

Preclinical Characterization of the Anti-Leukemia Activity of the CD33/CD16a/NKG2D Immune-Modulating TriNKET® CC-96191

 , , , and
, , , and  add
Show full author list
add
Show full author list
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Therapeutics
2.2. Lentiviral Expression Vectors
2.3. Surface Plasmon Resonance (SPR)
2.4. Peripheral Blood Mononuclear Cells and Primary Human NK Cells
2.5. Secreted Cytokine Measurements
2.6. Human NK and Acute Leukemia Cell Lines
2.7. Quantification of CD33 Expression
2.8. Assessment of Binding of CC-96191 to CD33FL and CD33∆E2
2.9. Quantification of Drug-Induced Cytotoxicity
2.10. Frequency of CD33 SNPs
2.11. Primary AML Aspirates
2.12. Statistical Considerations
3. Results
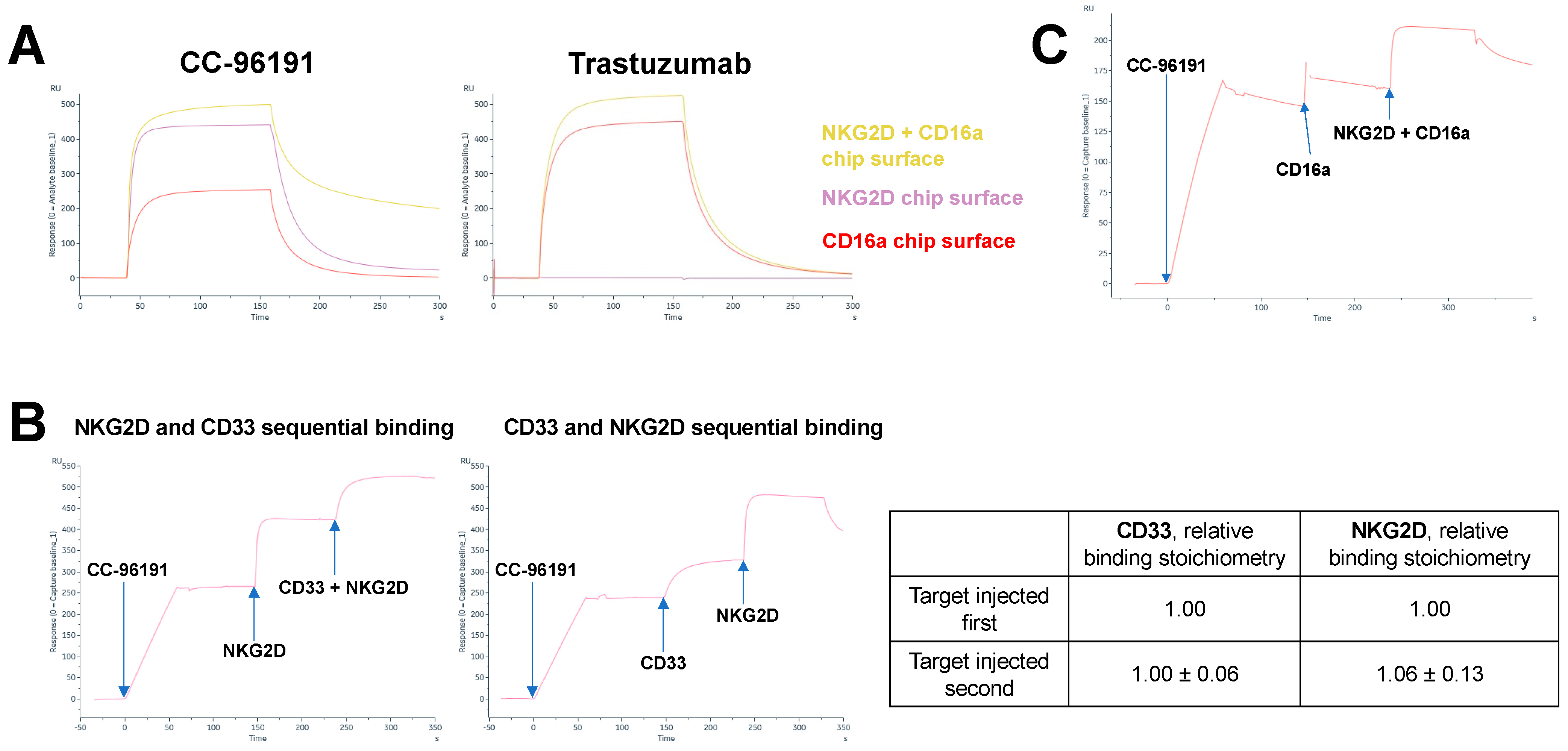
3.1. CC-96191 Simultaneously Binds CD33, NKG2D, and CD16a with Greater Strength Than Single Binding to NKG2D or CD16a
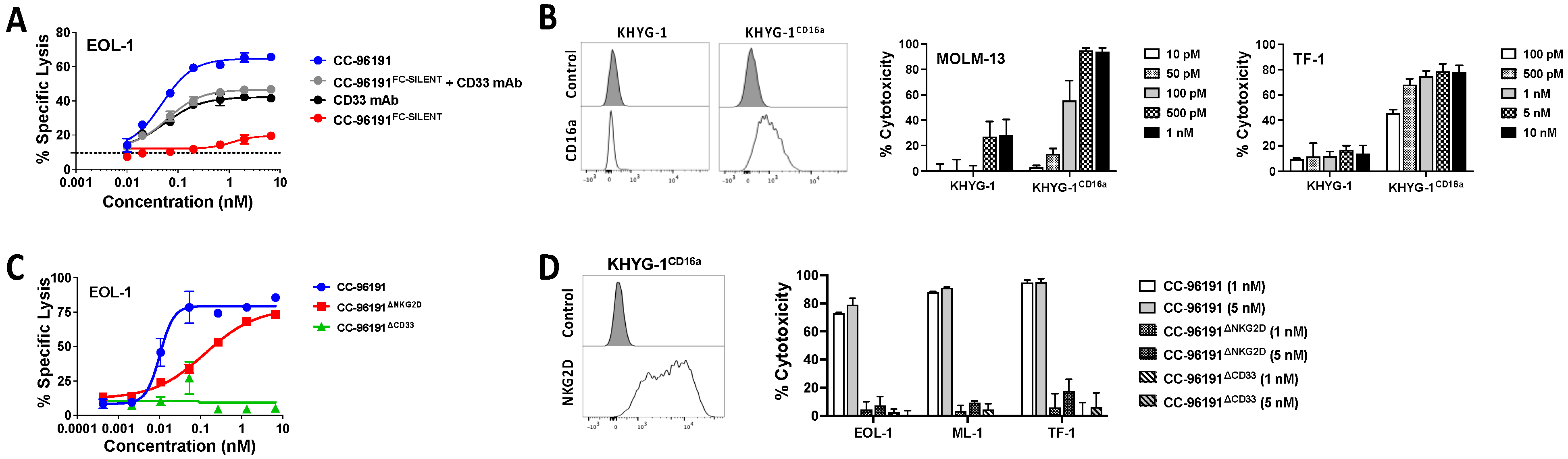
3.2. Co-engagement of CD16a, NKG2D, and CD33 Is Required for Optimal Cytolytic Activity of CC-96191 against CD33+ AML Cells
3.3. Effect of CD33 Expression and CD33 SNPs on CC-96191-Induced Cytotoxicity
3.4. Impact of Soluble MICA and CD33 on CC-96191-Induced Cytotoxicity
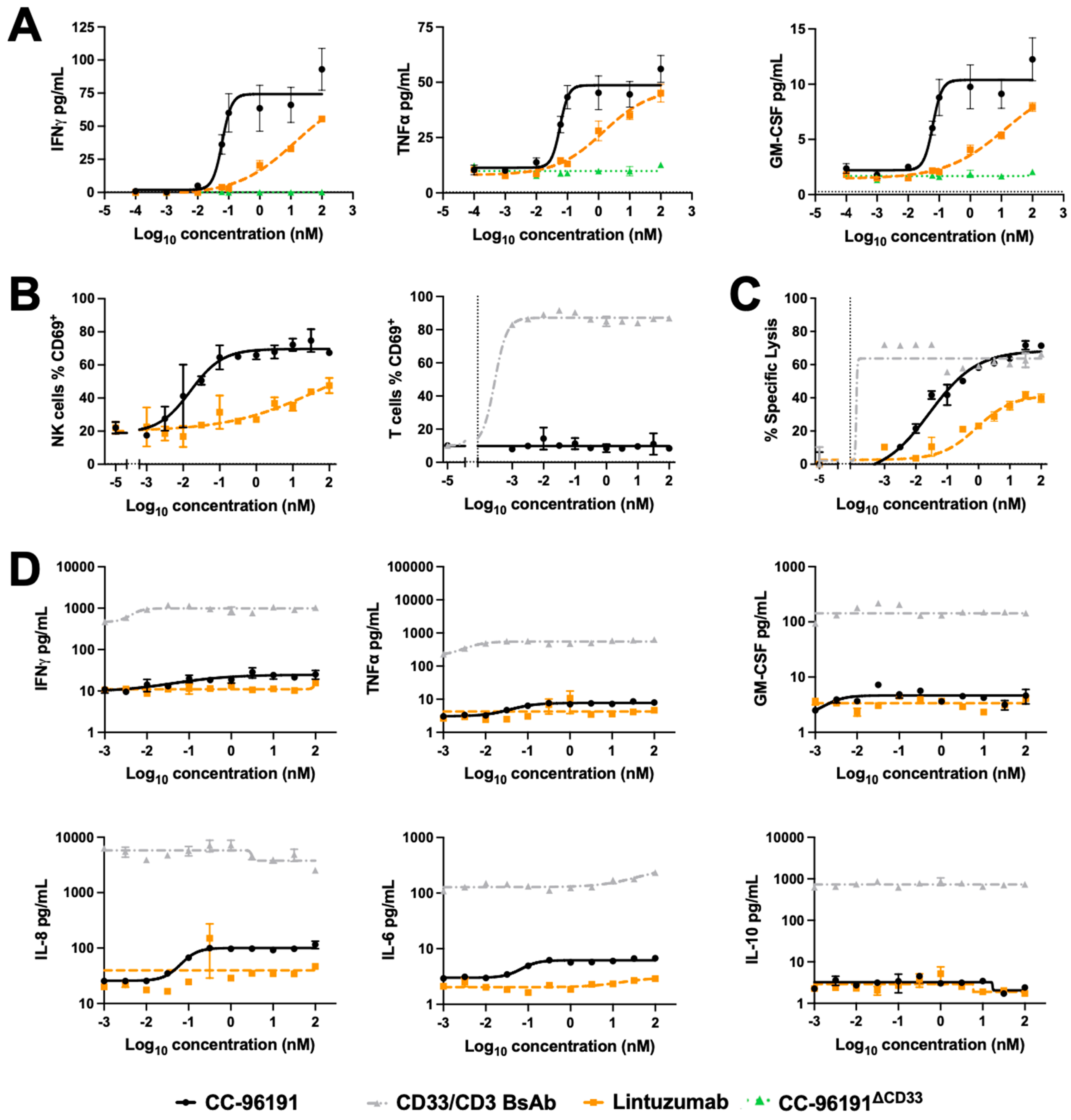
3.5. CC-96191-Induced Cytokine Secretion, Cellular Activation, and Cytolysis by Human NK Cells and PBMCs
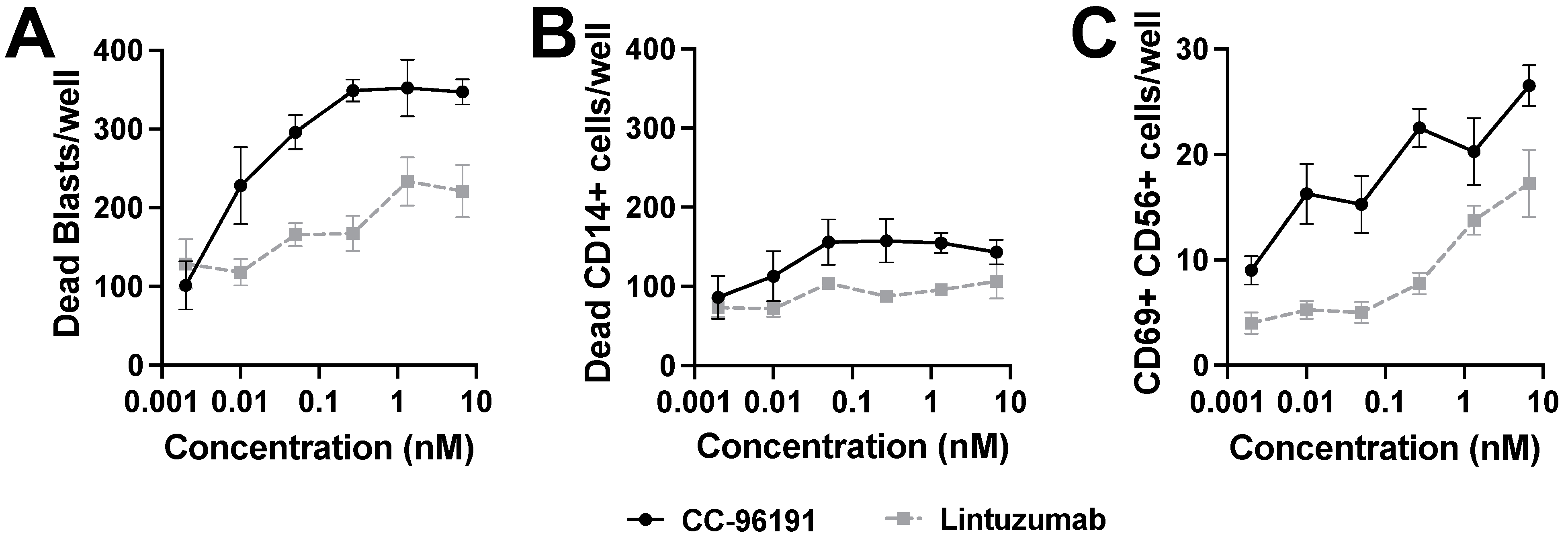
3.6. CC-96191-Induced Selective Destruction of Leukemia Cells in Bone Marrow Samples from Patients with AML
3.7. Effect of ABC Transporter Protein Expression on CC-96191-Induced Cytotoxicity
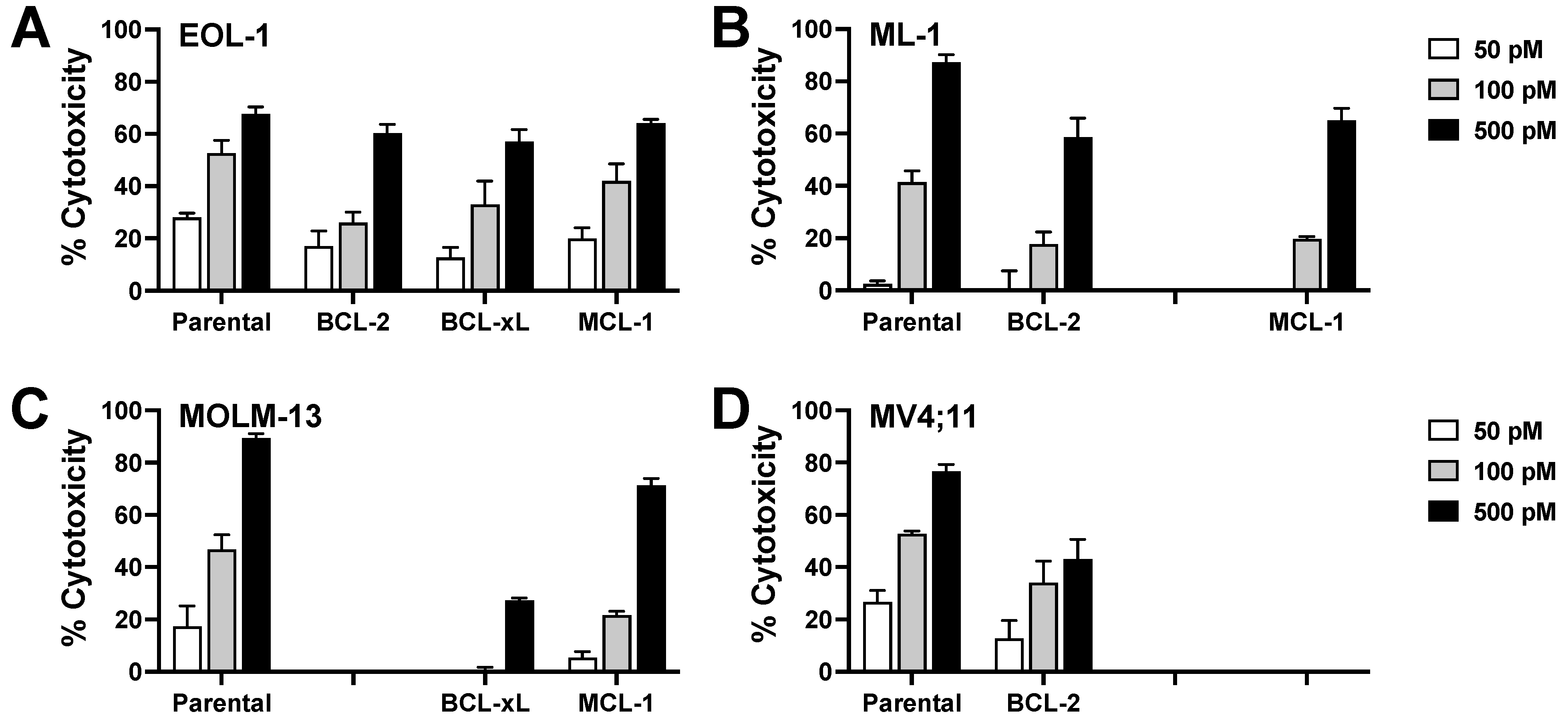
3.8. Effect of Anti-Apoptotic BCL-2 Family Proteins on CC-96191-Induced Cytotoxicity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Döhner, H.; Wei, A.H.; Löwenberg, B. Towards precision medicine for AML. Nat. Rev. Clin. Oncol. 2021, 18, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef] [PubMed]
- Shimony, S.; Stahl, M.; Stone, R.M. Acute myeloid leukemia: 2023 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2023, 98, 502–526. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Erba, H.P.; Freeman, S.D.; Wei, A.H. Acute myeloid leukaemia. Lancet 2023, 401, 2073–2086. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Pollard, J.A.; Alonzo, T.A.; Loken, M.; Gerbing, R.B.; Ho, P.A.; Bernstein, I.D.; Raimondi, S.C.; Hirsch, B.; Franklin, J.; Walter, R.B.; et al. Correlation of CD33 expression level with disease characteristics and response to gemtuzumab ozogamicin containing chemotherapy in childhood AML. Blood 2012, 119, 3705–3711. [Google Scholar] [CrossRef]
- Krupka, C.; Kufer, P.; Kischel, R.; Zugmaier, G.; Bögeholz, J.; Köhnke, T.; Lichtenegger, F.S.; Schneider, S.; Metzeler, K.H.; Fiegl, M.; et al. CD33 target validation and sustained depletion of AML blasts in long-term cultures by the bispecific T-cell-engaging antibody AMG 330. Blood 2014, 123, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Perna, F.; Berman, S.H.; Soni, R.K.; Mansilla-Soto, J.; Eyquem, J.; Hamieh, M.; Hendrickson, R.C.; Brennan, C.W.; Sadelain, M. Integrating proteomics and transcriptomics for systematic combinatorial chimeric antigen receptor therapy of AML. Cancer Cell 2017, 32, 506–519.e5. [Google Scholar] [CrossRef]
- Khan, N.; Hills, R.K.; Virgo, P.; Couzens, S.; Clark, N.; Gilkes, A.; Richardson, P.; Knapper, S.; Grimwade, D.; Russell, N.H.; et al. Expression of CD33 is a predictive factor for effect of gemtuzumab ozogamicin at different doses in adult acute myeloid leukaemia. Leukemia 2017, 31, 1059–1068. [Google Scholar] [CrossRef]
- Walter, R.B.; Appelbaum, F.R.; Estey, E.H.; Bernstein, I.D. Acute myeloid leukemia stem cells and CD33-targeted immunotherapy. Blood 2012, 119, 6198–6208. [Google Scholar] [CrossRef]
- Godwin, C.D.; Gale, R.P.; Walter, R.B. Gemtuzumab ozogamicin in acute myeloid leukemia. Leukemia 2017, 31, 1855–1868. [Google Scholar] [CrossRef]
- Grossbard, M.L.; Press, O.W.; Appelbaum, F.R.; Bernstein, I.D.; Nadler, L.M. Monoclonal antibody-based therapies of leukemia and lymphoma. Blood 1992, 80, 863–878. [Google Scholar] [CrossRef]
- Feldman, E.J.; Brandwein, J.; Stone, R.; Kalaycio, M.; Moore, J.; O’Connor, J.; Wedel, N.; Roboz, G.J.; Miller, C.; Chopra, R.; et al. Phase III randomized multicenter study of a humanized anti-CD33 monoclonal antibody, lintuzumab, in combination with chemotherapy, versus chemotherapy alone in patients with refractory or first-relapsed acute myeloid leukemia. J. Clin. Oncol. 2005, 23, 4110–4116. [Google Scholar] [CrossRef]
- Sekeres, M.A.; Lancet, J.E.; Wood, B.L.; Grove, L.E.; Sandalic, L.; Sievers, E.L.; Jurcic, J.G. Randomized phase IIb study of low-dose cytarabine and lintuzumab versus low-dose cytarabine and placebo in older adults with untreated acute myeloid leukemia. Haematologica 2013, 98, 119–128. [Google Scholar] [CrossRef]
- Vasu, S.; Altman, J.K.; Uy, G.L.; Tallman, M.S.; Gojo, I.; Lozanski, G.; Burkard, U.; Osswald, A.; James, P.; Rüter, B.; et al. A phase I study of the fully human, fragment crystallizable-engineered, anti-CD-33 monoclonal antibody BI 836858 in patients with previously-treated acute myeloid leukemia. Haematologica 2022, 107, 770–773. [Google Scholar] [CrossRef]
- Hills, R.K.; Castaigne, S.; Appelbaum, F.R.; Delaunay, J.; Petersdorf, S.; Othus, M.; Estey, E.H.; Dombret, H.; Chevret, S.; Ifrah, N.; et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: A meta-analysis of individual patient data from randomised controlled trials. Lancet. Oncol. 2014, 15, 986–996. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Zeidan, A.M.; Bewersdorf, J.P. BiTEs, DARTS, BiKEs and TriKEs-are antibody based therapies changing the future treatment of AML? Life 2021, 11, 465. [Google Scholar] [CrossRef] [PubMed]
- Böhme, M.; Kayser, S. Immune-based therapeutic strategies for acute myeloid leukemia. Cancers 2021, 14, 105. [Google Scholar] [CrossRef] [PubMed]
- Haddad, F.; Zeidan, A.M.; Daver, N. Checkpoint Inhibitors and other immune-based therapies in acute myeloid leukemia. Cancer J. 2022, 28, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Angenendt, L.; Mikesch, J.H.; Schliemann, C. Emerging antibody-based therapies for the treatment of acute myeloid leukemia. Cancer Treat. Rev. 2022, 108, 102409. [Google Scholar] [CrossRef]
- Morse, J.W.; Rios, M.; Ye, J.; Rios, A.; Zhang, C.C.; Daver, N.G.; DiNardo, C.D.; Zhang, N.; An, Z. Antibody therapies for the treatment of acute myeloid leukemia: Exploring current and emerging therapeutic targets. Expert Opin. Investig. Drugs 2023, 32, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Shimasaki, N.; Jain, A.; Campana, D. NK cells for cancer immunotherapy. Nat. Rev. Drug Discov. 2020, 19, 200–218. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, J.; Miller, J.S. Recent progress in and challenges in cellular therapy using NK cells for hematological malignancies. Blood Rev. 2020, 44, 100678. [Google Scholar] [CrossRef] [PubMed]
- Phung, S.K.; Miller, J.S.; Felices, M. Bi-specific and tri-specific NK cell engagers: The new avenue of targeted NK cell immunotherapy. Mol. Diagn. Ther. 2021, 25, 577–592. [Google Scholar] [CrossRef] [PubMed]
- Allison, M.; Mathews, J.; Gilliland, T.; Mathew, S.O. Natural killer cell-mediated immunotherapy for leukemia. Cancers 2022, 14, 843. [Google Scholar] [CrossRef] [PubMed]
- Merino, A.; Maakaron, J.; Bachanova, V. Advances in NK cell therapy for hematologic malignancies: NK source, persistence and tumor targeting. Blood Rev. 2023, 60, 101073. [Google Scholar] [CrossRef] [PubMed]
- Bryceson, Y.T.; March, M.E.; Ljunggren, H.G.; Long, E.O. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood 2006, 107, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Laszlo, G.S.; Gudgeon, C.J.; Harrington, K.H.; Dell’Aringa, J.; Newhall, K.J.; Means, G.D.; Sinclair, A.M.; Kischel, R.; Frankel, S.R.; Walter, R.B. Cellular determinants for preclinical activity of a novel CD33/CD3 bispecific T-cell engager (BiTE) antibody, AMG 330, against human AML. Blood 2014, 123, 554–561. [Google Scholar] [CrossRef]
- Godwin, C.D.; Laszlo, G.S.; Wood, B.L.; Correnti, C.E.; Bates, O.M.; Garling, E.E.; Mao, Z.J.; Beddoe, M.E.; Lunn, M.C.; Humbert, O.; et al. The CD33 splice isoform lacking exon 2 as therapeutic target in human acute myeloid leukemia. Leukemia 2020, 34, 2479–2483. [Google Scholar] [CrossRef]
- Godwin, C.D.; Bates, O.M.; Jean, S.R.; Laszlo, G.S.; Garling, E.E.; Beddoe, M.E.; Cardone, M.H.; Walter, R.B. Anti-apoptotic BCL-2 family proteins confer resistance to calicheamicin-based antibody-drug conjugate therapy of acute leukemia. Leuk. Lymphoma 2020, 61, 2990–2994. [Google Scholar] [CrossRef]
- Godwin, C.D.; Laszlo, G.S.; Fiorenza, S.; Garling, E.E.; Phi, T.D.; Bates, O.M.; Correnti, C.E.; Hoffstrom, B.G.; Lunn, M.C.; Humbert, O.; et al. Targeting the membrane-proximal C2-set domain of CD33 for improved CD33-directed immunotherapy. Leukemia 2021, 35, 2496–2507. [Google Scholar] [CrossRef] [PubMed]
- Snijder, B.; Vladimer, G.I.; Krall, N.; Miura, K.; Schmolke, A.S.; Kornauth, C.; Lopez de la Fuente, O.; Choi, H.S.; van der Kouwe, E.; Gültekin, S.; et al. Image-based ex-vivo drug screening for patients with aggressive haematological malignancies: Interim results from a single-arm, open-label, pilot study. Lancet Haematol. 2017, 4, e595–e606. [Google Scholar] [CrossRef] [PubMed]
- Vladimer, G.I.; Snijder, B.; Krall, N.; Bigenzahn, J.W.; Huber, K.V.M.; Lardeau, C.H.; Sanjiv, K.; Ringler, A.; Berglund, U.W.; Sabler, M.; et al. Global survey of the immunomodulatory potential of common drugs. Nat. Chem. Biol. 2017, 13, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Krall, N.; Superti-Furga, G.; Vladimer, G.I. Patient-derived model systems and the development of next-generation anticancer therapeutics. Curr. Opin. Chem. Biol. 2020, 56, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Beum, P.V.; Lindorfer, M.A.; Taylor, R.P. Within peripheral blood mononuclear cells, antibody-dependent cellular cytotoxicity of rituximab-opsonized Daudi cells is promoted by NK cells and inhibited by monocytes due to shaving. J. Immunol. 2008, 181, 2916–2924. [Google Scholar] [CrossRef] [PubMed]
- Walter, R.B. Investigational CD33-targeted therapeutics for acute myeloid leukemia. Expert Opin. Investig. Drugs 2018, 27, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Mortland, L.; Alonzo, T.A.; Walter, R.B.; Gerbing, R.B.; Mitra, A.K.; Pollard, J.A.; Loken, M.R.; Hirsch, B.; Raimondi, S.; Franklin, J.; et al. Clinical significance of CD33 nonsynonymous single-nucleotide polymorphisms in pediatric patients with acute myeloid leukemia treated with gemtuzumab-ozogamicin-containing chemotherapy. Clin. Cancer Res. 2013, 19, 1620–1627. [Google Scholar] [CrossRef]
- Hilpert, J.; Grosse-Hovest, L.; Grunebach, F.; Buechele, C.; Nuebling, T.; Raum, T.; Steinle, A.; Salih, H.R. Comprehensive analysis of NKG2D ligand expression and release in leukemia: Implications for NKG2D-mediated NK cell responses. J. Immunol. 2012, 189, 1360–1371. [Google Scholar] [CrossRef]
- Biedermann, B.; Gil, D.; Bowen, D.T.; Crocker, P.R. Analysis of the CD33-related siglec family reveals that Siglec-9 is an endocytic receptor expressed on subsets of acute myeloid leukemia cells and absent from normal hematopoietic progenitors. Leuk. Res. 2007, 31, 211–220. [Google Scholar] [CrossRef]
- Wu, J.D.; Higgins, L.M.; Steinle, A.; Cosman, D.; Haugk, K.; Plymate, S.R. Prevalent expression of the immunostimulatory MHC class I chain-related molecule is counteracted by shedding in prostate cancer. J. Clin. Investig. 2004, 114, 560–568. [Google Scholar] [CrossRef]
- Groh, V.; Wu, J.; Yee, C.; Spies, T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 2002, 419, 734–738. [Google Scholar] [CrossRef] [PubMed]
- Salih, H.R.; Rammensee, H.G.; Steinle, A. Cutting edge: Down-regulation of MICA on human tumors by proteolytic shedding. J. Immunol. 2002, 169, 4098–4102. [Google Scholar] [CrossRef] [PubMed]
- Salih, H.R.; Antropius, H.; Gieseke, F.; Lutz, S.Z.; Kanz, L.; Rammensee, H.G.; Steinle, A. Functional expression and release of ligands for the activating immunoreceptor NKG2D in leukemia. Blood 2003, 102, 1389–1396. [Google Scholar] [CrossRef] [PubMed]
- Poggi, A.; Catellani, S.; Garuti, A.; Pierri, I.; Gobbi, M.; Zocchi, M.R. Effective in vivo induction of NKG2D ligands in acute myeloid leukaemias by all-trans-retinoic acid or sodium valproate. Leukemia 2009, 23, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, F.C.; de Souza, P.S.; Hancio, T.; de Faria, F.C.C.; Maia, R.C. Update on drug transporter proteins in acute myeloid leukemia: Pathological implication and clinical setting. Crit. Rev. Oncol. Hematol. 2021, 160, 103281. [Google Scholar] [CrossRef]
- Lowdell, M.W.; Craston, R.; Samuel, D.; Wood, M.E.; O’Neill, E.; Saha, V.; Prentice, H.G. Evidence that continued remission in patients treated for acute leukaemia is dependent upon autologous natural killer cells. Br. J. Haematol. 2002, 117, 821–827. [Google Scholar] [CrossRef]
- Hsu, K.C.; Keever-Taylor, C.A.; Wilton, A.; Pinto, C.; Heller, G.; Arkun, K.; O’Reilly, R.J.; Horowitz, M.M.; Dupont, B. Improved outcome in HLA-identical sibling hematopoietic stem-cell transplantation for acute myelogenous leukemia predicted by KIR and HLA genotypes. Blood 2005, 105, 4878–4884. [Google Scholar] [CrossRef]
- Ruggeri, L.; Aversa, F.; Martelli, M.F.; Velardi, A. Allogeneic hematopoietic transplantation and natural killer cell recognition of missing self. Immunol. Rev. 2006, 214, 202–218. [Google Scholar] [CrossRef]
- Cooley, S.; Weisdorf, D.J.; Guethlein, L.A.; Klein, J.P.; Wang, T.; Le, C.T.; Marsh, S.G.E.; Geraghty, D.; Spellman, S.; Haagenson, M.D.; et al. Donor selection for natural killer cell receptor genes leads to superior survival after unrelated transplantation for acute myelogenous leukemia. Blood 2010, 116, 2411–2419. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, J.E.; Giglio, F.; Gooley, T.A.; Stevenson, P.A.; Le Luduec, J.B.; Shaffer, B.C.; Rajalingam, R.; Hou, L.; Hurley, C.K.; Noreen, H.; et al. KIR3DL1/HLA-B subtypes govern acute myelogenous leukemia relapse after hematopoietic cell transplantation. J. Clin. Oncol. 2017, 35, 2268–2278. [Google Scholar] [CrossRef] [PubMed]
- Chretien, A.S.; Fauriat, C.; Orlanducci, F.; Rey, J.; Borg, G.B.; Gautherot, E.; Granjeaud, S.; Demerle, C.; Hamel, J.F.; Cerwenka, A.; et al. NKp30 expression is a prognostic immune biomarker for stratification of patients with intermediate-risk acute myeloid leukemia. Oncotarget 2017, 8, 49548–49563. [Google Scholar] [CrossRef] [PubMed]
- Chretien, A.S.; Devillier, R.; Fauriat, C.; Orlanducci, F.; Harbi, S.; Le Roy, A.; Rey, J.; Bouvier Borg, G.; Gautherot, E.; Hamel, J.F.; et al. NKp46 expression on NK cells as a prognostic and predictive biomarker for response to allo-SCT in patients with AML. Oncoimmunology 2017, 6, e1307491. [Google Scholar] [CrossRef] [PubMed]
- Mastaglio, S.; Wong, E.; Perera, T.; Ripley, J.; Blombery, P.; Smyth, M.J.; Koldej, R.; Ritchie, D. Natural killer receptor ligand expression on acute myeloid leukemia impacts survival and relapse after chemotherapy. Blood Adv. 2018, 2, 335–346. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Montesinos, P.; Ivanov, V.; DiNardo, C.D.; Novak, J.; Laribi, K.; Kim, I.; Stevens, D.A.; Fiedler, W.; Pagoni, M.; et al. Venetoclax plus LDAC for newly diagnosed AML ineligible for intensive chemotherapy: A phase 3 randomized placebo-controlled trial. Blood 2020, 135, 2137–2145. [Google Scholar] [CrossRef]
- Wei, A.H.; Panayiotidis, P.; Montesinos, P.; Laribi, K.; Ivanov, V.; Kim, I.; Novak, J.; Champion, R.; Fiedler, W.; Pagoni, M.; et al. Long-term follow-up of VIALE-C in patients with untreated AML ineligible for intensive chemotherapy. Blood 2022, 140, 2754–2756. [Google Scholar] [CrossRef]
- Pan, R.; Ryan, J.; Pan, D.; Wucherpfennig, K.W.; Letai, A. Augmenting NK cell-based immunotherapy by targeting mitochondrial apoptosis. Cell 2022, 185, 1521–1538.e18. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | CD33 Expression | KHYG-1CD16a Cytotoxicity | CC-96191 Specific Cytotoxicity | ||||
|---|---|---|---|---|---|---|---|
| 10 pM | 50 pM | 500 pM | 1 nM | 5 nM | |||
| EOL-1 | ++ | 26.0 ± 15.4 | ND | ND | 52.7 ± 3.2 | 62.3 ± 3.8 | 69.0 ± 4.6 |
| HL-60 | ++ | 8.2 ± 3.5 | ND | 2.0 ± 4.2 | 14.0 ± 3.6 | 30.7 ± 5.4 | 46.7 ± 3.5 |
| K562 | + | 91.4 ± 4.9 | ND | ND | ND | ND | ND |
| KG-1 | + | 12.4 ± 1.8 | ND | ND | No effect | 7.3 ± 8.3 | 5.0 ± 6.5 |
| ML-1 | ++ | 32.6 ± 8.5 | No effect | No effect | 34.3 ± 22.2 | 62.0 ± 15.6 | ND |
| ML1CD33KO | − | 22.8 ± 12.7 | No effect | No effect | No effect | No effect | No effect |
| MOLM-13 | ++ | 25.2 ± 13.8 | 2.7 ± 1.8 | 13.3 ± 4.5 | 94.7 ± 2.4 | 94.0 ± 3.1 | ND |
| MV4;11 | ++ | 34.9 ± 9.4 | ND | ND | 45.7 ± 17.3 | 51.0 ± 19.1 | 53.3 ± 18.7 |
| OCI-AML3 | + | 22.5 ± 12.9 | No effect | 5.7 ± 1.3 | 6.0 ± 3.1 | 1.3 ± 3.4 | ND |
| REH | − | 25.7 ± 13.2 | No effect | No effect | No effect | ND | ND |
| REHCD33FL | ++++ | 16.7 ± 7.9 | 21.0 ± 4.4 | 73.0 ± 7.8 | 89.0 ± 4.0 | ND | ND |
| TF-1 | +++ | 28.6 ± 1.6 | ND | ND | 68.0 ± 4.6 | 74.7 ± 4.3 | 78.3 ± 6.1 |
| TF-1CD33KO | − | 31.6 ± 6.5 | ND | ND | 0.3 ± 3.3 | No effect | No effect |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lunn-Halbert, M.C.; Laszlo, G.S.; Erraiss, S.; Orr, M.T.; Jessup, H.K.; Thomas, H.J.; Chan, H.; Jahromi, M.A.; Lloyd, J.; Cheung, A.F.; et al. Preclinical Characterization of the Anti-Leukemia Activity of the CD33/CD16a/NKG2D Immune-Modulating TriNKET® CC-96191. Cancers 2024, 16, 877. https://doi.org/10.3390/cancers16050877
Lunn-Halbert MC, Laszlo GS, Erraiss S, Orr MT, Jessup HK, Thomas HJ, Chan H, Jahromi MA, Lloyd J, Cheung AF, et al. Preclinical Characterization of the Anti-Leukemia Activity of the CD33/CD16a/NKG2D Immune-Modulating TriNKET® CC-96191. Cancers. 2024; 16(5):877. https://doi.org/10.3390/cancers16050877
Chicago/Turabian StyleLunn-Halbert, Margaret C., George S. Laszlo, Sarah Erraiss, Mark T. Orr, Heidi K. Jessup, Heather J. Thomas, Henry Chan, Mahan A. Jahromi, Jonathan Lloyd, Ann F. Cheung, and et al. 2024. "Preclinical Characterization of the Anti-Leukemia Activity of the CD33/CD16a/NKG2D Immune-Modulating TriNKET® CC-96191" Cancers 16, no. 5: 877. https://doi.org/10.3390/cancers16050877
APA StyleLunn-Halbert, M. C., Laszlo, G. S., Erraiss, S., Orr, M. T., Jessup, H. K., Thomas, H. J., Chan, H., Jahromi, M. A., Lloyd, J., Cheung, A. F., Chang, G. P., Dichwalkar, T., Fallon, D., Grinberg, A., Rodríguez-Arbolí, E., Lim, S. Y. T., Kehret, A. R., Huo, J., Cole, F. M., ... Walter, R. B. (2024). Preclinical Characterization of the Anti-Leukemia Activity of the CD33/CD16a/NKG2D Immune-Modulating TriNKET® CC-96191. Cancers, 16(5), 877. https://doi.org/10.3390/cancers16050877