation of c-SRC activity and function by the adapter protein CAS. Mol Cell Biol. 2000, 20, 5865–5878. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xing, L.; Ge, C.; Zeltser, R.; Maskevitch, G.; Mayer, B.J.; Alexandropoulos, K. c-Src signaling induced by the adapters Sin and Cas is mediated by Rap1 GTPase. Mol. Cell Biol. 2000, 20, 7363–7377. [Google Scholar] [CrossRef]
- Pellicena, P.; Miller, W.T. Processive phosphorylation of p130Cas by Src depends on SH3-polyproline interactions. J. Biol. Chem. 2001, 276, 28190–28196. [Google Scholar] [CrossRef]
- Riggins, R.B.; Quilliam, L.A.; Bouton, A.H. Synergistic promotion of c-Src activation and cell migration by Cas and AND-34/BCAR3. J. Biol. Chem. 2003, 278, 28264–28273. [Google Scholar] [CrossRef] [PubMed]
- Arthur, W.T.; Quilliam, L.A.; Cooper, J.A. Rap1 promotes cell spreading by localizing Rac guanine nucleotide exchange factors. J. Cell. Biol. 2004, 167, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Casanova, A.G.; Roth, G.S.; Hausmann, S.; Lu, X.; Belmudes, L.; Bourova-Flin, E.; Flores, N.M.; Benitez, A.M.; Caporicci, M.; Vayr, J.; et al. Cytoskeleton remodeling induced by SMYD2 methyltransferase drives breast cancer metastasis. bioRxiv 2023. [Google Scholar] [CrossRef] [PubMed]
- Garron, M.L.; Arsenieva, D.; Zhong, J.; Bloom, A.B.; Lerner, A.; O’Neill, G.M.; Arold, S.T. Structural insights into the association between BCAR3 and Cas family members, an atypical complex implicated in anti-oestrogen resistance. J. Mol. Biol. 2009, 386, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, R.; Lemoine, A.; Bertoglio, J.; Raingeaud, J. Human enhancer of filamentation 1-induced colorectal cancer cell migration: Role of serine phosphorylation and interaction with the breast cancer anti-estrogen resistance 3 protein. Int. J. Biochem. Cell Biol. 2015, 64, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.L.; Schrecengost, R.S.; Guerrero, M.S.; Thomas, K.S.; Bouton, A.H. Breast cancer antiestrogen resistance 3 (BCAR3) promotes cell motility by regulating actin cytoskeletal and adhesion remodeling in invasive breast cancer cells. PLoS ONE 2013, 8, e65678. [Google Scholar] [CrossRef] [PubMed]
- Samavat, H.; Kurzer, M.S. Estrogen metabolism and breast cancer. Cancer Lett. 2015, 356 Pt A, 231–243. [Google Scholar] [CrossRef]
- Liang, J.; Shang, Y. Estrogen and cancer. Annu. Rev. Physiol. 2013, 75, 225–240. [Google Scholar] [CrossRef] [PubMed]
- Briest, S.; Stearns, V. Tamoxifen metabolism and its effect on endocrine treatment of breast cancer. Clin. Adv. Hematol. Oncol. 2009, 7, 185–192. [Google Scholar]
- Love, R.R. Tamoxifen therapy in primary breast cancer: Biology, efficacy, and side effects. J. Clin. Oncol. 1989, 7, 803–815. [Google Scholar] [CrossRef]
- Bender, L.M.; Nahta, R. Her2 cross talk and therapeutic resistance in breast cancer. Front. Biosci. 2008, 13, 3906–3912. [Google Scholar] [CrossRef] [PubMed]
- Nunnery, S.E.; Mayer, I.A. Targeting the PI3K/AKT/mTOR Pathway in Hormone-Positive Breast Cancer. Drugs 2020, 80, 1685–1697. [Google Scholar] [CrossRef]
- Anbalagan, M.; Rowan, B.G. Estrogen receptor alpha phosphorylation and its functional impact in human breast cancer. Mol. Cell Endocrinol. 2015, 418 Pt 3, 264–272. [Google Scholar] [CrossRef]
- Rogatsky, I.; Trowbridge, J.M.; Garabedian, M.J. Potentiation of human estrogen receptor alpha transcriptional activation through phosphorylation of serines 104 and 106 by the cyclin A-CDK2 complex. J. Biol. Chem. 1999, 274, 22296–22302. [Google Scholar] [CrossRef] [PubMed]
- Le Goff, P.; Montano, M.M.; Schodin, D.J.; Katzenellenbogen, B.S. Phosphorylation of the human estrogen receptor. Identification of hormone-regulated sites and examination of their influence on transcriptional activity. J. Biol. Chem. 1994, 269, 4458–4466. [Google Scholar] [CrossRef] [PubMed]
- Joel, P.B.; Traish, A.M.; Lannigan, D.A. Estradiol and phorbol ester cause phosphorylation of serine 118 in the human estrogen receptor. Mol. Endocrinol. 1995, 9, 1041–1052. [Google Scholar] [PubMed]
- Reese, J.C.; Katzenellenbogen, B.S. Examination of the DNA-binding ability of estrogen receptor in whole cells: Implications for hormone-independent transactivation and the actions of antiestrogens. Mol. Cell Biol. 1992, 12, 4531–4538. [Google Scholar] [PubMed]
- Ali, S.; Metzger, D.; Bornert, J.M.; Chambon, P. Modulation of transcriptional activation by ligand-dependent phosphorylation of the human oestrogen receptor A/B region. Embo J. 1993, 12, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Michalides, R.; Griekspoor, A.; Balkenende, A.; Verwoerd, D.; Janssen, L.; Jalink, K.; Floore, A.; Velds, A.; van’t Veer, L.; Neefjes, J. Tamoxifen resistance by a conformational arrest of the estrogen receptor alpha after PKA activation in breast cancer. Cancer Cell 2004, 5, 597–605. [Google Scholar] [CrossRef]
- Arnold, S.F.; Melamed, M.; Vorojeikina, D.P.; Notides, A.C.; Sasson, S. Estradiol-binding mechanism and binding capacity of the human estrogen receptor is regulated by tyrosine phosphorylation. Mol. Endocrinol. 1997, 11, 48–53. [Google Scholar] [CrossRef]
- Weis, K.E.; Ekena, K.; Thomas, J.A.; Lazennec, G.; Katzenellenbogen, B.S. Constitutively active human estrogen receptors containing amino acid substitutions for tyrosine 537 in the receptor protein. Mol. Endocrinol. 1996, 10, 1388–1398. [Google Scholar] [PubMed]
- Gompel, A. Hormone and breast cancer. Presse Med. 2019, 48, 1085–1091. [Google Scholar] [CrossRef] [PubMed]
- Parl, F.F.; Dawling, S.; Roodi, N.; Crooke, P.S. Estrogen metabolism and breast cancer: A risk model. Ann N. Y. Acad. Sci. 2009, 1155, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Jordan, V.C. The role of tamoxifen in the treatment and prevention of breast cancer. Curr. Probl. Cancer 1992, 16, 129–176. [Google Scholar] [PubMed]
- Osborne, C.K. Tamoxifen in the treatment of breast cancer. N. Engl. J. Med. 1998, 339, 1609–1618. [Google Scholar] [CrossRef] [PubMed]
- Goss, P.E.; Strasser, K. Tamoxifen resistant and refractory breast cancer: The value of aromatase inhibitors. Drugs 2002, 62, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.R. Acquired tamoxifen resistance in human breast cancer--potential mechanisms and clinical implications. Anticancer Drugs 1997, 8, 911–930. [Google Scholar] [CrossRef] [PubMed]
- Near, R.I.; Zhang, Y.; Makkinje, A.; Vanden Borre, P.; Lerner, A. AND-34/BCAR3 differs from other NSP homologs in induction of anti-estrogen resistance, cyclin D1 promoter activation and altered breast cancer cell morphology. J. Cell Physiol. 2007, 212, 655–665. [Google Scholar] [CrossRef]
- Makkinje, A.; Near, R.I.; Infusini, G.; Vanden Borre, P.; Bloom, A.; Cai, D.; Costello, C.E.; Lerner, A. AND-34/BCAR3 regulates adhesion-dependent p130Cas serine phosphorylation and breast cancer cell growth pattern. Cell Signal 2009, 21, 1423–1435. [Google Scholar] [CrossRef]
- Dorssers, L.C.; van Agthoven, T.; Brinkman, A.; Veldscholte, J.; Smid, M.; Dechering, K.J. Breast cancer oestrogen independence mediated by BCAR1 or BCAR3 genes is transmitted through mechanisms distinct from the oestrogen receptor signalling pathway or the epidermal growth factor receptor signalling pathway. Breast Cancer Res. 2005, 7, R82–R92. [Google Scholar] [CrossRef]
- Oh, M.J.; Yi, S.J.; Kim, H.S.; Kim, J.H.; Jeong, Y.H.; van Agthoven, T.; Jhun, B.H. Functional roles of BCAR3 in the signaling pathways of insulin leading to DNA synthesis, membrane ruffling and GLUT4 translocation. Biochem. Biophys. Res. Commun. 2013, 441, 911–916. [Google Scholar] [CrossRef] [PubMed]
- Felekkis, K.; Quilliam, L.A.; Lerner, A. Characterization of AND-34 function and signaling. Methods Enzymol. 2006, 407, 55–63. [Google Scholar] [PubMed]
- Bouton, A.H.; Riggins, R.B.; Bruce-Staskal, P.J. Functions of the adapter protein Cas: Signal convergence and the determination of cellular responses. Oncogene 2001, 20, 6448–6458. [Google Scholar] [CrossRef] [PubMed]
- Defilippi, P.; Di Stefano, P.; Cabodi, S. p130Cas: A versatile scaffold in signaling networks. Trends Cell Biol. 2006, 16, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Planas-Silva, M.D.; Hamilton, K.N. Targeting c-Src kinase enhances tamoxifen’s inhibitory effect on cell growth by modulating expression of cell cycle and survival proteins. Cancer Chemother. Pharmacol. 2007, 60, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.S.; Sarwar, N.; Phoenix, F.; Coombes, R.C.; Ali, S. Phosphorylation at serines 104 and 106 by Erk1/2 MAPK is important for estrogen receptor-alpha activity. J. Mol. Endocrinol. 2008, 40, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Washbrook, E.; Sarwar, N.; Bates, G.J.; Pace, P.E.; Thirunuvakkarasu, V.; Taylor, J.; Epstein, R.J.; Fuller-Pace, F.V.; Egly, J.M.; et al. Phosphorylation of human estrogen receptor alpha at serine 118 by two distinct signal transduction pathways revealed by phosphorylation-specific antisera. Oncogene 2002, 21, 4921–4931. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.C.; Basu, A.; El-Gharbawy, A.; Carrier, L.M.; Smith, C.L.; Rowan, B.G. Identification of four novel phosphorylation sites in estrogen receptor alpha: Impact on receptor-dependent gene expression and phosphorylation by protein kinase CK2. BMC Biochem. 2009, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Butt, A.J.; McNeil, C.M.; Musgrove, E.A.; Sutherland, R.L. Downstream targets of growth factor and oestrogen signalling and endocrine resistance: The potential roles of c-Myc, cyclin D1 and cyclin E. Endocr. Relat. Cancer 2005, 12 (Suppl. S1), S47–S59. [Google Scholar] [CrossRef]
- Kilker, R.L.; Hartl, M.W.; Rutherford, T.M.; Planas-Silva, M.D. Cyclin D1 expression is dependent on estrogen receptor function in tamoxifen-resistant breast cancer cells. J. Steroid Biochem. Mol. Biol. 2004, 92, 63–71. [Google Scholar] [CrossRef]
- Neuman, E.; Ladha, M.H.; Lin, N.; Upton, T.M.; Miller, S.J.; DiRenzo, J.; Pestell, R.G.; Hinds, P.W.; Dowdy, S.F.; Brown, M.; et al. Cyclin D1 stimulation of estrogen receptor transcriptional activity independent of cdk4. Mol. Cell Biol. 1997, 17, 5338–5347. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Felekkis, K.N.; Near, R.I.; O’Neill, G.M.; van Seventer, J.M.; Golemis, E.A.; Lerner, A. The GDP exchange factor AND-34 is expressed in B cells, associates with HEF1, and activates Cdc42. J. Immunol. 2003, 170, 969–978. [Google Scholar] [CrossRef]
- Burack, W.R.; Shaw, A.S. Signal transduction: Hanging on a scaffold. Curr. Opin. Cell Biol. 2000, 12, 211–216. [Google Scholar] [CrossRef]
- Pruitt, K.; Der, C.J. Ras and Rho regulation of the cell cycle and oncogenesis. Cancer Lett. 2001, 171, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Khosravi-Far, R.; Campbell, S.; Rossman, K.L.; Der, C.J. Increasing complexity of Ras signal transduction: Involvement of Rho family proteins. Adv. Cancer Res. 1998, 72, 57–107. [Google Scholar]
- Bar-Sagi, D.; Hall, A. Ras and Rho GTPases: A family reunion. Cell 2000, 103, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Chou, M.M.; Masuda-Robens, J.M.; Gupta, M.L. Cdc42 promotes G1 progression through p70 S6 kinase-mediated induction of cyclin E expression. J. Biol. Chem. 2003, 278, 35241–35247. [Google Scholar] [CrossRef]
- Pugacheva, E.N.; Golemis, E.A. The focal adhesion scaffolding protein HEF1 regulates activation of the Aurora-A and Nek2 kinases at the centrosome. Nat. Cell Biol. 2005, 7, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Fritz, G.; Kaina, B. Rho GTPases: Promising cellular targets for novel anticancer drugs. Curr. Cancer Drug Targets 2006, 6, 1–14. [Google Scholar]
- Maddox, A.S.; Burridge, K. RhoA is required for cortical retraction and rigidity during mitotic cell rounding. J. Cell Biol. 2003, 160, 255–265. [Google Scholar] [CrossRef]
- O’Keefe, L.; Somers, W.G.; Harley, A.; Saint, R. The pebble GTP exchange factor and the control of cytokinesis. Cell Struct. Funct. 2001, 26, 619–626. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dadke, D.; Jarnik, M.; Pugacheva, E.N.; Singh, M.K.; Golemis, E.A. Deregulation of HEF1 impairs M-phase progression by disrupting the RhoA activation cycle. Mol. Biol. Cell 2006, 17, 1204–1217. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Kloeker, S.; Jensen, C.C.; Bockholt, S.; Honda, H.; Hirai, H.; Beckerle, M.C. Members of the Zyxin family of LIM proteins interact with members of the p130Cas family of signal transducers. J. Biol. Chem. 2002, 277, 9580–9589. [Google Scholar] [CrossRef] [PubMed]
- Hirota, T.; Morisaki, T.; Nishiyama, Y.; Marumoto, T.; Tada, K.; Hara, T.; Masuko, N.; Inagaki, M.; Hatakeyama, K.; Saya, H. Zyxin, a regulator of actin filament assembly, targets the mitotic apparatus by interacting with h-warts/LATS1 tumor suppressor. J. Cell Biol. 2000, 149, 1073–1086. [Google Scholar] [CrossRef]
- Law, S.F.; Zhang, Y.Z.; Klein-Szanto, A.J.; Golemis, E.A. Cell cycle-regulated processing of HEF1 to multiple protein forms differentially targeted to multiple subcellular compartments. Mol. Cell Biol. 1998, 18, 3540–3551. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Tang, H.; Li, L.; Wang, X.; Yu, Z.; Li, J. Peptide-based therapeutic cancer vaccine: Current trends in clinical application. Cell Prolif. 2021, 54, e13025. [Google Scholar] [CrossRef] [PubMed]
- Buonaguro, L.; Tagliamonte, M. Peptide-based vaccine for cancer therapies. Front. Immunol. 2023, 14, 1210044. [Google Scholar] [CrossRef] [PubMed]
- Colella, T.A.; Bullock, T.N.; Russell, L.B.; Mullins, D.W.; Overwijk, W.W.; Luckey, C.J.; Pierce, R.A.; Restifo, N.P.; Engelhard, V.H. Self-tolerance to the murine homologue of a tyrosinase-derived melanoma antigen: Implications for tumor immunotherapy. J. Exp. Med. 2000, 191, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Carreno, B.M.; Magrini, V.; Becker-Hapak, M.; Kaabinejadian, S.; Hundal, J.; Petti, A.A.; Ly, A.; Lie, W.R.; Hildebrand, W.H.; Mardis, E.R.; et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015, 348, 803–808. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Zarling, A.L.; Ficarro, S.B.; White, F.M.; Shabanowitz, J.; Hunt, D.F.; Engelhard, V.H. Phosphorylated peptides are naturally processed and presented by major histocompatibility complex class I molecules in vivo. J. Exp. Med. 2000, 192, 1755–1762. [Google Scholar] [CrossRef]
- Zarling, A.L.; Polefrone, J.M.; Evans, A.M.; Mikesh, L.M.; Shabanowitz, J.; Lewis, S.T.; Engelhard, V.H.; Hunt, D.F. Identification of class I MHC-associated phosphopeptides as targets for cancer immunotherapy. Proc. Natl. Acad. Sci. USA 2006, 103, 14889–14894. [Google Scholar] [CrossRef] [PubMed]
- Zarling, A.L.; Obeng, R.C.; Desch, A.N.; Pinczewski, J.; Cummings, K.L.; Deacon, D.H.; Conaway, M.; Slingluff, C.L., Jr.; Engelhard, V.H. MHC-restricted phosphopeptides from insulin receptor substrate-2 and CDC25b offer broad-based immunotherapeutic agents for cancer. Cancer Res. 2014, 74, 6784–6795. [Google Scholar] [CrossRef] [PubMed]
- Engelhard, V.H.; Obeng, R.C.; Cummings, K.L.; Petroni, G.R.; Ambakhutwala, A.L.; Chianese-Bullock, K.A.; Smith, K.T.; Lulu, A.; Varhegyi, N.; Smolkin, M.E.; et al. MHC-restricted phosphopeptide antigens: Preclinical validation and first-in-humans clinical trial in participants with high-risk melanoma. J. Immunother. Cancer 2020, 8, e000262. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Marchesi, F.; Garlanda, C. Macrophages as tools and targets in cancer therapy. Nat. Rev. Drug Discov. 2022, 21, 799–820. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.R.; Minutolo, N.G.; Gill, S.; Klichinsky, M. Macrophage-Based Approaches for Cancer Immunotherapy. Cancer Res. 2021, 81, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Mehla, K.; Singh, P.K. Metabolic Regulation of Macrophage Polarization in Cancer. Trends Cancer 2019, 5, 822–834. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Helming, L.; Milde, R.; Varin, A.; Melgert, B.N.; Draijer, C.; Thomas, B.; Fabbri, M.; Crawshaw, A.; Ho, L.P.; et al. Genetic programs expressed in resting and IL-4 alternatively activated mouse and human macrophages: Similarities and differences. Blood 2013, 121, e57–e69. [Google Scholar] [CrossRef]
- Zeng, C.; Wang, Q.; Liu, X.; Wang, K.; Wang, C.; Ju, X.; Wang, T.; Zhou, Q.; Fu, X.; Yu, J.; et al. Localized Administration of Bcar3 siRNA via Nano-Self-Assembly to Treat Idiopathic Pulmonary Fibrosis by Disrupting Macrophage-Fibroblast Crosstalk. Int. J. Nanomedicine 2024, 19, 1827–1842. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Shen, Y.; Wu, D.; Zhang, J.; Lin, C.; Wang, L.; Yu, C.; Yu, B.; Shen, W. CircBCAR3 accelerates esophageal cancer tumorigenesis and metastasis via sponging miR-27a-3p. Mol. Cancer 2022, 21, 145. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.J.; van Agthoven, T.; Choi, J.E.; Jeong, Y.J.; Chung, Y.H.; Kim, C.M.; Jhun, B.H. BCAR3 regulates EGF-induced DNA synthesis in normal human breast MCF-12A cells. Biochem. Biophys. Res. Commun. 2008, 375, 430–434. [Google Scholar] [CrossRef] [PubMed]
- Rufanova, V.A.; Sorokin, A. CrkII associates with BCAR3 in response to endothelin-1 in human glomerular mesangial cells. Exp. Biol. Med. 2006, 231, 752–756. [Google Scholar]
- Eggen, B.J.; Benus, G.F.; Folkertsma, S.; Jonk, L.J.; Kruijer, W. TAK1 activation of the mouse JunB promoter is mediated through a CCAAT box and NF-Y. FEBS Lett. 2001, 506, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Hackel, A.; Aksamit, A.; Bruderek, K.; Lang, S.; Brandau, S. TNF-α and IL-1β sensitize human MSC for IFN-γ signaling and enhance neutrophil recruitment. Eur. J. Immunol. 2021, 51, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Rolfe, P.A.; Foernzler, D.; O’Rourke, D.; Zhao, S.; Scheuenpflug, J.; Feng, Z. Comparison of Two Illumina Whole Transcriptome RNA Sequencing Library Preparation Methods Using Human Cancer FFPE Specimens. Technol. Cancer Res. Treat. 2022, 21, 15330338221076304. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Lionta, E.; Spyrou, G.; Vassilatis, D.K.; Cournia, Z. Structure-based virtual screening for drug discovery: Principles, applications and recent advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef]
- Abbott, M.; Ustoyev, Y. Cancer and the Immune System: The History and Background of Immunotherapy. Semin. Oncol. Nurs. 2019, 35, 150923. [Google Scholar] [CrossRef]
- Kumar, A.R.; Devan, A.R.; Nair, B.; Vinod, B.S.; Nath, L.R. Harnessing the immune system against cancer: Current immunotherapy approaches and therapeutic targets. Mol. Biol. Rep. 2021, 48, 8075–8095. [Google Scholar] [CrossRef]
- Huang, T.; Song, X.; Xu, D.; Tiek, D.; Goenka, A.; Wu, B.; Sastry, N.; Hu, B.; Cheng, S.Y. Stem cell programs in cancer initiation, progression, and therapy resistance. Theranostics 2020, 10, 8721–8743. [Google Scholar] [CrossRef]
- Ahmad, G.; Amiji, M.M. Cancer stem cell-targeted therapeutics and delivery strategies. Expert Opin. Drug Deliv. 2017, 14, 997–1008. [Google Scholar] [CrossRef]
- Li, W.; Ma, H.; Zhang, J.; Zhu, L.; Wang, C.; Yang, Y. Unraveling the roles of CD44/CD24 and ALDH1 as cancer stem cell markers in tumorigenesis and metastasis. Sci. Rep. 2017, 7, 13856. [Google Scholar] [CrossRef]
- Vikram, R.; Chou, W.C.; Hung, S.C.; Shen, C.Y. Tumorigenic and Metastatic Role of CD44(-/low)/CD24(-/low) Cells in Luminal Breast Cancer. Cancers 2020, 12, 1239. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model (Cell/Animal) | Methods | Cellular Function of BCAR3 | Ref. |
|---|---|---|---|
| Esophageal cancer cells and xenograft mouse model | Loss-of-function assays. Xenograft growth and lung metastasis studies. Bioinformatics analysis. | CircBCAR3 promotes esophageal cancer cell proliferation, migration, and invasion and inhibits ferroptosis in vitro. In vivo, it supports the growth and metastasis of esophageal xenografts. | [112] |
| Ovarian cancer cells | Deep sequencing. RNA binding assays. Proliferation assays. Use of tRF5-Glu mimics. | tRF5-Glu regulates BCAR3 expression by binding to its 3′UTR, leading to decreased BCAR3 levels. Lower BCAR3 expression is associated with suppressed ovarian cancer cell proliferation. | [14] |
| Breast cancer cell lines (MCF-12A cells) | Microinjection of anti-BCAR3 antibody. siRNAs targeting BCAR3 and SH2 domain of BCAR3. | BCAR3, particularly through its SH2 domain, plays a crucial role in EGF-induced DNA synthesis, indicating its involvement in cell cycle progression and mitogenic signaling pathways. | [113] |
| Head and neck squamous cell carcinoma (HNSCC) patients | RNA-sequencing and bioinformatics analysis. siRNA transfection. | BCAR3 is overexpressed in HNSCC and contributes to tumor growth. Its expression is associated with perineural invasion (PNI) and poor survival. | [10] |
| Breast cancer cell lines (estrogen-independent 578-T, estrogen-dependent MCF7, ZR-75-1) | Overexpression studies. F-actin analysis. Kinase activity assays. Luciferase assay for cyclin D1 promoter activation. | BCAR3 overexpression leads to activation of Rho family GTPases Cdc42 and Rac, resulting in changes in F-actin distribution, enhanced PAK1 autophosphorylation and kinase activity, and activation of the cyclin D1 promoter. | [15] |
| TNBC cells, mouse orthotopic tumor models | In-vivo and in-vitro studies. Analysis of RNA expression databases. Correlation analysis. Functional assays. | BCAR3 is significantly upregulated in TNBC and contributes to tumor growth and progression. Elevated BCAR3 levels are associated with poor patient survival. BCAR3, in conjunction with MET receptor signaling, regulates proliferation and migration of TNBC cells. | [7] |
| Mammary-epithelium specific SMYD2 ablation in mice, breast cancer cells, PDX (Patient-Derived Xenografts), genetically engineered mice | In-vivo survival studies. Identification of physiological substrates. Methylation signaling pathway analysis. In-vitro migration and invasiveness assays. | BCAR3 is identified as a physiological substrate of SMYD2, and its methylation at K334 is crucial for metastasis. Methylation of BCAR3 by SMYD2 recruits FMNL proteins to cell edges, influencing lamellipodia dynamics and promoting cancer cell migration and invasiveness. | [43] |
| Breast cancer cell lines (MCF7 cells) | Gain-and loss-of-function approaches. Adhesion signaling and spreading assays. | BCAR3 modulates c-Src activity and regulates the association between Src and BCAR1. This coordination is crucial for breast cancer cell adhesion signaling and spreading, contributing to aggressive and invasive tumor phenotypes. | [16] |
| Breast cancer cell lines (MCF7 cells) | Use of structure-based BCAR1 and BCAR3 mutants. ERK1/2 activity assays. Reverse-phase protein array. | BCAR3’s antiestrogen resistance critically depends on binding to BCAR1. The BCAR1/BCAR3 interaction enhances BCAR1 phosphorylation, potentiating antiestrogen resistance. This resistance is associated with increased ERK1/2 activity. | [21] |
| Breast cancer cells (MCF-7, MDA-MB-231, BT-549 and SK-BR-3 cells) | Coimmunoprecipitation. Automated imaging system for cell migration. | BCAR3 inhibits the TGFβ/Smad signaling pathway, leading to suppression of Smad activation, gene transcription, cell migration, and matrix digestion. | [26] |
| Human glomerular mesangial cells | Adenovirus-mediated gene transfer. Co-precipitation assays. | BCAR3 associates with CrkII in response to endothelin-1, a process that is enhanced by Pyk2 activity. This interaction is implicated in proliferative kidney pathologies and is part of the ET-1 signaling pathway, suggesting a role in cell proliferation, contraction, and extracellular matrix synthesis in the kidney. | [114] |
| PTPα-null MEF cells | Reconstitution assays. Localization studies. | BCAR3 acts as a connector between phospho-Tyr789 PTPα and BCAR1 at adhesion sites, facilitating BCAR1 interaction and phosphorylation by Src. This identifies a novel role of BCAR3 in promoting cell migration through the assembly and activation of integrin-induced adhesions. | [3] |
| Normal epithelial cells | Localization studies. Phosphorylation assays. Lamellipodia dynamics assessment. Cell migration assays. | BCAR3 is essential for regulating cytoskeletal dynamics during cell migration, necessary for Cas phosphorylation and lamellipodia dynamics, crucial for cell migration, and part of a positive-feedback loop with Cas for cellular localization and activation signaling. | [2] |
| Melanoma and Breast Cancer Cell Lines, Mice, Human Donors, Clinical Trial Participants | Clinical trial with vaccinations using pBCAR3126-134 and pIRS21097-1105 peptides in adjuvant and Hiltonol. Adverse events monitoring based on NCI CTCAE. Interferon-γ ELISpot assay for T-cell responses. | pBCAR3 peptides triggered immunogenicity in vivo and in vitro. T-cells specific for pBCAR3126-134 inhibited tumor xenograft growth., Identification of pIRS21097-1105 peptide in human tumors by mass spectrometry. Induction of T-cell responses in clinical trial participants with no severe AEs, DLTs, or deaths. Immune response to pIRS21097-1105 in 42% of patients and to pBCAR3126-134 in 17% of patients. | [105] |
| 1878 MM patients (1930 samples) from 7 independent datasets | Comparative analysis of BCAR3 expression in different stages and molecular subtypes. Analysis of 1q21 amplification. Assessment of event-free survival (EFS) and overall survival (OS). Therapeutic response evaluation to bortezomib and dexamethasone. | Predictor of better prognosis in MM patients, associated with higher EFS and OS. Indicative of prognosis post-relapse. Independent prognostic factor. Potential biomarker. | [11] |
| 32 cases of ectopic and eutopic endometrium in endometriosis, 31 controls | Real-time PCR. Immunohistochemistry. Western blotting. Lentivirus overexpression. Vector knockdown. CCK-8 assay. Transwell experiments. Estrogen intervention experiments. | Promotes migration and invasion of endometrial cells in endometriosis. Associated with higher expression in advanced stages of endometriosis. Does not induce EMT directly. Regulates anti-estrogen effects. | [13] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moon, D.O. Deciphering the Role of BCAR3 in Cancer Progression: Gene Regulation, Signal Transduction, and Therapeutic Implications. Cancers 2024, 16, 1674. https://doi.org/10.3390/cancers16091674
Moon DO. Deciphering the Role of BCAR3 in Cancer Progression: Gene Regulation, Signal Transduction, and Therapeutic Implications. Cancers. 2024; 16(9):1674. https://doi.org/10.3390/cancers16091674
Chicago/Turabian StyleMoon, Dong Oh. 2024. "Deciphering the Role of BCAR3 in Cancer Progression: Gene Regulation, Signal Transduction, and Therapeutic Implications" Cancers 16, no. 9: 1674. https://doi.org/10.3390/cancers16091674
APA StyleMoon, D. O. (2024). Deciphering the Role of BCAR3 in Cancer Progression: Gene Regulation, Signal Transduction, and Therapeutic Implications. Cancers, 16(9), 1674. https://doi.org/10.3390/cancers16091674