

Transcription Factors and Markers Related to Epithelial–Mesenchymal Transition and Their Role in Resistance to Therapies in Head and Neck Cancers

{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
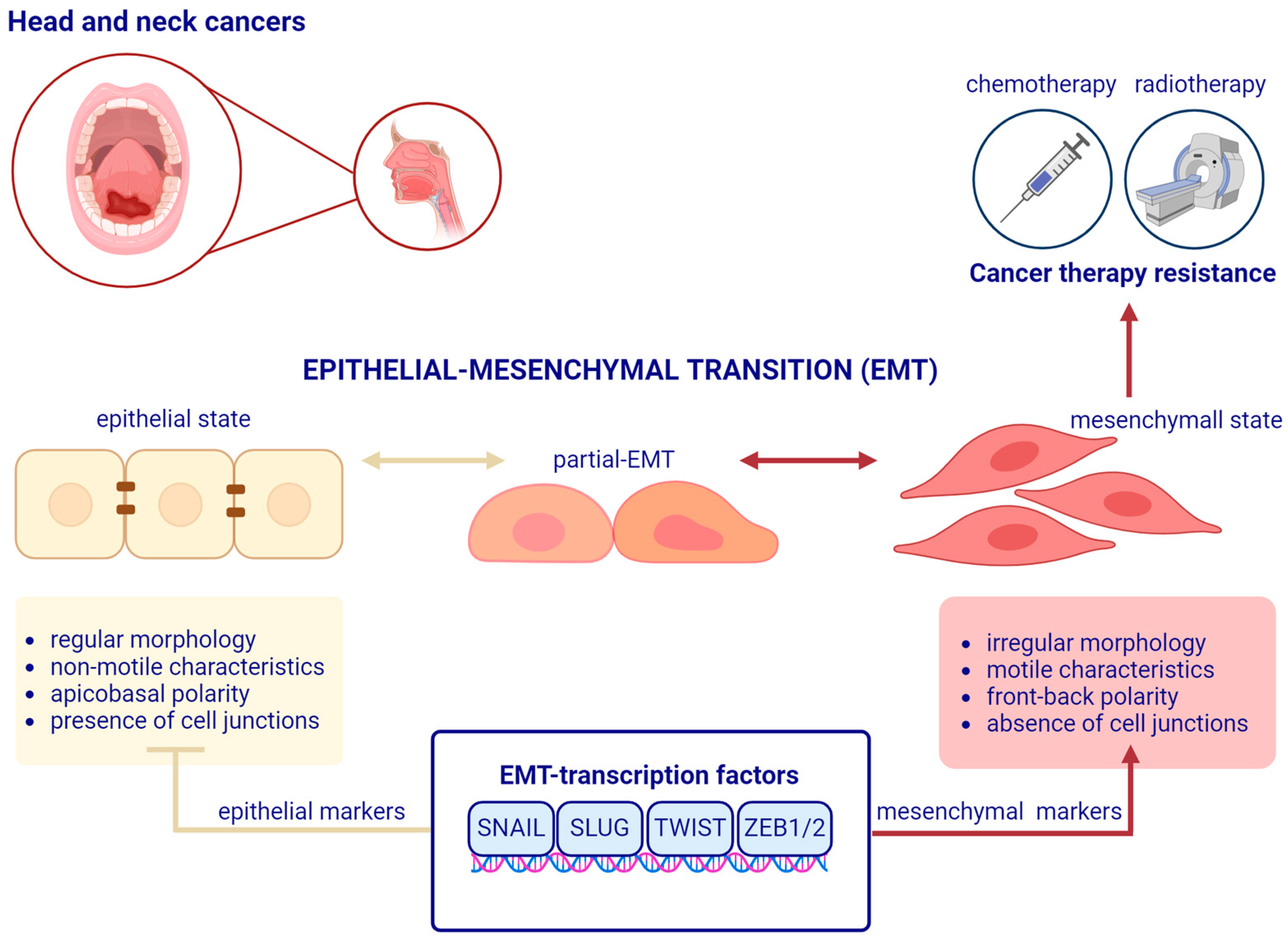
1. Introduction
2. A Brief Overview of Epithelial–Mesenchymal Transition
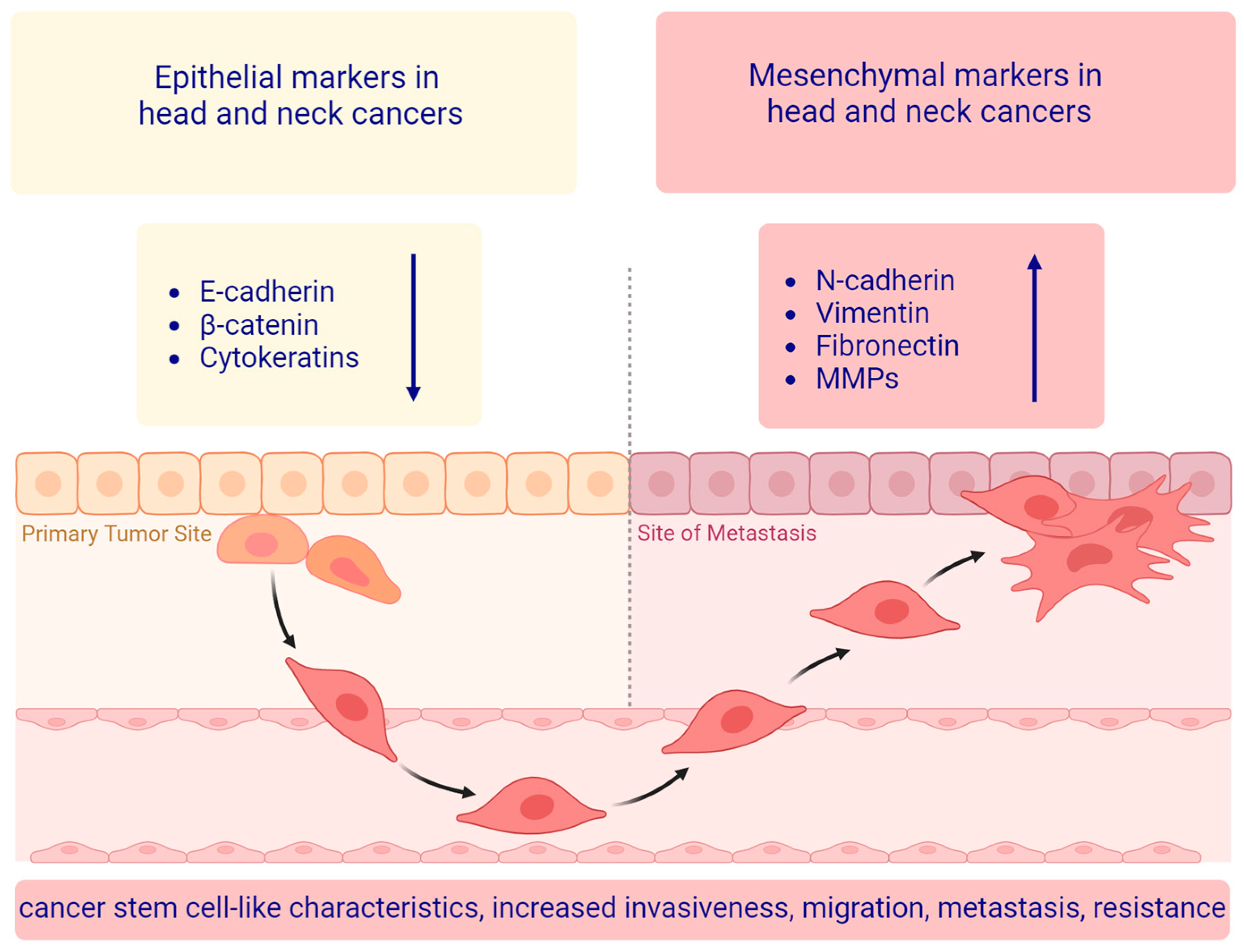
3. The Main EMT Markers in Head and Neck Squamous Cell Carcinoma
3.1. Epithelial Markers
3.1.1. E-Cadherin
3.1.2. β-Catenin
3.1.3. Cytokeratins
3.2. Mesenchymal Markers
3.2.1. Vimentin
3.2.2. Fibronectin-1
3.2.3. N-Cadherin
3.3. Other Protein Markers Associated with the EMT
Matrix Metalloproteinases
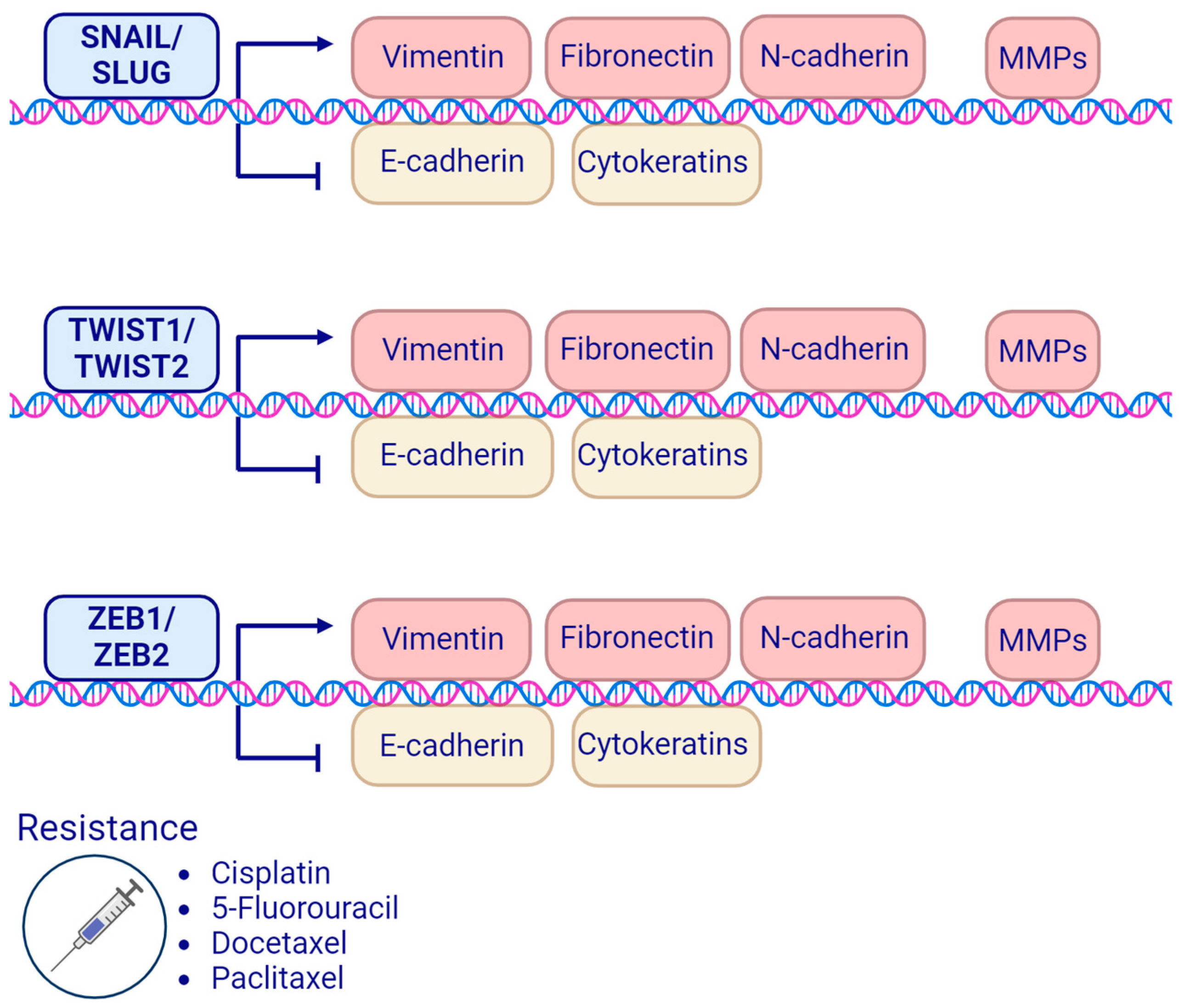
4. EMT-Related Transcription Factors and Treatment Resistance in Head and Neck Squamous Cell Carcinoma
4.1. SNAIL Family Transcription Factors
4.1.1. SNAIL Transcription Factor
4.1.2. SLUG Transcription Factor
4.2. TWIST Family Transcription Factors
4.3. ZEB Homeobox Family Transcription Factors
4.3.1. ZEB1 Transcription Factor
4.3.2. ZEB2 Transcription Factor
5. Summary of the Current Knowledge of EMT, Transcription Factors, and Markers in HNSCC
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AJCC | American Joint Committee on Cancer |
| ALDH | Aldehyde dehydrogenase |
| APC | Adenomatous polyposis coli |
| BRD4 | Bromodomain-containing protein 4 |
| CAFs | Cancer-associated fibroblasts |
| CBP | CREB-binding protein |
| CDDP | Cis-diamminedichloroplatinum(II) (Cisplatin) |
| CHK1 | Checkpoint kinase 1 |
| CHT | Chemotherapy |
| CK | Cytokeratin |
| cRNA | Circular RNA |
| CSC | Cancer stem cells |
| DDR | DNA damage response |
| deltaEF1 | Delta-crystallin enhancer-binding factor 1 |
| EBV | Epstein–Barr virus |
| ECM | Extracellular matrix |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial–mesenchymal transition |
| ERCC1 | Excision repair cross-complementation group 1 |
| FAK | Focal adhesion kinase |
| 5-FU | 5-Fluorouracil |
| HADAC | Histone deacetylase |
| HIF-1α | Hypoxia-inducible factor 1-alpha |
| HNC | Head and neck cancer |
| HNSCC | Head and neck squamous cell carcinoma |
| HPV | Human papillomavirus |
| IF | Immunofluorescence |
| IHC | Immunohistochemistry |
| LI | Labeling index |
| lncRNA | Long non-coding RNA |
| MAP | Mitogen-activated protein |
| MAPK | Mitogen-activated protein kinase |
| MDR | Multidrug resistance |
| MDR1 | Multidrug resistance protein 1 |
| MET | Mesenchymal–epithelial transition |
| miRNA | MicroRNA |
| MMP | Matrix metalloproteinase |
| mTORC1 | Mechanistic target of rapamycin complex 1 |
| ncRNA | Non-coding RNA |
| NER | Nucleotide excision repair |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NPC | Nasopharyngeal carcinoma cells |
| NSCLC | Non-small cell lung carcinoma |
| PCD | Programmed cell death |
| PTMs | Post-translational modifications |
| p-EMT | Partial epithelial–mesenchymal transition |
| P-gp | P-glycoprotein |
| RMHNC | Recurrent/metastatic head and neck carcinoma |
| ROCK2 | Rho-associated coiled-coil-containing protein kinase 2 |
| RTH | Radiotherapy |
| RT-PCR | Reverse transcription PCR |
| siRNA | Small interfering RNA |
| TCF-LEF | T-cell and lymphoid enhancer factors |
| TF | Transcription factor |
| TGF-β1 | Transforming growth factor-beta 1 |
| TJ | Tight junction |
| TKI | Tyrosine kinase inhibitor |
| TNF-R | Tumor necrosis factor receptor |
| TNF-α | Tumor necrosis factor α-induced |
| USP7 | Ubiquitin-specific protease 7 |
| ZFH | Zinc finger E-box-binding homeobox protein family |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Picon, H.; Guddati, A.K. Mechanisms of Resistance in Head and Neck Cancer. Am. J. Cancer Res. 2020, 10, 2742–2751. [Google Scholar] [PubMed]
- Guidi, A.; Codecà, C.; Ferrari, D. Chemotherapy and Immunotherapy for Recurrent and Metastatic Head and Neck Cancer: A Systematic Review. Med. Oncol. 2018, 35, 37. [Google Scholar] [CrossRef]
- Bhat, G.R.; Hyole, R.G.; Li, J. Head and Neck Cancer: Current Challenges and Future Perspectives. Adv. Cancer Res. 2021, 152, 67–102. [Google Scholar]
- Mody, M.D.; Rocco, J.W.; Yom, S.S.; Haddad, R.I.; Saba, N.F. Head and Neck Cancer. Lancet 2021, 398, 2289–2299. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.Q.M. Head and Neck Cancer. N. Engl. J. Med. 2020, 382, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.B.; Edge, S.B.; Greene, F.L.; Byrd, D.R.; Brookland, R.K.; Washington, M.K.; Gershenwald, J.E.; Compton, C.C.; Hess, K.R.; Sullivan, D.C.; et al. AJCC Cancer Staging Manual; Springer International Publishing: Berlin/Heidelberg, Germany, 2018; ISBN 9783319406176. [Google Scholar]
- Pfister, D.G.; Spencer, S.; Adelstein, D.; Adkins, D.; Anzai, Y.; Brizel, D.M.; Bruce, J.Y.; Busse, P.M.; Caudell, J.J.; Cmelak, A.J.; et al. Head and Neck Cancers, Version 2.2020, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2020, 18, 873–898. [Google Scholar] [CrossRef] [PubMed]
- Atashi, F.; Vahed, N.; Emamverdizadeh, P.; Fattahi, S.; Paya, L. Drug Resistance against 5-Fluorouracil and Cisplatin in the Treatment of Head and Neck Squamous Cell Carcinoma: A Systematic Review. J. Dent. Res. Dent. Clin. Dent. Prospect. 2021, 15, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Noguti, J.; De Moura, C.F.G.; De Jesus, G.P.P.; Da Silva, V.H.P.; Hossaka, T.A.; Oshima, C.T.F.; Ribeiro, D.A. Metastasis from Oral Cancer: An Overview. Cancer Genom. Proteom. 2012, 9, 329–335. [Google Scholar]
- Takes, R.P.; Rinaldo, A.; Silver, C.E.; Haigentz, M., Jr.; Woolgar, J.A.; Triantafyllou, A.; Mondin, V.; Paccagnella, D.; de Bree, R.; Shaha, A.R.; et al. Distant Metastases from Head and Neck Squamous Cell Carcinoma. Part I. Basic Aspects. Oral Oncol. 2012, 48, 775–779. [Google Scholar] [CrossRef]
- Göppel, J.; Möckelmann, N.; Münscher, A.; Sauter, G.; Schumacher, U. Expression of Epithelial–Mesenchymal Transition Regulating Transcription Factors in Head and Neck Squamous Cell Carcinomas. Anticancer Res. 2017, 37, 5435–5440. [Google Scholar] [PubMed]
- Lin, X.; Zhou, W.; Liu, Z.; Cao, W.; Lin, C. Targeting Cellular Metabolism in Head and Neck Cancer Precision Medicine Era: A Promising Strategy to Overcome Therapy Resistance. Oral Dis. 2023, 29, 3101–3120. [Google Scholar] [CrossRef] [PubMed]
- Siqueira, J.M.; Heguedusch, D.; Rodini, C.O.; Nunes, F.D.; Rodrigues, M.F.S.D. Mechanisms Involved in Cancer Stem Cell Resistance in Head and Neck Squamous Cell Carcinoma. Cancer Drug Resist. 2023, 6, 116–137. [Google Scholar] [CrossRef]
- Debnath, P.; Huirem, R.S.; Dutta, P.; Palchaudhuri, S. Epithelial-Mesenchymal Transition and Its Transcription Factors. Biosci. Rep. 2022, 42, BSR20211754. [Google Scholar] [CrossRef] [PubMed]
- Hay, E.D. Organization and Fine Structure of Epithelium and Mesenchyme in the Developing Chick Embryo. In Epithelial-Mesenchymal Interactions: 18th Hahnemann Symposium; Williams and Wilkins: Baltimore, MD, USA, 1968. [Google Scholar]
- Hay, E.D. An Overview of Epithelio-Mesenchymal Transformation. Acta Anat. 1995, 154, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The Basics of Epithelial-Mesenchymal Transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial-Mesenchymal Transitions in Tumour Progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Weinberg, R.A. Epithelial-to-Mesenchymal Transition in Cancer: Complexity and Opportunities. Front. Med. 2018, 12, 361–373. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Aiello, N.M.; Kang, Y. Context-Dependent EMT Programs in Cancer Metastasis. J. Exp. Med. 2019, 216, 1016–1026. [Google Scholar] [CrossRef]
- Samatov, T.R.; Tonevitsky, A.G.; Schumacher, U. Epithelial-Mesenchymal Transition: Focus on Metastatic Cascade, Alternative Splicing, Non-Coding RNAs and Modulating Compounds. Mol. Cancer 2013, 12, 107. [Google Scholar] [CrossRef] [PubMed]
- Den Hollander, P.; Maddela, J.J.; Mani, S.A. Spatial and Temporal Relationship between Epithelial-Mesenchymal Transition (EMT) and Stem Cells in Cancer. Clin. Chem. 2024, 70, 190–205. [Google Scholar] [CrossRef]
- Serrano-Gomez, S.J.; Maziveyi, M.; Alahari, S.K. Regulation of Epithelial-Mesenchymal Transition through Epigenetic and Post-Translational Modifications. Mol. Cancer 2016, 15, 18. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Fang, J. Epigenetic Regulation of Epithelial–mesenchymal Transition. Cell. Mol. Life Sci. 2016, 73, 4493–4515. [Google Scholar] [CrossRef] [PubMed]
- Shang, B.-Q.; Li, M.-L.; Quan, H.-Y.; Hou, P.-F.; Li, Z.-W.; Chu, S.-F.; Zheng, J.-N.; Bai, J. Functional Roles of Circular RNAs during Epithelial-to-Mesenchymal Transition. Mol. Cancer 2019, 18, 138. [Google Scholar] [CrossRef] [PubMed]
- Cavallari, I.; Ciccarese, F.; Sharova, E.; Urso, L.; Raimondi, V.; Silic-Benussi, M.; D’Agostino, D.M.; Ciminale, V. The miR-200 Family of microRNAs: Fine Tuners of Epithelial-Mesenchymal Transition and Circulating Cancer Biomarkers. Cancers 2021, 13, 5874. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Taniue, K.; Ono, Y.; Fujiya, M.; Mizukami, Y.; Okumura, T. Long Non-Coding RNAs in Epithelial-Mesenchymal Transition of Pancreatic Cancer. Front. Mol. Biosci. 2021, 8, 717890. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and Definitions for Research on Epithelial–mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Taki, M.; Abiko, K.; Ukita, M.; Murakami, R.; Yamanoi, K.; Yamaguchi, K.; Hamanishi, J.; Baba, T.; Matsumura, N.; Mandai, M. Tumor Immune Microenvironment during Epithelial-Mesenchymal Transition. Clin. Cancer Res. 2021, 27, 4669–4679. [Google Scholar] [CrossRef]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in Cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef]
- Derynck, R.; Weinberg, R.A. EMT and Cancer: More Than Meets the Eye. Dev. Cell 2019, 49, 313–316. [Google Scholar] [CrossRef]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624.e24. [Google Scholar] [CrossRef] [PubMed]
- Parikh, A.S.; Puram, S.V.; Faquin, W.C.; Richmon, J.D.; Emerick, K.S.; Deschler, D.G.; Varvares, M.A.; Tirosh, I.; Bernstein, B.E.; Lin, D.T. Immunohistochemical Quantification of Partial-EMT in Oral Cavity Squamous Cell Carcinoma Primary Tumors Is Associated with Nodal Metastasis. Oral Oncol. 2019, 99, 104458. [Google Scholar] [CrossRef] [PubMed]
- Tada, H.; Takahashi, H.; Ida, S.; Nagata, Y.; Chikamatsu, K. Epithelial–Mesenchymal Transition Status of Circulating Tumor Cells Is Associated with Tumor Relapse in Head and Neck Squamous Cell Carcinoma. Anticancer Res. 2020, 40, 3559–3564. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.-J.; Song, P.-P.; Zhou, H.; Shen, X.-H.; Wang, J.-G.; Ma, X.-F.; Gu, Y.-J.; Liu, D.-D.; Feng, A.-N.; Qian, X.-Y.; et al. Role of Epithelial-Mesenchymal Transition Markers E-Cadherin, N-Cadherin, β-Catenin and ZEB2 in Laryngeal Squamous Cell Carcinoma. Oncol. Lett. 2018, 15, 3472–3481. [Google Scholar] [CrossRef] [PubMed]
- Baumeister, P.; Zhou, J.; Canis, M.; Gires, O. Epithelial-to-Mesenchymal Transition-Derived Heterogeneity in Head and Neck Squamous Cell Carcinomas. Cancers 2021, 13, 5355. [Google Scholar] [CrossRef]
- Ling, Z.; Cheng, B.; Tao, X. Epithelial-to-Mesenchymal Transition in Oral Squamous Cell Carcinoma: Challenges and Opportunities. Int. J. Cancer 2021, 148, 1548–1561. [Google Scholar] [CrossRef] [PubMed]
- Hoch, C.C.; Stögbauer, F.; Wollenberg, B. Unraveling the Role of Epithelial-Mesenchymal Transition in Adenoid Cystic Carcinoma of the Salivary Glands: A Comprehensive Review. Cancers 2023, 15, 2886. [Google Scholar] [CrossRef]
- Chen, C.; Zimmermann, M.; Tinhofer, I.; Kaufmann, A.M.; Albers, A.E. Epithelial-to-Mesenchymal Transition and Cancer Stem(-Like) Cells in Head and Neck Squamous Cell Carcinoma. Cancer Lett. 2013, 338, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Barrett, T.F.; Paolini, R.; Parikh, A.; Puram, S.V. Partial EMT in Head and Neck Cancer Biology: A Spectrum instead of a Switch. Oncogene 2021, 40, 5049–5065. [Google Scholar] [CrossRef]
- Mandal, M.; Myers, J.N.; Lippman, S.M.; Johnson, F.M.; Williams, M.D.; Rayala, S.; Ohshiro, K.; Rosenthal, D.I.; Weber, R.S.; Gallick, G.E.; et al. Epithelial to Mesenchymal Transition in Head and Neck Squamous Carcinoma. Cancer 2008, 112, 2088–2100. [Google Scholar] [CrossRef] [PubMed]
- Nijkamp, M.M.; Span, P.N.; Hoogsteen, I.J.; van der Kogel, A.J.; Kaanders, J.H.A.M.; Bussink, J. Expression of E-Cadherin and Vimentin Correlates with Metastasis Formation in Head and Neck Squamous Cell Carcinoma Patients. Radiother. Oncol. 2011, 99, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Loh, C.-Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef] [PubMed]
- Ingruber, J.; Dudás, J.; Savic, D.; Schweigl, G.; Steinbichler, T.B.; Greier, M.D.C.; Santer, M.; Carollo, S.; Trajanoski, Z.; Riechelmann, H. EMT-Related Transcription Factors and Protein Stabilization Mechanisms Involvement in Cadherin Switch of Head and Neck Squamous Cell Carcinoma. Exp. Cell Res. 2022, 414, 113084. [Google Scholar] [CrossRef] [PubMed]
- Heuberger, J.; Birchmeier, W. Interplay of Cadherin-Mediated Cell Adhesion and Canonical Wnt Signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002915. [Google Scholar] [CrossRef]
- Clevers, H. Wnt/beta-Catenin Signaling in Development and Disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef]
- Kaur, J.; Sawhney, M.; DattaGupta, S.; Shukla, N.K.; Srivastava, A.; Walfish, P.G.; Ralhan, R. Clinical Significance of Altered Expression of β-Catenin and E-Cadherin in Oral Dysplasia and Cancer: Potential Link with ALCAM Expression. PLoS ONE 2013, 8, e67361. [Google Scholar] [CrossRef]
- Angadi, P.V.; Patil, P.V.; Angadi, V.; Mane, D.; Shekar, S.; Hallikerimath, S.; Kale, A.D.; Kardesai, S.G. Immunoexpression of Epithelial Mesenchymal Transition Proteins E-Cadherin, β-Catenin, and N-Cadherin in Oral Squamous Cell Carcinoma. Int. J. Surg. Pathol. 2016, 24, 696–703. [Google Scholar] [CrossRef]
- Kakinouchi, K.; Yoshie, S.; Tsuji, S.; Murono, S.; Hazama, A. Dysfunction of Cl- Channels Promotes Epithelial to Mesenchymal Transition in Oral Squamous Cell Carcinoma via Activation of Wnt/β-Catenin Signaling Pathway. Biochem. Biophys. Res. Commun. 2021, 555, 95–101. [Google Scholar] [CrossRef]
- Reed, E.R.; Jankowski, S.A.; Spinella, A.J.; Noonan, V.; Haddad, R.; Nomoto, K.; Matsui, J.; Bais, M.V.; Varelas, X.; Kukuruzinska, M.A.; et al. β-catenin/CBP Activation of mTORC1 Signaling Promotes Partial Epithelial-Mesenchymal States in Head and Neck Cancer. Transl. Res. 2023, 260, 46–60. [Google Scholar] [CrossRef]
- Schaafsma, H.E.; Van Der Velden, L.A.; Manni, J.J.; Peters, H.; Link, M.; Rutter, D.J.; Ramaekers, F.C. Increased Expression of Cytokeratins 8, 18 and Vimentin in the Invasion Front of Mucosal Squamous Cell Carcinoma. J. Pathol. 1993, 170, 77–86. [Google Scholar] [CrossRef]
- Hembrough, T.A.; Vasudevan, J.; Allietta, M.M.; Glass, W.F., 2nd; Gonias, S.L. A Cytokeratin 8-like Protein with Plasminogen-Binding Activity Is Present on the External Surfaces of Hepatocytes, HepG2 Cells and Breast Carcinoma Cell Lines. J. Cell Sci. 1995, 108 Pt 3, 1071–1082. [Google Scholar] [CrossRef]
- Xu, X.C.; Lee, J.S.; Lippman, S.M.; Ro, J.Y.; Hong, W.K.; Lotan, R. Increased Expression of Cytokeratins CK8 and CK19 Is Associated with Head and Neck Carcinogenesis. Cancer Epidemiol. Biomark. Prev. 1995, 4, 871–876. [Google Scholar]
- Andratschke, M.; Hagedorn, H.; Nerlich, A. Expression of the Epithelial Cell Adhesion Molecule and Cytokeratin 8 in Head and Neck Squamous Cell Cancer: A Comparative Study. Anticancer Res. 2015, 35, 3953–3960. [Google Scholar]
- Gires, O.; Münz, M.; Schaffrik, M.; Kieu, C.; Rauch, J.; Ahlemann, M.; Eberle, D.; Mack, B.; Wollenberg, B.; Lang, S.; et al. Profile Identification of Disease-Associated Humoral Antigens Using AMIDA, a Novel Proteomics-Based Technology. Cell. Mol. Life Sci. 2004, 61, 1198–1207. [Google Scholar] [CrossRef]
- Rauch, J.; Ahlemann, M.; Schaffrik, M.; Mack, B.; Ertongur, S.; Andratschke, M.; Zeidler, R.; Lang, S.; Gires, O. Allogenic Antibody-Mediated Identification of Head and Neck Cancer Antigens. Biochem. Biophys. Res. Commun. 2004, 323, 156–162. [Google Scholar] [CrossRef]
- Tanaka, S.; Kawano, S.; Hattori, T.; Matsubara, R.; Sakamoto, T.; Hashiguchi, Y.; Kaneko, N.; Mikami, Y.; Morioka, M.; Maruse, Y.; et al. Cytokeratin 19 as a Biomarker of Highly Invasive Oral Squamous Cell Carcinoma with Metastatic Potential. J. Oral Maxillofac. Surg. Med. Pathol. 2020, 32, 1–7. [Google Scholar] [CrossRef]
- Sakamoto, K.; Aragaki, T.; Morita, K.-I.; Kawachi, H.; Kayamori, K.; Nakanishi, S.; Omura, K.; Miki, Y.; Okada, N.; Katsube, K.-I.; et al. Down-Regulation of Keratin 4 and Keratin 13 Expression in Oral Squamous Cell Carcinoma and Epithelial Dysplasia: A Clue for Histopathogenesis. Histopathology 2011, 58, 531–542. [Google Scholar] [CrossRef]
- Mikami, T.; Cheng, J.; Maruyama, S.; Kobayashi, T.; Funayama, A.; Yamazaki, M.; Adeola, H.A.; Wu, L.; Shingaki, S.; Saito, C.; et al. Emergence of Keratin 17 vs. Loss of Keratin 13: Their Reciprocal Immunohistochemical Profiles in Oral Carcinoma in Situ. Oral Oncol. 2011, 47, 497–503. [Google Scholar] [CrossRef]
- Pandey, S.; Søland, T.M.; Bjerkli, I.H.; Sand, L.P.; Petersen, F.C.; Costea, D.E.; Senguven, B.; Sapkota, D. Combined Loss of Expression of Involucrin and Cytokeratin 13 Is Associated with Poor Prognosis in Squamous Cell Carcinoma of Mobile Tongue. Head Neck 2021, 43, 3374–3385. [Google Scholar] [CrossRef]
- Liu, S.; Liu, L.; Ye, W.; Ye, D.; Wang, T.; Guo, W.; Liao, Y.; Xu, D.; Song, H.; Zhang, L.; et al. High Vimentin Expression Associated with Lymph Node Metastasis and Predicated a Poor Prognosis in Oral Squamous Cell Carcinoma. Sci. Rep. 2016, 6, 38834. [Google Scholar] [CrossRef]
- Liu, P.-F.; Kang, B.-H.; Wu, Y.-M.; Sun, J.-H.; Yen, L.-M.; Fu, T.-Y.; Lin, Y.-C.; Liou, H.-H.; Lin, Y.-S.; Sie, H.-C.; et al. Vimentin Is a Potential Prognostic Factor for Tongue Squamous Cell Carcinoma among Five Epithelial-Mesenchymal Transition-Related Proteins. PLoS ONE 2017, 12, e0178581. [Google Scholar] [CrossRef]
- Evans, R.M. Vimentin: The Conundrum of the Intermediate Filament Gene Family. Bioessays 1998, 20, 79–86. [Google Scholar] [CrossRef]
- Hol, E.M.; Capetanaki, Y. Type III Intermediate Filaments Desmin, Glial Fibrillary Acidic Protein (GFAP), Vimentin, and Peripherin. Cold Spring Harb. Perspect. Biol. 2017, 9, a021642. [Google Scholar] [CrossRef]
- Satelli, A.; Li, S. Vimentin in Cancer and Its Potential as a Molecular Target for Cancer Therapy. Cell. Mol. Life Sci. 2011, 68, 3033–3046. [Google Scholar] [CrossRef]
- Ivaska, J.; Pallari, H.-M.; Nevo, J.; Eriksson, J.E. Novel Functions of Vimentin in Cell Adhesion, Migration, and Signaling. Exp. Cell Res. 2007, 313, 2050–2062. [Google Scholar] [CrossRef]
- Lowery, J.; Kuczmarski, E.R.; Herrmann, H.; Goldman, R.D. Intermediate Filaments Play a Pivotal Role in Regulating Cell Architecture and Function. J. Biol. Chem. 2015, 290, 17145–17153. [Google Scholar] [CrossRef]
- Wu, S.; Du, Y.; Beckford, J.; Alachkar, H. Upregulation of the EMT Marker Vimentin Is Associated with Poor Clinical Outcome in Acute Myeloid Leukemia. J. Transl. Med. 2018, 16, 170. [Google Scholar] [CrossRef]
- Gundamaraju, R.; Lu, W.; Paul, M.K.; Jha, N.K.; Gupta, P.K.; Ojha, S.; Chattopadhyay, I.; Rao, P.V.; Ghavami, S. Autophagy and EMT in Cancer and Metastasis: Who Controls Whom? Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166431. [Google Scholar] [CrossRef]
- Ridge, K.M.; Eriksson, J.E.; Pekny, M.; Goldman, R.D. Roles of Vimentin in Health and Disease. Genes Dev. 2022, 36, 391–407. [Google Scholar] [CrossRef]
- Gilles, C.; Polette, M.; Mestdagt, M.; Nawrocki-Raby, B.; Ruggeri, P.; Birembaut, P.; Foidart, J.-M. Transactivation of Vimentin by β-Catenin in Human Breast Cancer Cells1. Cancer Res. 2003, 63, 2658–2664. [Google Scholar]
- Wang, X.; Ji, S.; Ma, Y.; Xing, X.; Zhou, Y.; Xu, X.; Song, J.; Wang, S.; Jiang, W.; Wang, X.; et al. Vimentin Plays an Important Role in the Promotion of Breast Cancer Cell Migration and Invasion by Leucine Aminopeptidase 3. Cytotechnology 2020, 72, 639–647. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, P.; Su, X.-J.; Zhang, B. The Ubiquitin Ligase TRIM56 Inhibits Ovarian Cancer Progression by Targeting Vimentin. J. Cell. Physiol. 2018, 233, 2420–2425. [Google Scholar] [CrossRef]
- Wei, J.; Xu, G.; Wu, M.; Zhang, Y.; Li, Q.; Liu, P.; Zhu, T.; Song, A.; Zhao, L.; Han, Z.; et al. Overexpression of Vimentin Contributes to Prostate Cancer Invasion and Metastasis via Src Regulation. Anticancer Res. 2008, 28, 327–334. [Google Scholar]
- Lang, S.H.; Hyde, C.; Reid, I.N.; Hitchcock, I.S.; Hart, C.A.; Bryden, A.A.G.; Villette, J.-M.; Stower, M.J.; Maitland, N.J. Enhanced Expression of Vimentin in Motile Prostate Cell Lines and in Poorly Differentiated and Metastatic Prostate Carcinoma. Prostate 2002, 52, 253–263. [Google Scholar] [CrossRef]
- Tadokoro, A.; Kanaji, N.; Liu, D.; Yokomise, H.; Haba, R.; Ishii, T.; Takagi, T.; Watanabe, N.; Kita, N.; Kadowaki, N.; et al. Vimentin Regulates Invasiveness and Is a Poor Prognostic Marker in Non-Small Cell Lung Cancer. Anticancer Res. 2016, 36, 1545–1551. [Google Scholar]
- Wang, Q.; Zhu, G.; Lin, C.; Lin, P.; Chen, H.; He, R.; Huang, Y.; Yang, S.; Ye, J. Vimentin Affects Colorectal Cancer Proliferation, Invasion, and Migration via Regulated by Activator Protein 1. J. Cell. Physiol. 2021, 236, 7591–7604. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, L.; Dong, X.; Liu, L.; Huo, L.; Chen, H. High Expression of Vimentin Is Associated with Progression and a Poor Outcome in Glioblastoma. Appl. Immunohistochem. Mol. Morphol. 2018, 26, 337–344. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, X.; Xiao, Y.; Wu, L.; Peng, Y.; Tang, W.; Liu, G.; Sun, Y.; Wang, J.; Zhu, H.; et al. Coexpression of FOXK1 and Vimentin Promotes EMT, Migration, and Invasion in Gastric Cancer Cells. J. Mol. Med. 2019, 97, 163–176. [Google Scholar] [CrossRef]
- Hendrix, M.J.; Seftor, E.A.; Chu, Y.W.; Seftor, R.E.; Nagle, R.B.; McDaniel, K.M.; Leong, S.P.; Yohem, K.H.; Leibovitz, A.M.; Meyskens, F.L., Jr. Coexpression of Vimentin and Keratins by Human Melanoma Tumor Cells: Correlation with Invasive and Metastatic Potential. J. Natl. Cancer Inst. 1992, 84, 165–174. [Google Scholar] [CrossRef]
- Li, M.; Zhang, B.; Sun, B.; Wang, X.; Ban, X.; Sun, T.; Liu, Z.; Zhao, X. A Novel Function for Vimentin: The Potential Biomarker for Predicting Melanoma Hematogenous Metastasis. J. Exp. Clin. Cancer Res. 2010, 29, 109. [Google Scholar] [CrossRef]
- Liu, C.-Y.; Lin, H.-H.; Tang, M.-J.; Wang, Y.-K. Vimentin Contributes to Epithelial-Mesenchymal Transition Cancer Cell Mechanics by Mediating Cytoskeletal Organization and Focal Adhesion Maturation. Oncotarget 2015, 6, 15966–15983. [Google Scholar] [CrossRef]
- Huang, Y.; Tong, J.; He, F.; Yu, X.; Fan, L.; Hu, J.; Tan, J.; Chen, Z. miR-141 Regulates TGF-β1-Induced Epithelial-Mesenchymal Transition through Repression of HIPK2 Expression in Renal Tubular Epithelial Cells. Int. J. Mol. Med. 2015, 35, 311–318. [Google Scholar] [CrossRef]
- Sun, B.O.; Fang, Y.; Li, Z.; Chen, Z.; Xiang, J. Role of Cellular Cytoskeleton in Epithelial-Mesenchymal Transition Process during Cancer Progression. Biomed. Rep. 2015, 3, 603–610. [Google Scholar] [CrossRef]
- Liu, X.; Wang, C.; Chen, Z.; Jin, Y.; Wang, Y.; Kolokythas, A.; Dai, Y.; Zhou, X. MicroRNA-138 Suppresses Epithelial-Mesenchymal Transition in Squamous Cell Carcinoma Cell Lines. Biochem. J. 2011, 440, 23–31. [Google Scholar] [CrossRef]
- Haddad, Y.; Choi, W.; McConkey, D.J. Delta-Crystallin Enhancer Binding Factor 1 Controls the Epithelial to Mesenchymal Transition Phenotype and Resistance to the Epidermal Growth Factor Receptor Inhibitor Erlotinib in Human Head and Neck Squamous Cell Carcinoma Lines. Clin. Cancer Res. 2009, 15, 532–542. [Google Scholar] [CrossRef]
- Albanell, J.; Rojo, F.; Baselga, J. Pharmacodynamic Studies with the Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor ZD1839. Semin. Oncol. 2001, 28, 56–66. [Google Scholar] [CrossRef]
- Lin, T.-C.; Yang, C.-H.; Cheng, L.-H.; Chang, W.-T.; Lin, Y.-R.; Cheng, H.-C. Fibronectin in Cancer: Friend or Foe. Cells 2019, 9, 27. [Google Scholar] [CrossRef]
- Singh, P.; Carraher, C.; Schwarzbauer, J.E. Assembly of Fibronectin Extracellular Matrix. Annu. Rev. Cell Dev. Biol. 2010, 26, 397–419. [Google Scholar] [CrossRef]
- Warawdekar, U.M.; Zingde, S.M.; Iyer, K.S.N.; Jagannath, P.; Mehta, A.R.; Mehta, N.G. Elevated Levels and Fragmented Nature of Cellular Fibronectin in the Plasma of Gastrointestinal and Head and Neck Cancer Patients. Clin. Chim. Acta 2006, 372, 83–93. [Google Scholar] [CrossRef]
- Sponziello, M.; Rosignolo, F.; Celano, M.; Maggisano, V.; Pecce, V.; De Rose, R.F.; Lombardo, G.E.; Durante, C.; Filetti, S.; Damante, G.; et al. Fibronectin-1 Expression Is Increased in Aggressive Thyroid Cancer and Favors the Migration and Invasion of Cancer Cells. Mol. Cell. Endocrinol. 2016, 431, 123–132. [Google Scholar] [CrossRef]
- Pang, X.; Tang, Y.; Liang, X. Transforming Growth Factor-β Signaling in Head and Neck Squamous Cell Carcinoma: Insights into Cellular Responses (Review). Oncol. Lett. 2018, 16, 4799–4806. [Google Scholar] [CrossRef]
- Zhang, W.; Li, J.; Wu, Y.; Ge, H.; Song, Y.; Wang, D.; Yuan, H.; Jiang, H.; Wang, Y.; Cheng, J. TEAD4 Overexpression Promotes Epithelial-Mesenchymal Transition and Associates with Aggressiveness and Adverse Prognosis in Head Neck Squamous Cell Carcinoma. Cancer Cell Int. 2018, 18, 178. [Google Scholar] [CrossRef]
- Kim, N.; Ryu, H.; Kim, S.; Joo, M.; Jeon, H.J.; Lee, M.-W.; Song, I.-C.; Kim, M.-N.; Kim, J.-M.; Lee, H.J. CXCR7 Promotes Migration and Invasion in Head and Neck Squamous Cell Carcinoma by Upregulating TGF-β1/Smad2/3 Signaling. Sci. Rep. 2019, 9, 18100. [Google Scholar] [CrossRef]
- Jensen, D.H.; Dabelsteen, E.; Specht, L.; Fiehn, A.M.K.; Therkildsen, M.H.; Jønson, L.; Vikesaa, J.; Nielsen, F.C.; von Buchwald, C. Molecular Profiling of Tumour Budding Implicates TGFβ-Mediated Epithelial-Mesenchymal Transition as a Therapeutic Target in Oral Squamous Cell Carcinoma. J. Pathol. 2015, 236, 505–516. [Google Scholar] [CrossRef]
- Jiang, J.; Zheng, M.; Zhang, M.; Yang, X.; Li, L.; Wang, S.-S.; Wu, J.-S.; Yu, X.-H.; Wu, J.-B.; Pang, X.; et al. PRRX1 Regulates Cellular Phenotype Plasticity and Dormancy of Head and Neck Squamous Cell Carcinoma Through miR-642b-3p. Neoplasia 2019, 21, 216–229. [Google Scholar] [CrossRef]
- Zhang, J.; Cheng, Q.; Zhou, Y.; Wang, Y.; Chen, X. Slug Is a Key Mediator of Hypoxia Induced Cadherin Switch in HNSCC: Correlations with Poor Prognosis. Oral Oncol. 2013, 49, 1043–1050. [Google Scholar] [CrossRef]
- Cappellesso, R.; Marioni, G.; Crescenzi, M.; Giacomelli, L.; Guzzardo, V.; Mussato, A.; Staffieri, A.; Martini, A.; Blandamura, S.; Fassina, A. The Prognostic Role of the Epithelial-Mesenchymal Transition Markers E-Cadherin and Slug in Laryngeal Squamous Cell Carcinoma. Histopathology 2015, 67, 491–500. [Google Scholar] [CrossRef]
- Katafiasz, D.; Smith, L.M.; Wahl, J.K., 3rd. Slug (SNAI2) Expression in Oral SCC Cells Results in Altered Cell-Cell Adhesion and Increased Motility. Cell Adhes. Migr. 2011, 5, 315–322. [Google Scholar] [CrossRef]
- Hakim, S.G.; Taubitz, C.; Hoppe, S.; Steller, D.; Rades, D.; Ribbat-Idel, J.; Alsharif, U.; Falougy, M. Prognostic Impact of the Loss of E-Cadherin and de Novo Expression of N-Cadherin at the Invasive Front of Primary and Recurrent Oral Squamous Cell Carcinoma. Front. Oncol. 2023, 13, 1151879. [Google Scholar] [CrossRef]
- Bachir, A.I.; Horwitz, A.R.; Nelson, W.J.; Bianchini, J.M. Actin-Based Adhesion Modules Mediate Cell Interactions with the Extracellular Matrix and Neighboring Cells. Cold Spring Harb. Perspect. Biol. 2017, 9, a023234. [Google Scholar] [CrossRef]
- Essid, N.; Chambard, J.C.; Elgaaïed, A.B. Induction of Epithelial-Mesenchymal Transition (EMT) and Gli1 Expression in Head and Neck Squamous Cell Carcinoma (HNSCC) Spheroid Cultures. Bosn. J. Basic Med. Sci. 2018, 18, 336–346. [Google Scholar] [CrossRef]
- Verma, R.P.; Hansch, C. Matrix Metalloproteinases (MMPs): Chemical-Biological Functions and (Q)SARs. Bioorg. Med. Chem. 2007, 15, 2223–2268. [Google Scholar] [CrossRef]
- Jobin, P.G.; Butler, G.S.; Overall, C.M. New Intracellular Activities of Matrix Metalloproteinases Shine in the Moonlight. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2043–2055. [Google Scholar] [CrossRef]
- Scheau, C.; Badarau, I.A.; Costache, R.; Caruntu, C.; Mihai, G.L.; Didilescu, A.C.; Constantin, C.; Neagu, M. The Role of Matrix Metalloproteinases in the Epithelial-Mesenchymal Transition of Hepatocellular Carcinoma. Anal. Cell. Pathol. 2019, 2019, 9423907. [Google Scholar] [CrossRef]
- Li, Z.; Wei, J.; Chen, B.; Wang, Y.; Yang, S.; Wu, K.; Meng, X. The Role of MMP-9 and MMP-9 Inhibition in Different Types of Thyroid Carcinoma. Molecules 2023, 28, 3705. [Google Scholar] [CrossRef]
- Christopoulos, T.A.; Papageorgakopoulou, N.; Ravazoula, P.; Mastronikolis, N.S.; Papadas, T.A.; Theocharis, D.A.; Vynios, D.H. Expression of Metalloproteinases and Their Tissue Inhibitors in Squamous Cell Laryngeal Carcinoma. Oncol. Rep. 2007, 18, 855–860. [Google Scholar] [CrossRef][Green Version]
- Grzelczyk, W.L.; Szemraj, J.; Józefowicz-Korczyńska, M. The Matrix Metalloproteinase in Larynx Cancer. Postep. Hig. Med. Dosw. 2016, 70, 1190–1197. [Google Scholar]
- Nissinen, L.; Kähäri, V.-M. Matrix Metalloproteinases in Inflammation. Biochim. Biophys. Acta 2014, 1840, 2571–2580. [Google Scholar] [CrossRef]
- Canel, M.; Secades, P.; Garzón-Arango, M.; Allonca, E.; Suarez, C.; Serrels, A.; Frame, M.; Brunton, V.; Chiara, M.-D. Involvement of Focal Adhesion Kinase in Cellular Invasion of Head and Neck Squamous Cell Carcinomas via Regulation of MMP-2 Expression. Br. J. Cancer 2008, 98, 1274–1284. [Google Scholar] [CrossRef]
- Aparna, M.; Rao, L.; Kunhikatta, V.; Radhakrishnan, R. The Role of MMP-2 and MMP-9 as Prognostic Markers in the Early Stages of Tongue Squamous Cell Carcinoma. J. Oral Pathol. Med. 2015, 44, 345–352. [Google Scholar] [CrossRef]
- Cho, H.Y.; Lee, S.W.; Jeon, Y.H.; Lee, D.H.; Kim, G.W.; Yoo, J.; Kim, S.Y.; Kwon, S.H. Combination of ACY-241 and JQ1 Synergistically Suppresses Metastasis of HNSCC via Regulation of MMP-2 and MMP-9. Int. J. Mol. Sci. 2020, 21, 6873. [Google Scholar] [CrossRef]
- Thierauf, J.; Veit, J.A.; Hess, J. Epithelial-to-Mesenchymal Transition in the Pathogenesis and Therapy of Head and Neck Cancer. Cancers 2017, 9, 76. [Google Scholar] [CrossRef]
- Wan, Y.; Liu, H.; Zhang, M.; Huang, Z.; Zhou, H.; Zhu, Y.; Tao, Y.; Xie, N.; Liu, X.; Hou, J.; et al. Prognostic Value of Epithelial-Mesenchymal Transition-Inducing Transcription Factors in Head and Neck Squamous Cell Carcinoma: A Meta-Analysis. Head Neck 2020, 42, 1067–1076. [Google Scholar] [CrossRef]
- Manzanares, M.; Blanco, M.J.; Nieto, M.A. Snail3 Orthologues in Vertebrates: Divergent Members of the Snail Zinc-Finger Gene Family. Dev. Genes Evol. 2004, 214, 47–53. [Google Scholar] [CrossRef]
- Scanlon, C.S.; Van Tubergen, E.A.; Inglehart, R.C.; D’Silva, N.J. Biomarkers of Epithelial-Mesenchymal Transition in Squamous Cell Carcinoma. J. Dent. Res. 2013, 92, 114–121. [Google Scholar] [CrossRef]
- Bradley, C.K.; Norton, C.R.; Chen, Y.; Han, X.; Booth, C.J.; Yoon, J.K.; Krebs, L.T.; Gridley, T. The Snail Family Gene snai3 Is Not Essential for Embryogenesis in Mice. PLoS ONE 2013, 8, e65344. [Google Scholar] [CrossRef]
- Dennis, M.; Wang, G.; Luo, J.; Lin, Y.; Dohadwala, M.; Abemayor, E.; Elashoff, D.A.; Sharma, S.; Dubinett, S.M.; St John, M.A. Snail Controls the Mesenchymal Phenotype and Drives Erlotinib Resistance in Oral Epithelial and Head and Neck Squamous Cell Carcinoma Cells. Otolaryngol. Head Neck Surg. 2012, 147, 726–732. [Google Scholar] [CrossRef]
- Maseki, S.; Ijichi, K.; Tanaka, H.; Fujii, M.; Hasegawa, Y.; Ogawa, T.; Murakami, S.; Kondo, E.; Nakanishi, H. Acquisition of EMT Phenotype in the Gefitinib-Resistant Cells of a Head and Neck Squamous Cell Carcinoma Cell Line through Akt/GSK-3β/snail Signalling Pathway. Br. J. Cancer 2012, 106, 1196–1204. [Google Scholar] [CrossRef]
- Chen, Y.-Q.; Hung, C.-Y.; Wei, M.-T.; Kuo, J.-C.; Yang, M.-H.; Cheng, H.-Y.; Chiou, A. Snail Augments Nuclear Deformability to Promote Lymph Node Metastasis of Head and Neck Squamous Cell Carcinoma. Front. Cell Dev. Biol. 2022, 10, 809738. [Google Scholar] [CrossRef]
- Masui, T.; Ota, I.; Yook, J.-I.; Mikami, S.; Yane, K.; Yamanaka, T.; Hosoi, H. Snail-Induced Epithelial-Mesenchymal Transition Promotes Cancer Stem Cell-like Phenotype in Head and Neck Cancer Cells. Int. J. Oncol. 2014, 44, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Wu, C.; Sun, W.; Liu, D.; Luo, M.; Su, B.; Zhang, L.; Mei, Q.; Hu, G. Snail-Mediated Cancer Stem Cell-like Phenotype in Human CNE2 Nasopharyngeal Carcinoma Cell. Head Neck 2018, 40, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Ling, J.; Huang, Y.; Chang, A.; Zhuo, X. The Expression and Clinical Significance of an Epithelial-Mesenchymal Transition Inducer, SNAI1, in Head and Neck Carcinoma. J. Oral Pathol. Med. 2021, 50, 145–154. [Google Scholar] [CrossRef]
- Hsu, D.S.-S.; Lan, H.-Y.; Huang, C.-H.; Tai, S.-K.; Chang, S.-Y.; Tsai, T.-L.; Chang, C.-C.; Tzeng, C.-H.; Wu, K.-J.; Kao, J.-Y.; et al. Regulation of Excision Repair Cross-Complementation Group 1 by Snail Contributes to Cisplatin Resistance in Head and Neck Cancer. Clin. Cancer Res. 2010, 16, 4561–4571. [Google Scholar] [CrossRef] [PubMed]
- Puram, S.V.; Parikh, A.S.; Tirosh, I. Single Cell RNA-Seq Highlights a Role for a Partial EMT in Head and Neck Cancer. Mol. Cell. Oncol. 2018, 5, e1448244. [Google Scholar] [CrossRef] [PubMed]
- Steinbichler, T.B.; Dudas, J.; Ingruber, J.; Glueckert, R.; Sprung, S.; Fleischer, F.; Cidlinsky, N.; Dejaco, D.; Kofler, B.; Giotakis, A.I.; et al. Slug Is A Surrogate Marker of Epithelial to Mesenchymal Transition (EMT) in Head and Neck Cancer. J. Clin. Med. Res. 2020, 9, 2061. [Google Scholar] [CrossRef]
- Schinke, H.; Pan, M.; Akyol, M.; Zhou, J.; Shi, E.; Kranz, G.; Libl, D.; Quadt, T.; Simon, F.; Canis, M.; et al. SLUG-Related Partial Epithelial-to-Mesenchymal Transition Is a Transcriptomic Prognosticator of Head and Neck Cancer Survival. Mol. Oncol. 2022, 16, 347–367. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.H.; Lee, S.H.; Lim, Y.C. Wnt/β-catenin/Slug Pathway Contributes to Tumor Invasion and Lymph Node Metastasis in Head and Neck Squamous Cell Carcinoma. Clin. Exp. Metastasis 2021, 38, 163–174. [Google Scholar] [CrossRef]
- Riechelmann, H.; Steinbichler, T.B.; Sprung, S.; Santer, M.; Runge, A.; Ganswindt, U.; Gamerith, G.; Dudas, J. The Epithelial-Mesenchymal Transcription Factor Slug Predicts Survival Benefit of Up-Front Surgery in Head and Neck Cancer. Cancers 2021, 13, 772. [Google Scholar] [CrossRef]
- Moon, J.H.; Lee, S.H.; Koo, B.S.; Kim, J.M.; Huang, S.; Cho, J.H.; Eun, Y.G.; Shin, H.A.; Lim, Y.C. Slug Is a Novel Molecular Target for Head and Neck Squamous Cell Carcinoma Stem-like Cells. Oral Oncol. 2020, 111, 104948. [Google Scholar] [CrossRef]
- Luanpitpong, S.; Li, J.; Manke, A.; Brundage, K.; Ellis, E.; McLaughlin, S.L.; Angsutararux, P.; Chanthra, N.; Voronkova, M.; Chen, Y.C.; et al. SLUG Is Required for SOX9 Stabilization and Functions to Promote Cancer Stem Cells and Metastasis in Human Lung Carcinoma. Oncogene 2016, 35, 2824–2833. [Google Scholar] [CrossRef]
- Ciehanover, A.; Hod, Y.; Hershko, A. A Heat-Stable Polypeptide Component of an ATP-Dependent Proteolytic System from Reticulocytes. Biochem. Biophys. Res. Commun. 1978, 81, 1100–1105. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A.; Heller, H.; Haas, A.L.; Rose, I.A. Proposed Role of ATP in Protein Breakdown: Conjugation of Protein with Multiple Chains of the Polypeptide of ATP-Dependent Proteolysis. Proc. Natl. Acad. Sci. USA 1980, 77, 1783–1786. [Google Scholar] [CrossRef]
- Bae, W.-J.; Lee, S.-H.; Rho, Y.-S.; Koo, B.-S.; Lim, Y.-C. Transforming Growth Factor β1 Enhances Stemness of Head and Neck Squamous Cell Carcinoma Cells through Activation of Wnt Signaling. Oncol. Lett. 2016, 12, 5315–5320. [Google Scholar] [CrossRef]
- Nakamura, R.; Ishii, H.; Endo, K.; Hotta, A.; Fujii, E.; Miyazawa, K.; Saitoh, M. Reciprocal Expression of Slug and Snail in Human Oral Cancer Cells. PLoS ONE 2018, 13, e0199442. [Google Scholar] [CrossRef]
- Franco, H.L.; Casasnovas, J.; Rodríguez-Medina, J.R.; Cadilla, C.L. Redundant or Separate Entities?—Roles of Twist1 and Twist2 as Molecular Switches during Gene Transcription. Nucleic Acids Res. 2011, 39, 1177–1186. [Google Scholar] [CrossRef]
- Simpson, P. Maternal-Zygotic Gene Interactions during Formation of the Dorsoventral Pattern in Drosophila Embryos. Genetics 1983, 105, 615–632. [Google Scholar] [CrossRef]
- Nüsslein-Volhard, C.; Wieschaus, E.; Kluding, H. Mutations Affecting the Pattern of the Larval Cuticle in Drosophila Melanogaster: I. Zygotic Loci on the Second Chromosome. Wilehm Roux Arch. Dev. Biol. 1984, 193, 267–282. [Google Scholar] [CrossRef]
- Li, L.; Cserjesi, P.; Olson, E.N. Dermo-1: A Novel Twist-Related bHLH Protein Expressed in the Developing Dermis. Dev. Biol. 1995, 172, 280–292. [Google Scholar] [CrossRef]
- Kim, J.Y.; Park, M.; Ohn, J.; Seong, R.H.; Chung, J.H.; Kim, K.H.; Jo, S.J.; Kwon, O. Twist2-Driven Chromatin Remodeling Governs the Postnatal Maturation of Dermal Fibroblasts. Cell Rep. 2022, 39, 110821. [Google Scholar] [CrossRef]
- Puisieux, A.; Valsesia-Wittmann, S.; Ansieau, S. A Twist for Survival and Cancer Progression. Br. J. Cancer 2006, 94, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.Z.; Zhang, W.; Wang, L.-H. Regulation of Cancer Cell Survival, Migration, and Invasion by Twist: AKT2 Comes to Interplay. Cancer Res. 2008, 68, 957–960. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.Z.; Zhang, W.Z.; Sun, M.; Wang, Q.; Coppola, D.; Mansour, M.; Xu, L.M.; Costanzo, C.; Cheng, J.Q.; Wang, L.-H. Twist Is Transcriptionally Induced by Activation of STAT3 and Mediates STAT3 Oncogenic Function. J. Biol. Chem. 2008, 283, 14665–14673. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Qin, L.; He, T.; Qin, J.; Hong, J.; Wong, J.; Liao, L.; Xu, J. The TWIST/Mi2/NuRD Protein Complex and Its Essential Role in Cancer Metastasis. Cell Res. 2011, 21, 275–289. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Natsugoe, S.; Ishigami, S.; Matsumoto, M.; Okumura, H.; Setoyama, T.; Uchikado, Y.; Kita, Y.; Tamotsu, K.; Sakamoto, A.; et al. Significance of Twist Expression and Its Association with E-Cadherin in Esophageal Squamous Cell Carcinoma. J. Exp. Clin. Cancer Res. 2009, 28, 158. [Google Scholar] [CrossRef] [PubMed]
- Ardalan Khales, S.; Ebrahimi, E.; Jahanzad, E.; Ardalan Khales, S.; Forghanifard, M.M. MAML1 and TWIST1 Co-Overexpression Promote Invasion of Head and Neck Squamous Cell Carcinoma. Asia Pac. J. Clin. Oncol. 2018, 14, e434–e441. [Google Scholar] [CrossRef]
- Lu, S.; Yu, L.; Mu, Y.; Ma, J.; Tian, J.; Xu, W.; Wang, H. Role and Mechanism of Twist1 in Modulating the Chemosensitivity of FaDu Cells. Mol. Med. Rep. 2014, 10, 53–60. [Google Scholar] [CrossRef][Green Version]
- Li, R.; Wu, C.; Liang, H.; Zhao, Y.; Lin, C.; Zhang, X.; Ye, C. Knockdown of TWIST Enhances the Cytotoxicity of Chemotherapeutic Drugs in Doxorubicin-Resistant HepG2 Cells by Suppressing MDR1 and EMT. Int. J. Oncol. 2018, 53, 1763–1773. [Google Scholar] [CrossRef]
- Pham, C.G.; Bubici, C.; Zazzeroni, F.; Knabb, J.R.; Papa, S.; Kuntzen, C.; Franzoso, G. Upregulation of Twist-1 by NF-kappaB Blocks Cytotoxicity Induced by Chemotherapeutic Drugs. Mol. Cell. Biol. 2007, 27, 3920–3935. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Ang, H.L.; Moghadam, E.R.; Mohammadi, S.; Zarrin, V.; Hushmandi, K.; Samarghandian, S.; Zarrabi, A.; Najafi, M.; Mohammadinejad, R.; et al. MicroRNAs and Their Influence on the ZEB Family: Mechanistic Aspects and Therapeutic Applications in Cancer Therapy. Biomolecules 2020, 10, 1040. [Google Scholar] [CrossRef]
- Mohammadinejad, R.; Sassan, H.; Pardakhty, A.; Hashemabadi, M.; Ashrafizadeh, M.; Dehshahri, A.; Mandegary, A. ZEB1 and ZEB2 Gene Editing Mediated by CRISPR/Cas9 in A549 Cell Line. Bratisl. Lek. Listy 2020, 121, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Tilló, E.; Siles, L.; de Barrios, O.; Cuatrecasas, M.; Vaquero, E.C.; Castells, A.; Postigo, A. Expanding Roles of ZEB Factors in Tumorigenesis and Tumor Progression. Am. J. Cancer Res. 2011, 1, 897–912. [Google Scholar] [PubMed]
- Park, M.K.; Lee, H.; Lee, C.H. Post-Translational Modification of ZEB Family Members in Cancer Progression. Int. J. Mol. Sci. 2022, 23, 15127. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, S.; Bindea, G.; Albu, R.I.; Mlecnik, B.; Machiels, J.-P. Cetuximab Promotes Epithelial to Mesenchymal Transition and Cancer Associated Fibroblasts in Patients with Head and Neck Cancer. Oncotarget 2015, 6, 34288–34299. [Google Scholar] [CrossRef] [PubMed]
- Lima de Oliveira, J.; Moré Milan, T.; Longo Bighetti-Trevisan, R.; Fernandes, R.R.; Machado Leopoldino, A.; Oliveira de Almeida, L. Epithelial-Mesenchymal Transition and Cancer Stem Cells: A Route to Acquired Cisplatin Resistance through Epigenetics in HNSCC. Oral Dis. 2023, 29, 1991–2005. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Lei, D.; Wang, L.; Yang, X.; Jia, S.; Yang, Z.; Shan, C.; Yang, X.; Zhang, C.; Lu, B. MiRNA-101 Inhibits Oral Squamous-Cell Carcinoma Growth and Metastasis by Targeting Zinc Finger E-Box Binding Homeobox 1. Am. J. Cancer Res. 2016, 6, 1396–1407. [Google Scholar] [PubMed]
- Kuo, S.Z.; Blair, K.J.; Rahimy, E.; Kiang, A.; Abhold, E.; Fan, J.-B.; Wang-Rodriguez, J.; Altuna, X.; Ongkeko, W.M. Salinomycin Induces Cell Death and Differentiation in Head and Neck Squamous Cell Carcinoma Stem Cells despite Activation of Epithelial-Mesenchymal Transition and Akt. BMC Cancer 2012, 12, 556. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Zhang, L.; Chen, M.; Zhu, J.; Meng, Z.; Liang, L. A de Novo Triplication on 2q22.3 Including the Entire ZEB2 Gene Associated with Global Developmental Delay, Multiple Congenital Anomalies and Behavioral Abnormalities. Mol. Cytogenet. 2015, 8, 99. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Ma, L.; Wu, Z.; Wang, G.; Huang, Q.; Shen, Z.; Yu, R. Zinc Finger E-box Binding Homeobox 2 Functions as an Oncogene in Human Laryngeal Squamous Cell Carcinoma. Mol. Med. Rep. 2019, 19, 4545–4552. [Google Scholar] [CrossRef]
- Kong, Y.H.; Syed Zanaruddin, S.N.; Lau, S.H.; Ramanathan, A.; Kallarakkal, T.G.; Vincent-Chong, V.K.; Wan Mustafa, W.M.; Abraham, M.T.; Abdul Rahman, Z.A.; Zain, R.B.; et al. Co-Expression of TWIST1 and ZEB2 in Oral Squamous Cell Carcinoma Is Associated with Poor Survival. PLoS ONE 2015, 10, e0134045. [Google Scholar] [CrossRef]
- Geng, D.-M.; Kan, X.-M.; Zhang, W.-W. Effect of ZEB2 Silencing on Cisplatin Resistance in Gastric Cancer. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 1746–1752. [Google Scholar] [PubMed]
- Jiang, T.; Dong, P.; Li, L.; Ma, X.; Xu, P.; Zhu, H.; Wang, Y.; Yang, B.; Liu, K.; Liu, J.; et al. MicroRNA-200c Regulates Cisplatin Resistance by Targeting ZEB2 in Human Gastric Cancer Cells. Oncol. Rep. 2017, 38, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.-J.; Feng, W.; Li, Y. HOTTIP-miR-205-ZEB2 Axis Confers Cisplatin Resistance to Ovarian Cancer Cells. Front. Cell Dev. Biol. 2021, 9, 707424. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Han, Y.; Li, S. Oncogenic Circular RNA circ_0007534 Contributes to Paclitaxel Resistance in Endometrial Cancer by Sponging miR-625 and Promoting ZEB2 Expression. Front. Oncol. 2022, 12, 985470. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Zhou, Y.; Yang, H.; Li, L.; Deng, X.; Cheng, C.; Xie, Y.; Luo, X.; Fang, W.; Liu, Z. A Directly Negative Interaction of miR-203 and ZEB2 Modulates Tumor Stemness and Chemotherapy Resistance in Nasopharyngeal Carcinoma. Oncotarget 2016, 7, 67288–67301. [Google Scholar] [CrossRef] [PubMed]
- Chu, P.-Y.; Hu, F.-W.; Yu, C.-C.; Tsai, L.-L.; Yu, C.-H.; Wu, B.-C.; Chen, Y.-W.; Huang, P.-I.; Lo, W.-L. Epithelial-Mesenchymal Transition Transcription Factor ZEB1/ZEB2 Co-Expression Predicts Poor Prognosis and Maintains Tumor-Initiating Properties in Head and Neck Cancer. Oral Oncol. 2013, 49, 34–41. [Google Scholar] [CrossRef]
- Oshimori, N.; Oristian, D.; Fuchs, E. TGF-β Promotes Heterogeneity and Drug Resistance in Squamous Cell Carcinoma. Cell 2015, 160, 963–976. [Google Scholar] [CrossRef]
- Steinbichler, T.B.; Savic, D.; Dejaco, D.; Romani, A.; Kofler, B.; Skvortsova, I.I.; Riechelmann, H.; Dudas, J. Pleiotropic Effects of Epithelial Mesenchymal Crosstalk on Head and Neck Cancer: EMT and beyond. Cancer Microenviron. 2019, 12, 67–76. [Google Scholar] [CrossRef] [PubMed]
- de Morais, E.F.; Rolim, L.S.A.; de Melo Fernandes Almeida, D.R.; de Farias Morais, H.G.; de Souza, L.B.; de Almeida Freitas, R. Biological Role of Epithelial-Mesenchymal-Transition-Inducing Transcription Factors in Head and Neck Squamous Cell Carcinoma: A Systematic Review. Arch. Oral Biol. 2020, 119, 104904. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, Y.; Xia, C.; Ding, L.; Pu, Y.; Hu, X.; Cai, H.; Hu, Q. Integrated Analysis of Single-Cell RNA-Seq and Bulk RNA-Seq Reveals Distinct Cancer-Associated Fibroblasts in Head and Neck Squamous Cell Carcinoma. Ann. Transl. Med. 2021, 9, 1017. [Google Scholar] [CrossRef]
- Deshmukh, A.P.; Vasaikar, S.V.; Tomczak, K.; Tripathi, S.; den Hollander, P.; Arslan, E.; Chakraborty, P.; Soundararajan, R.; Jolly, M.K.; Rai, K.; et al. Identification of EMT Signaling Cross-Talk and Gene Regulatory Networks by Single-Cell RNA Sequencing. Proc. Natl. Acad. Sci. USA 2021, 118, e2102050118. [Google Scholar] [CrossRef] [PubMed]
- Quah, H.S.; Cao, E.Y.; Suteja, L.; Li, C.H.; Leong, H.S.; Chong, F.T.; Gupta, S.; Arcinas, C.; Ouyang, J.F.; Ang, V.; et al. Single Cell Analysis in Head and Neck Cancer Reveals Potential Immune Evasion Mechanisms during Early Metastasis. Nat. Commun. 2023, 14, 1680. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pawlicka, M.; Gumbarewicz, E.; Błaszczak, E.; Stepulak, A. Transcription Factors and Markers Related to Epithelial–Mesenchymal Transition and Their Role in Resistance to Therapies in Head and Neck Cancers. Cancers 2024, 16, 1354. https://doi.org/10.3390/cancers16071354
Pawlicka M, Gumbarewicz E, Błaszczak E, Stepulak A. Transcription Factors and Markers Related to Epithelial–Mesenchymal Transition and Their Role in Resistance to Therapies in Head and Neck Cancers. Cancers. 2024; 16(7):1354. https://doi.org/10.3390/cancers16071354
Chicago/Turabian StylePawlicka, Marta, Ewelina Gumbarewicz, Ewa Błaszczak, and Andrzej Stepulak. 2024. "Transcription Factors and Markers Related to Epithelial–Mesenchymal Transition and Their Role in Resistance to Therapies in Head and Neck Cancers" Cancers 16, no. 7: 1354. https://doi.org/10.3390/cancers16071354
APA StylePawlicka, M., Gumbarewicz, E., Błaszczak, E., & Stepulak, A. (2024). Transcription Factors and Markers Related to Epithelial–Mesenchymal Transition and Their Role in Resistance to Therapies in Head and Neck Cancers. Cancers, 16(7), 1354. https://doi.org/10.3390/cancers16071354