
scRNAseq and High-Throughput Spatial Analysis of Tumor and Normal Microenvironment in Solid Tumors Reveal a Possible Origin of Circulating Tumor Hybrid Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. scRNAseq and Spatial Datasets
2.2. scRNAseq and Spatial Data Processing
2.3. Doublet-Scoring Analysis
2.4. Hybrid Cell Identification
2.5. Differential Gene Expression
2.6. Functional Gene Set Enrichment Analysis
2.7. Statistical Analysis
3. Results
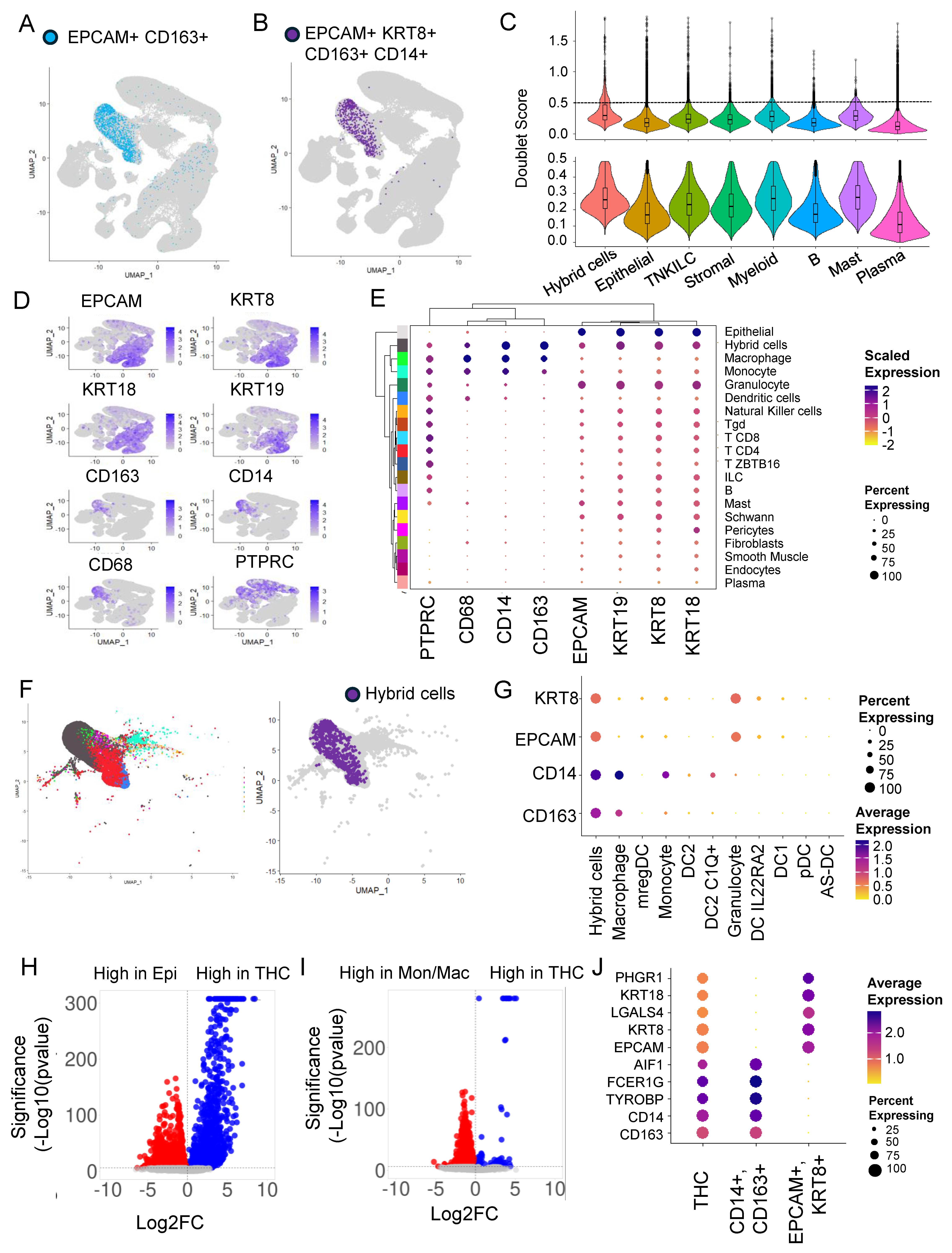
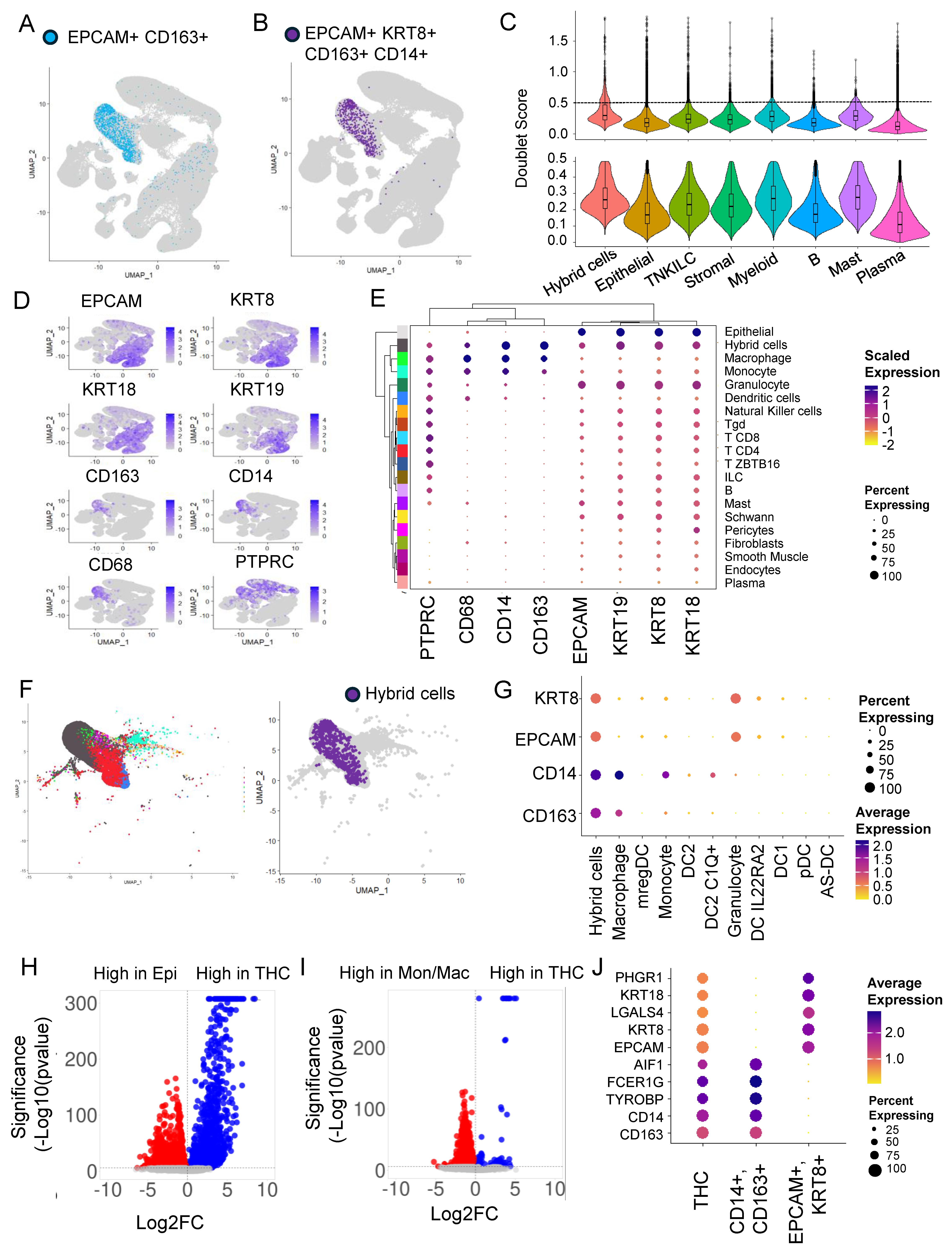
3.1. Hybrid Cells in Colon Cancer and Normal Biopsies Are More Frequently Found in Myeloid Cell Clusters
3.2. Doublets Analysis Further Refines Hybrid Cells’ Identity
3.3. Hybrid Cells in Colon Cancer and Normal Biopsies Are More Frequently Found with Macrophage/Monocyte Clusters
3.4. Differential Gene Expression Analysis Revealed an Extensive Hybrid Transcriptome
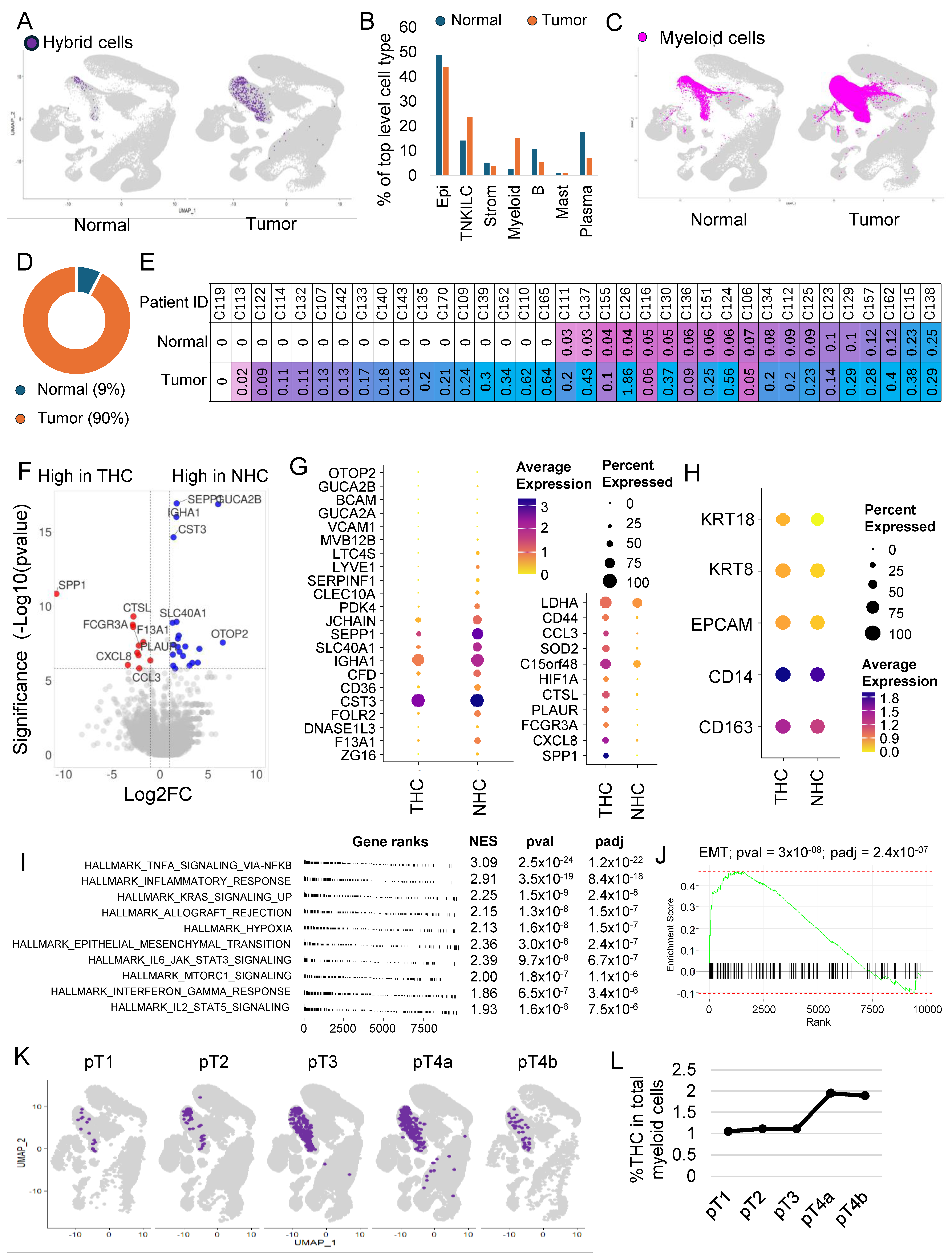
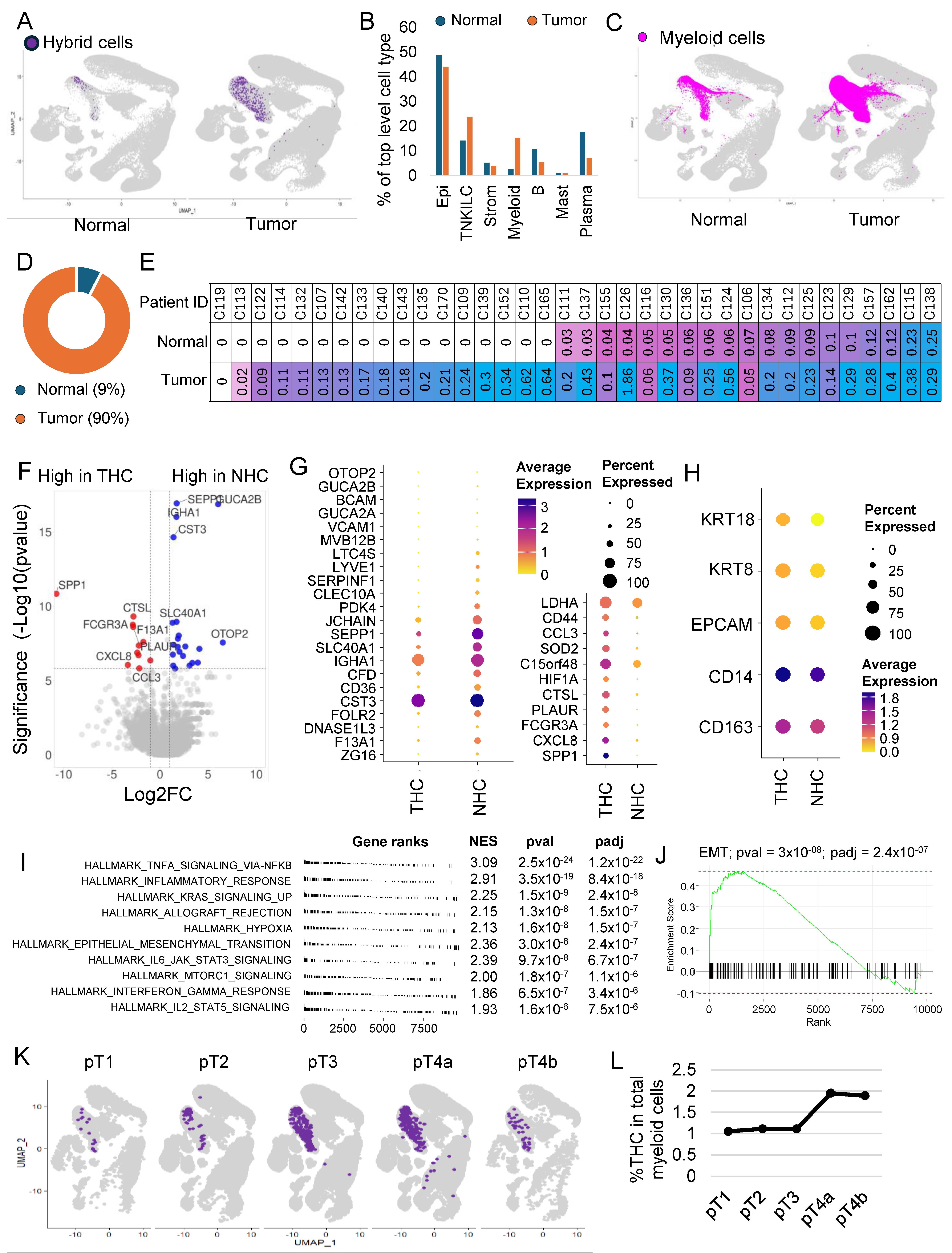
3.5. The Majority of the Hybrid Cells Were Found in Tumor Tissue Compared to Normal
3.6. THC Shows a Distinct Gene Expression Profile
3.7. THC Number within the Tumor and Their Relation to Clinical Features
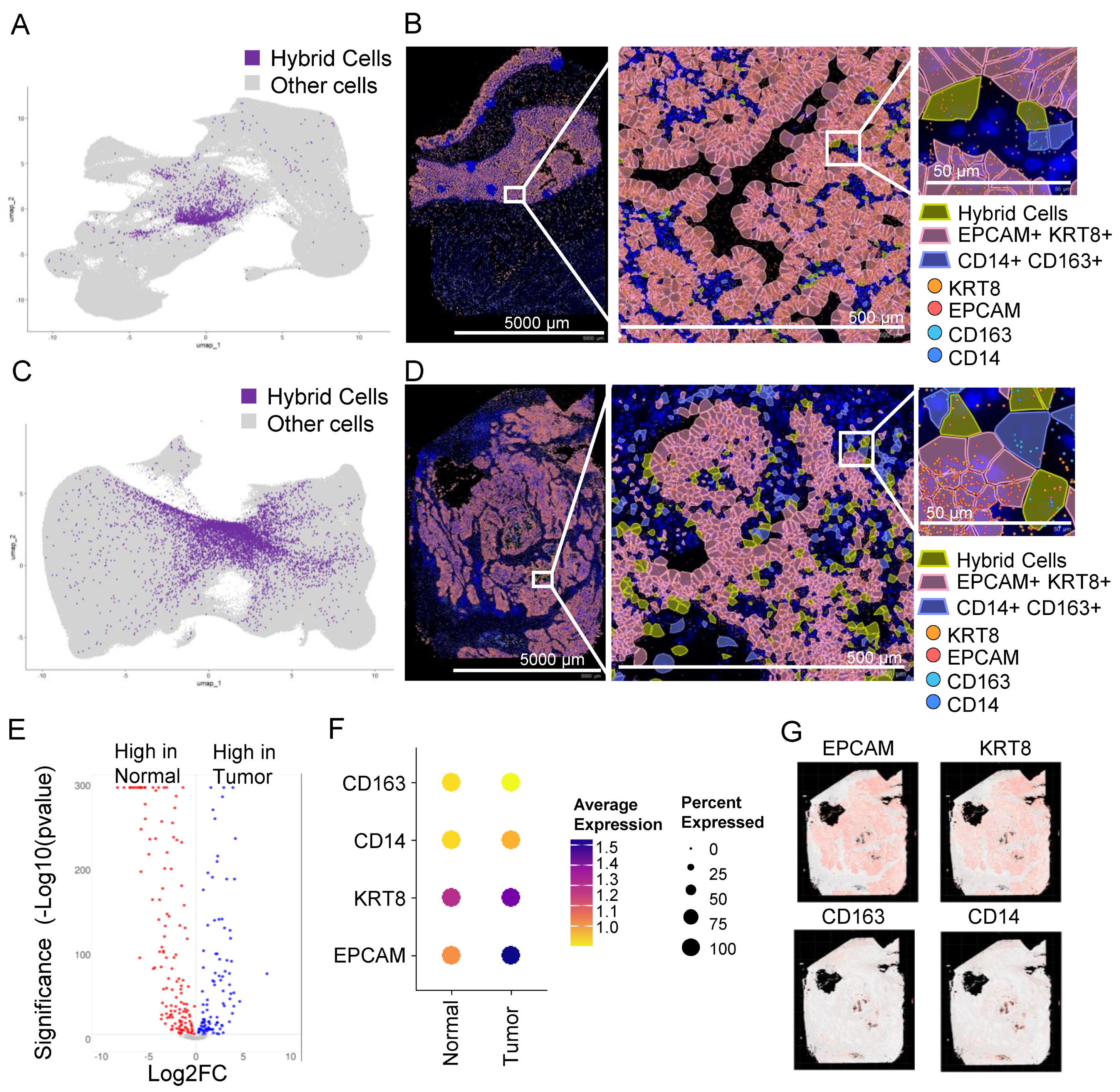
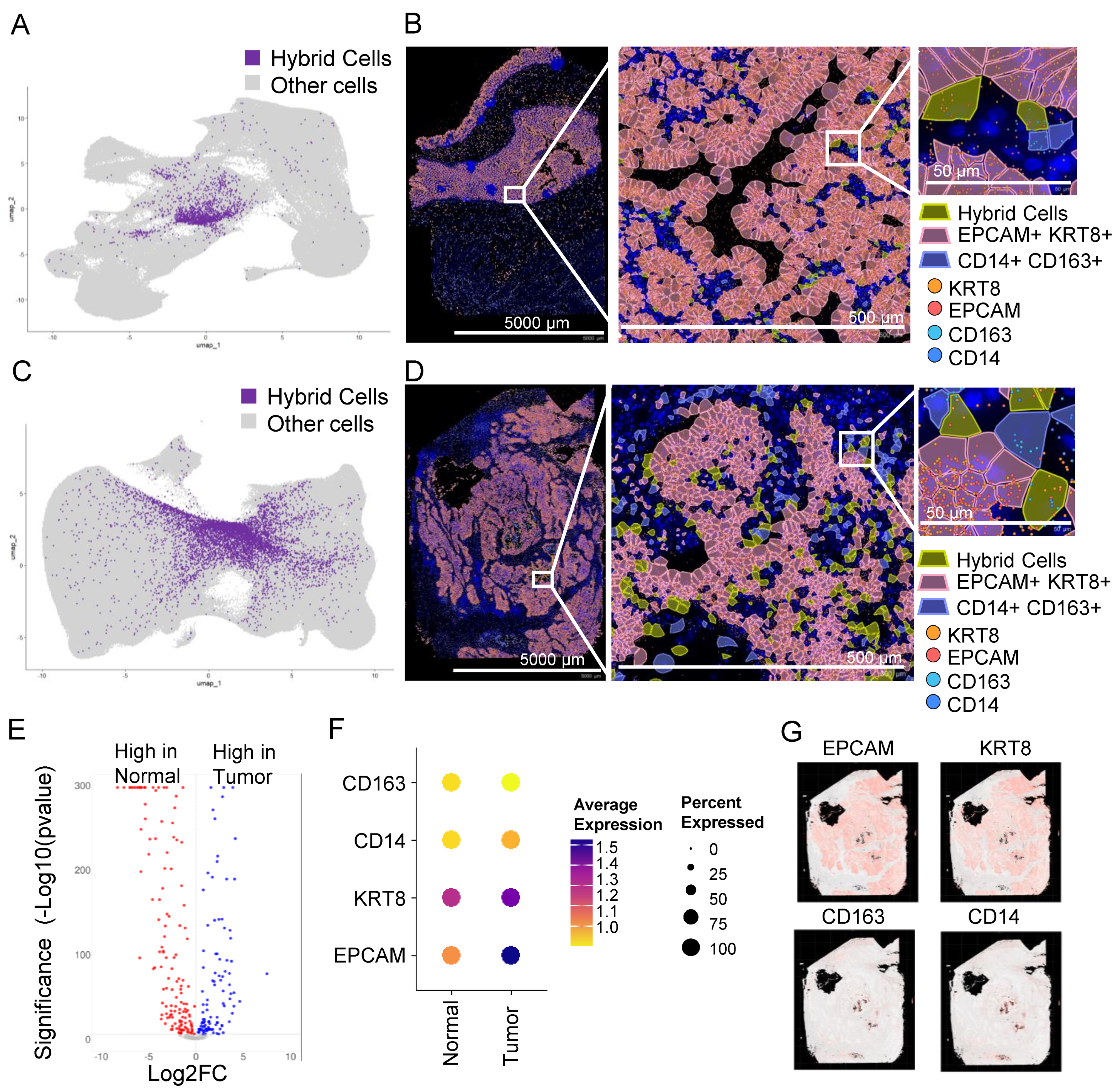
3.8. In Situ High-Resolution Spatial Mapping Confirms the Presence of THCs in Tumor Sections
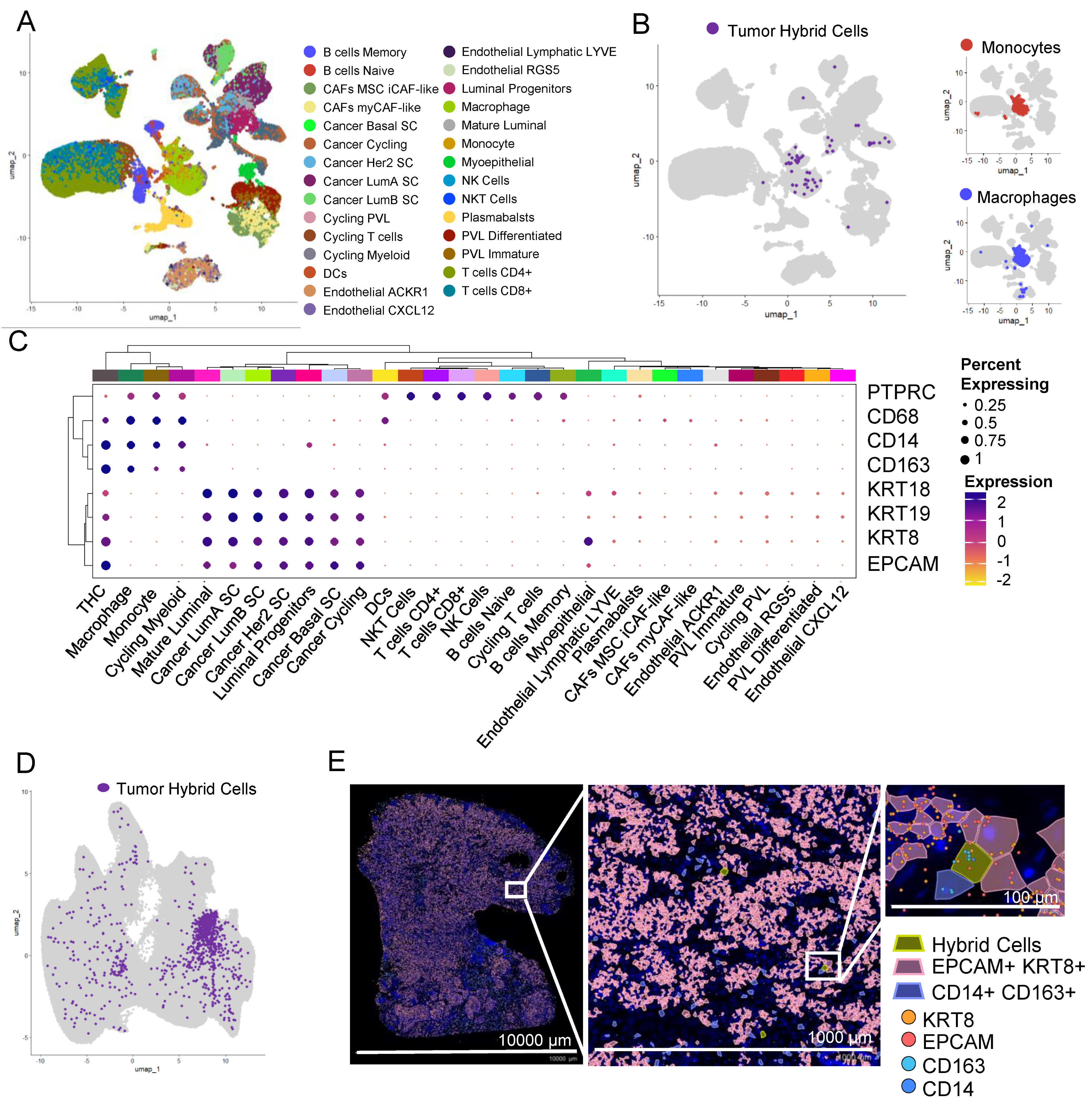
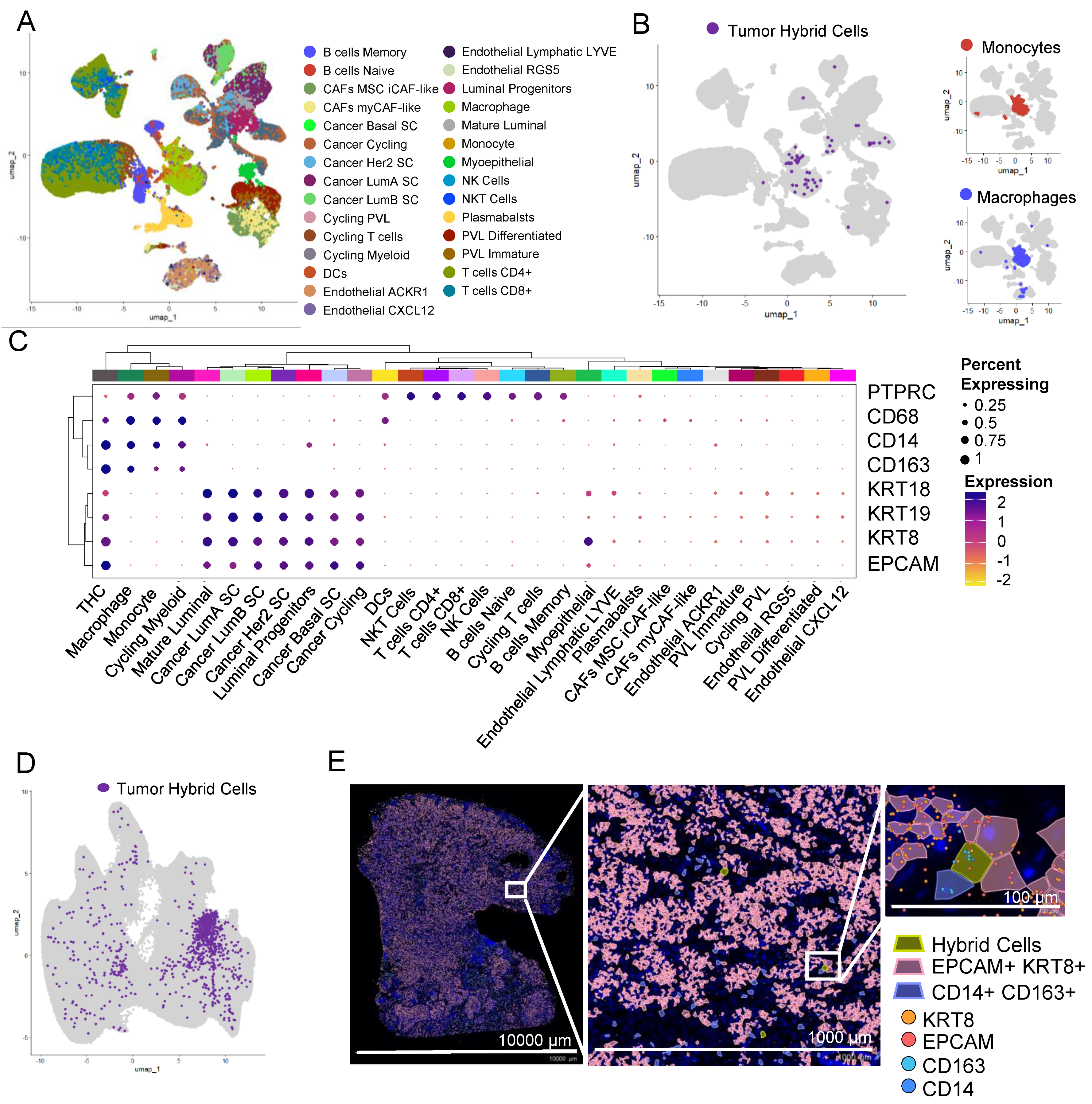
3.9. THCs Are Found in scRNAseq and In Situ Spatial Data from Breast Cancer
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Code Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mehlen, P.; Puisieux, A. Metastasis: A question of life or death. Nat. Rev. Cancer 2006, 6, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Castaneda, M.; den Hollander, P.; Kuburich, N.A.; Rosen, J.M.; Mani, S.A. Mechanisms of cancer metastasis. Semin. Cancer Biol. 2022, 87, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Aichel, O. Eine neue Hypothese über Ursachen und Wesen bösartiger Geschwülste; Universo: Santiago, Chile, 1908. [Google Scholar]
- Adams, D.L.; Adams, D.K.; Alpaugh, R.K.; Cristofanilli, M.; Martin, S.S.; Chumsri, S.; Tang, C.M.; Marks, J.R. Circulating Cancer-Associated Macrophage-Like Cells Differentiate Malignant Breast Cancer and Benign Breast Conditions. Cancer Epidemiol. Biomarkers Prev. 2016, 25, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.L.; Martin, S.S.; Alpaugh, R.K.; Charpentier, M.; Tsai, S.; Bergan, R.C.; Ogden, I.M.; Catalona, W.; Chumsri, S.; Tang, C.M.; et al. Circulating giant macrophages as a potential biomarker of solid tumors. Proc. Natl. Acad. Sci. USA 2014, 111, 3514–3519. [Google Scholar] [CrossRef] [PubMed]
- Augustyn, A.; Adams, D.L.; He, J.; Qiao, Y.; Verma, V.; Liao, Z.; Tang, C.M.; Heymach, J.V.; Tsao, A.S.; Lin, S.H. Giant Circulating Cancer-Associated Macrophage-Like Cells Are Associated With Disease Recurrence and Survival in Non-Small-Cell Lung Cancer Treated With Chemoradiation and Atezolizumab. Clin. Lung Cancer 2021, 22, e451–e465. [Google Scholar] [CrossRef] [PubMed]
- Gironda, D.J.; Adams, D.L.; He, J.; Xu, T.; Gao, H.; Qiao, Y.; Komaki, R.; Reuben, J.M.; Liao, Z.; Blum-Murphy, M.; et al. Cancer associated macrophage-like cells and prognosis of esophageal cancer after chemoradiation therapy. J. Transl. Med. 2020, 18, 413. [Google Scholar] [CrossRef] [PubMed]
- Gast, C.E.; Silk, A.D.; Zarour, L.; Riegler, L.; Burkhart, J.G.; Gustafson, K.T.; Parappilly, M.S.; Roh-Johnson, M.; Goodman, J.R.; Olson, B.; et al. Cell fusion potentiates tumor heterogeneity and reveals circulating hybrid cells that correlate with stage and survival. Sci. Adv. 2018, 4, eaat7828. [Google Scholar] [CrossRef] [PubMed]
- Sutton, T.L.; Patel, R.K.; Anderson, A.N.; Bowden, S.G.; Whalen, R.; Giske, N.R.; Wong, M.H. Circulating Cells with Macrophage-like Characteristics in Cancer: The Importance of Circulating Neoplastic-Immune Hybrid Cells in Cancer. Cancers 2022, 14, 3871. [Google Scholar] [CrossRef] [PubMed]
- Manjunath, Y.; Mitchem, J.B.; Suvilesh, K.N.; Avella, D.M.; Kimchi, E.T.; Staveley-O’Carroll, K.F.; Deroche, C.B.; Pantel, K.; Li, G.; Kaifi, J.T. Circulating Giant Tumor-Macrophage Fusion Cells Are Independent Prognosticators in Patients With NSCLC. J. Thorac. Oncol. 2020, 15, 1460–1471. [Google Scholar] [CrossRef] [PubMed]
- Sutton, T.L.; Walker, B.S.; Wong, M.H. Circulating Hybrid Cells Join the Fray of Circulating Cellular Biomarkers. Cell. Mol. Gastroenterol. Hepatol. 2019, 8, 595–607. [Google Scholar] [CrossRef] [PubMed]
- Henn, T.E.; Anderson, A.N.; Hollett, Y.R.; Sutton, T.L.; Walker, B.S.; Swain, J.R.; Sauer, D.A.; Clayburgh, D.R.; Wong, M.H. Circulating hybrid cells predict presence of occult nodal metastases in oral cavity carcinoma. Head. Neck 2021, 43, 2193–2201. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.S.; Sutton, T.L.; Zarour, L.; Hunter, J.G.; Wood, S.G.; Tsikitis, V.L.; Herzig, D.O.; Lopez, C.D.; Chen, E.Y.; Mayo, S.C.; et al. Circulating Hybrid Cells: A Novel Liquid Biomarker of Treatment Response in Gastrointestinal Cancers. Ann. Surg. Oncol. 2021, 28, 8567–8578. [Google Scholar] [CrossRef] [PubMed]
- Ozel, C.; Seidel, J.; Meyer-Staeckling, S.; Brandt, B.H.; Niggemann, B.; Zanker, K.S.; Dittmar, T. Hybrid cells derived from breast epithelial cell/breast cancer cell fusion events show a differential RAF-AKT crosstalk. Cell Commun. Signal 2012, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Tosun, S.; Fried, S.; Niggemann, B.; Zanker, K.S.; Dittmar, T. Hybrid Cells Derived from Human Breast Cancer Cells and Human Breast Epithelial Cells Exhibit Differential TLR4 and TLR9 Signaling. Int. J. Mol. Sci. 2016, 17, 726. [Google Scholar] [CrossRef] [PubMed]
- Gauck, D.; Keil, S.; Niggemann, B.; Zanker, K.S.; Dittmar, T. Hybrid clone cells derived from human breast epithelial cells and human breast cancer cells exhibit properties of cancer stem/initiating cells. BMC Cancer 2017, 17, 515. [Google Scholar] [CrossRef] [PubMed]
- Clawson, G.A.; Matters, G.L.; Xin, P.; Imamura-Kawasawa, Y.; Du, Z.; Thiboutot, D.M.; Helm, K.F.; Neves, R.I.; Abraham, T. Macrophage-tumor cell fusions from peripheral blood of melanoma patients. PLoS ONE 2015, 10, e0134320. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, L.A.; Montalban-Hernandez, K.; Avendano-Ortiz, J.; Marin, E.; Lozano, R.; Toledano, V.; Sanchez-Maroto, L.; Terron, V.; Valentin, J.; Pulido, E.; et al. Tumor stem cells fuse with monocytes to form highly invasive tumor-hybrid cells. Oncoimmunology 2020, 9, 1773204. [Google Scholar] [CrossRef] [PubMed]
- Dietz, M.S.; Sutton, T.L.; Walker, B.S.; Gast, C.E.; Zarour, L.; Sengupta, S.K.; Swain, J.R.; Eng, J.; Parappilly, M.; Limbach, K.; et al. Relevance of circulating hybrid cells as a non-invasive biomarker for myriad solid tumors. Sci. Rep. 2021, 11, 13630. [Google Scholar] [CrossRef]
- Manjunath, Y.; Porciani, D.; Mitchem, J.B.; Suvilesh, K.N.; Avella, D.M.; Kimchi, E.T.; Staveley-O’Carroll, K.F.; Burke, D.H.; Li, G.; Kaifi, J.T. Tumor-Cell-Macrophage Fusion Cells as Liquid Biomarkers and Tumor Enhancers in Cancer. Int. J. Mol. Sci. 2020, 21, 1872. [Google Scholar] [CrossRef] [PubMed]
- Manjunath, Y.; Upparahalli, S.V.; Avella, D.M.; Deroche, C.B.; Kimchi, E.T.; Staveley-O’carroll, K.F.; Smith, C.J.; Li, G.; Kaifi, J.T. PD-L1 expression with epithelial mesenchymal transition of circulating tumor cells is associated with poor survival in curatively resected non-small cell lung cancer. Cancers 2019, 11, 806. [Google Scholar] [CrossRef] [PubMed]
- Shabo, I.; Midtbo, K.; Andersson, H.; Akerlund, E.; Olsson, H.; Wegman, P.; Gunnarsson, C.; Lindstrom, A. Macrophage traits in cancer cells are induced by macrophage-cancer cell fusion and cannot be explained by cellular interaction. BMC Cancer 2015, 15, 922. [Google Scholar] [CrossRef] [PubMed]
- Shabo, I.; Olsson, H.; Elkarim, R.; Sun, X.F.; Svanvik, J. Macrophage Infiltration in Tumor Stroma is Related to Tumor Cell Expression of CD163 in Colorectal Cancer. Cancer Microenviron. 2014, 7, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Shabo, I.; Olsson, H.; Stal, O.; Svanvik, J. Breast cancer expression of DAP12 is associated with skeletal and liver metastases and poor survival. Clin. Breast Cancer 2013, 13, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Shabo, I.; Olsson, H.; Sun, X.F.; Svanvik, J. Expression of the macrophage antigen CD163 in rectal cancer cells is associated with early local recurrence and reduced survival time. Int. J. Cancer 2009, 125, 1826–1831. [Google Scholar] [CrossRef] [PubMed]
- Shabo, I.; Stal, O.; Olsson, H.; Dore, S.; Svanvik, J. Breast cancer expression of CD163, a macrophage scavenger receptor, is related to early distant recurrence and reduced patient survival. Int. J. Cancer 2008, 123, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Shabo, I.; Svanvik, J. Expression of macrophage antigens by tumor cells. Adv. Exp. Med. Biol. 2011, 714, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Clawson, G.A.; Kimchi, E.; Patrick, S.D.; Xin, P.; Harouaka, R.; Zheng, S.; Berg, A.; Schell, T.; Staveley-O’Carroll, K.F.; Neves, R.I.; et al. Circulating tumor cells in melanoma patients. PLoS ONE 2012, 7, e0041052. [Google Scholar] [CrossRef] [PubMed]
- Clawson, G.A.; Matters, G.L.; Xin, P.; McGovern, C.; Wafula, E.; DePamphilis, C.; Meckley, M.; Wong, J.; Stewart, L.; D’Jamoos, C.; et al. “Stealth dissemination” of macrophage-tumor cell fusions cultured from blood of patients with pancreatic ductal adenocarcinoma. PLoS ONE 2017, 12, e0184451. [Google Scholar] [CrossRef] [PubMed]
- Kaifi, J.T.; Kunkel, M.; Das, A.; Harouaka, R.A.; Dicker, D.T.; Li, G.; Zhu, J.; Clawson, G.A.; Yang, Z.; Reed, M.F.; et al. Circulating tumor cell isolation during resection of colorectal cancer lung and liver metastases: A prospective trial with different detection techniques. Cancer Biol. Ther. 2015, 16, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.L.; Adams, D.K.; He, J.; Kalhor, N.; Zhang, M.; Xu, T.; Gao, H.; Reuben, J.M.; Qiao, Y.; Komaki, R.; et al. Sequential Tracking of PD-L1 Expression and RAD50 Induction in Circulating Tumor and Stromal Cells of Lung Cancer Patients Undergoing Radiotherapy. Clin. Cancer Res. 2017, 23, 5948–5958. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.L.; Stefansson, S.; Haudenschild, C.; Martin, S.S.; Charpentier, M.; Chumsri, S.; Cristofanilli, M.; Tang, C.-M.; Alpaugh, R.K. Cytometric characterization of Circulating Tumor Cells Captured by microfiltration and their correlation to the cellsearch® CTC test. Cytometry Part A 2015, 87, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.M.; Zhu, P.; Li, S.; Makarova, O.V.; Amstutz, P.T.; Adams, D.L. Blood-based biopsies-clinical utility beyond circulating tumor cells. Cytometry A 2018, 93, 1246–1250. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.M.; BenMohamed, F.; Decina, A.; Mukherjee, S.; Levi, S.; Garrido Castillo, L.N.; Brechot, D.; Jurcic, J.; Raza, A.; Paterlini Brechot, P. Circulating cancer giant cells with unique characteristics frequently found in patients with myelodysplastic syndromes (MDS). Med. Oncol. 2023, 40, 204. [Google Scholar] [CrossRef] [PubMed]
- Pelka, K.; Hofree, M.; Chen, J.H.; Sarkizova, S.; Pirl, J.D.; Jorgji, V.; Bejnood, A.; Dionne, D.; Ge, W.H.; Xu, K.H.; et al. Spatially organized multicellular immune hubs in human colorectal cancer. Cell 2021, 184, 4734–4752.e4720. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Z.; Al-Eryani, G.; Roden, D.L.; Junankar, S.; Harvey, K.; Andersson, A.; Thennavan, A.; Wang, C.; Torpy, J.R.; Bartonicek, N.; et al. A single-cell and spatially resolved atlas of human breast cancers. Nat. Genet. 2021, 53, 1334–1347. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Stuart, T.; Kowalski, M.H.; Choudhary, S.; Hoffman, P.; Hartman, A.; Srivastava, A.; Molla, G.; Madad, S.; Fernandez-Granda, C.; et al. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat. Biotechnol. 2023, 42, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Marsh, S.E. Samuel-Marsh/scCustomize, version 0.62; scCustomize: Custom Visualizations & Functions for Streamlined Analyses of Single Cell Sequencing; Zenodo: Geneva, Switzerland, 2021. [Google Scholar] [CrossRef]
- Bais, A.S.; Kostka, D. scds: Computational annotation of doublets in single-cell RNA sequencing data. Bioinformatics 2020, 36, 1150–1158. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, A.Z.; Swain, J.R.; Davies, P.S.; Bailey, A.S.; Decker, A.D.; Willenbring, H.; Grompe, M.; Fleming, W.H.; Wong, M.H. Bone marrow-derived cells fuse with normal and transformed intestinal stem cells. Proc. Natl. Acad. Sci. USA 2006, 103, 6321–6325. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular origins of cancer: Molecular basis of colorectal cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Sun, H.; Zhang, Y.; Wang, Z.; Xun, Z.; Li, Z.; Ding, X.; Bao, R.; Hong, L.; Jia, W.; et al. Single-cell and spatial analysis reveal interaction of FAP(+) fibroblasts and SPP1(+) macrophages in colorectal cancer. Nat. Commun. 2022, 13, 1742. [Google Scholar] [CrossRef] [PubMed]
- Bjerkvig, R.; Tysnes, B.B.; Aboody, K.S.; Najbauer, J.; Terzis, A.J. Opinion: The origin of the cancer stem cell: Current controversies and new insights. Nat. Rev. Cancer 2005, 5, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Dittmar, T.; Nagler, C.; Schwitalla, S.; Reith, G.; Niggemann, B.; Zanker, K.S. Recurrence cancer stem cells--made by cell fusion? Med. Hypotheses 2009, 73, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Zhang, Y.; Park, S.C.; Eun, J.R.; Nguyen, N.T.; Tschudy-Seney, B.; Jung, Y.J.; Theise, N.D.; Zern, M.A.; Duan, Y. CD34(+) Liver Cancer Stem Cells Were Formed by Fusion of Hepatobiliary Stem/Progenitor Cells with Hematopoietic Precursor-Derived Myeloid Intermediates. Stem Cells Dev. 2015, 24, 2467–2478. [Google Scholar] [CrossRef] [PubMed]
- Hass, R.; von der Ohe, J.; Ungefroren, H. Potential Role of MSC/Cancer Cell Fusion and EMT for Breast Cancer Stem Cell Formation. Cancers 2019, 11, 1432. [Google Scholar] [CrossRef] [PubMed]
- Hass, R.; von der Ohe, J.; Dittmar, T. Cancer Cell Fusion and Post-Hybrid Selection Process (PHSP). Cancers 2021, 13, 4636. [Google Scholar] [CrossRef] [PubMed]
- Laberge, G.S.; Duvall, E.; Haedicke, K.; Pawelek, J. Leukocyte(-)Cancer Cell Fusion-Genesis of a Deadly Journey. Cells 2019, 8, 170. [Google Scholar] [CrossRef] [PubMed]
- Lazova, R.; Chakraborty, A.; Pawelek, J.M. Leukocyte-cancer cell fusion: Initiator of the warburg effect in malignancy? Adv. Exp. Med. Biol. 2011, 714, 151–172. [Google Scholar] [CrossRef] [PubMed]
- Pawelek, J.; Chakraborty, A.; Lazova, R.; Yilmaz, Y.; Cooper, D.; Brash, D.; Handerson, T. Co-opting macrophage traits in cancer progression: A consequence of tumor cell fusion? Contrib. Microbiol. 2006, 13, 138–155. [Google Scholar] [CrossRef] [PubMed]
- Pawelek, J.M. Tumour-cell fusion as a source of myeloid traits in cancer. Lancet Oncol. 2005, 6, 988–993. [Google Scholar] [CrossRef] [PubMed]
- Pawelek, J.M. Cancer-cell fusion with migratory bone-marrow-derived cells as an explanation for metastasis: New therapeutic paradigms. Future Oncol. 2008, 4, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Shabo, I.; Svanvik, J.; Lindstrom, A.; Lechertier, T.; Trabulo, S.; Hulit, J.; Sparey, T.; Pawelek, J. Roles of cell fusion, hybridization and polyploid cell formation in cancer metastasis. World J. Clin. Oncol. 2020, 11, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Sieler, M.; Weiler, J.; Dittmar, T. Cell-Cell Fusion and the Roads to Novel Properties of Tumor Hybrid Cells. Cells 2021, 10, 1465. [Google Scholar] [CrossRef] [PubMed]
- Sutton, T.L.; Walker, B.S.; Wong, M.H. Digesting the Importance of Cell Fusion in the Intestine. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 299–302. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.F.; Xiang, W.; Xue, B.Z.; Wang, Y.H.; Yi, D.Y.; Jiang, X.B.; Zhao, H.Y.; Fu, P. Cell fusion in cancer hallmarks: Current research status and future indications. Oncol. Lett. 2021, 22, 530. [Google Scholar] [CrossRef] [PubMed]
- Weiler, J.; Dittmar, T. Cell Fusion in Human Cancer: The Dark Matter Hypothesis. Cells 2019, 8, 132. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.W.; Hung, C.N.; Chiu, C.H.; Tan, X.; Chen, M.; Chen, C.C.; Saeed, M.; Hsu, C.W.; Liss, M.A.; Wang, C.M.; et al. Phagocytosis-initiated tumor hybrid cells acquire a c-Myc-mediated quasi-polarization state for immunoevasion and distant dissemination. Nat. Commun. 2023, 14, 6569. [Google Scholar] [CrossRef] [PubMed]
- Pienta, K.J.; Hammarlund, E.U.; Brown, J.S.; Amend, S.R.; Axelrod, R.M. Cancer recurrence and lethality are enabled by enhanced survival and reversible cell cycle arrest of polyaneuploid cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2020838118. [Google Scholar] [CrossRef] [PubMed]
- Pienta, K.J.; Hammarlund, E.U.; Axelrod, R.; Brown, J.S.; Amend, S.R. Poly-aneuploid cancer cells promote evolvability, generating lethal cancer. Evol. Appl. 2020, 13, 1626–1634. [Google Scholar] [CrossRef] [PubMed]
- Amend, S.R.; Torga, G.; Lin, K.C.; Kostecka, L.G.; de Marzo, A.; Austin, R.H.; Pienta, K.J. Polyploid giant cancer cells: Unrecognized actuators of tumorigenesis, metastasis, and resistance. Prostate 2019, 79, 1489–1497. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Erenpreisa, J.; Sikora, E. Polyploid giant cancer cells: An emerging new field of cancer biology. Semin. Cancer Biol. 2022, 81, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.N.; Conley, P.; Klocke, C.D.; Sengupta, S.K.; Robinson, T.L.; Fan, Y.; Jones, J.A.; Gibbs, S.L.; Skalet, A.H.; Wu, G.; et al. Analysis of uveal melanoma scRNA sequencing data identifies neoplastic-immune hybrid cells that exhibit metastatic potential. bioRxiv 2023. [Google Scholar] [CrossRef]
- Lajoie, M.J.; Boyken, S.E.; Salter, A.I.; Bruffey, J.; Rajan, A.; Langan, R.A.; Olshefsky, A.; Muhunthan, V.; Bick, M.J.; Gewe, M.; et al. Designed protein logic to target cells with precise combinations of surface antigens. Science 2020, 369, 1637–1643. [Google Scholar] [CrossRef] [PubMed]
- Morsut, L.; Roybal, K.T.; Xiong, X.; Gordley, R.M.; Coyle, S.M.; Thomson, M.; Lim, W.A. Engineering Customized Cell Sensing and Response Behaviors Using Synthetic Notch Receptors. Cell 2016, 164, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Roybal, K.T.; Williams, J.Z.; Morsut, L.; Rupp, L.J.; Kolinko, I.; Choe, J.H.; Walker, W.J.; McNally, K.A.; Lim, W.A. Engineering T Cells with Customized Therapeutic Response Programs Using Synthetic Notch Receptors. Cell 2016, 167, 419–432.e416. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Feng, Z.; Ma, J.; Ling, S.; Cao, Y.; Gurung, B.; Wu, Y.; Katona, B.W.; O’Dwyer, K.P.; Siegel, D.L.; et al. Bispecific and split CAR T cells targeting CD13 and TIM3 eradicate acute myeloid leukemia. Blood 2020, 135, 713–723. [Google Scholar] [CrossRef]
- Cho, J.H.; Okuma, A.; Sofjan, K.; Lee, S.; Collins, J.J.; Wong, W.W. Engineering advanced logic and distributed computing in human CAR immune cells. Nat. Commun. 2021, 12, 792. [Google Scholar] [CrossRef] [PubMed]
- Angelici, B.; Shen, L.; Schreiber, J.; Abraham, A.; Benenson, Y. An AAV gene therapy computes over multiple cellular inputs to enable precise targeting of multifocal hepatocellular carcinoma in mice. Sci. Transl. Med. 2021, 13, eabh4456. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, A.M.; Raza, A. scRNAseq and High-Throughput Spatial Analysis of Tumor and Normal Microenvironment in Solid Tumors Reveal a Possible Origin of Circulating Tumor Hybrid Cells. Cancers 2024, 16, 1444. https://doi.org/10.3390/cancers16071444
Ali AM, Raza A. scRNAseq and High-Throughput Spatial Analysis of Tumor and Normal Microenvironment in Solid Tumors Reveal a Possible Origin of Circulating Tumor Hybrid Cells. Cancers. 2024; 16(7):1444. https://doi.org/10.3390/cancers16071444
Chicago/Turabian StyleAli, Abdullah Mahmood, and Azra Raza. 2024. "scRNAseq and High-Throughput Spatial Analysis of Tumor and Normal Microenvironment in Solid Tumors Reveal a Possible Origin of Circulating Tumor Hybrid Cells" Cancers 16, no. 7: 1444. https://doi.org/10.3390/cancers16071444
APA StyleAli, A. M., & Raza, A. (2024). scRNAseq and High-Throughput Spatial Analysis of Tumor and Normal Microenvironment in Solid Tumors Reveal a Possible Origin of Circulating Tumor Hybrid Cells. Cancers, 16(7), 1444. https://doi.org/10.3390/cancers16071444