

Inhibition of Enhancer of Zeste Homolog 2 Induces Blast Differentiation, Impairs Engraftment and Prolongs Survival in Murine Models of Acute Myeloid Leukemia

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemical Compounds
2.2. Cell Lines and Culture Conditions
2.3. Primary AML Samples and Culture Conditions
2.4. Antibodies
2.5. Cell Treatment and Immunoblots
2.6. Proliferation Assay
2.7. Colony Formation Unit (CFU) Assay
2.8. MOLM-13 Luciferase Xenograft Murine Model
2.9. Primary AML Differentiation
2.10. Patient-Derived Xenograft (PDX) Murine Studies
2.11. Statistical Analysis
3. Results
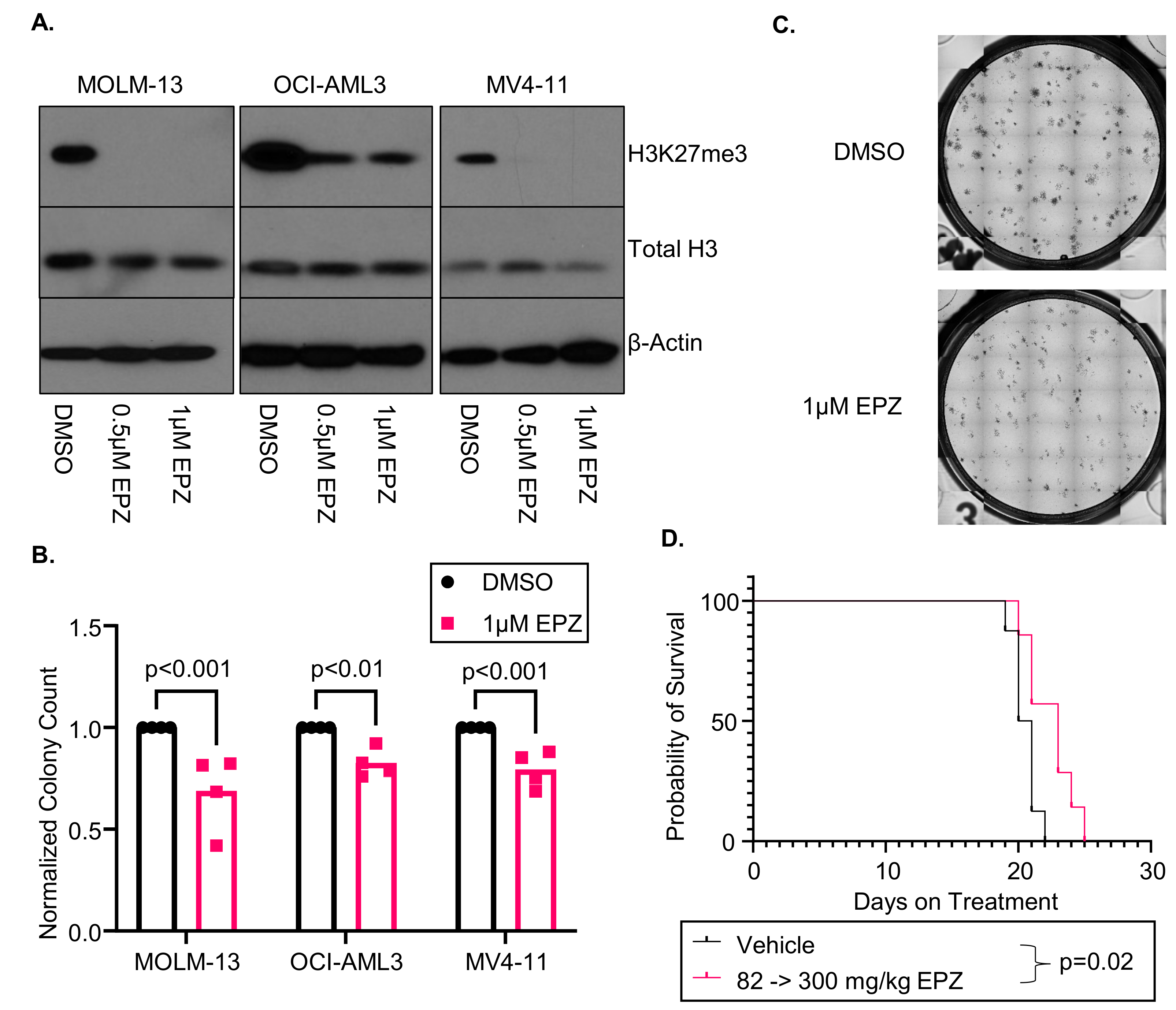
3.1. EPZ011989 Is an Effective Inhibitor of EZH2 Function in AML Cell Lines In Vitro and In Vivo
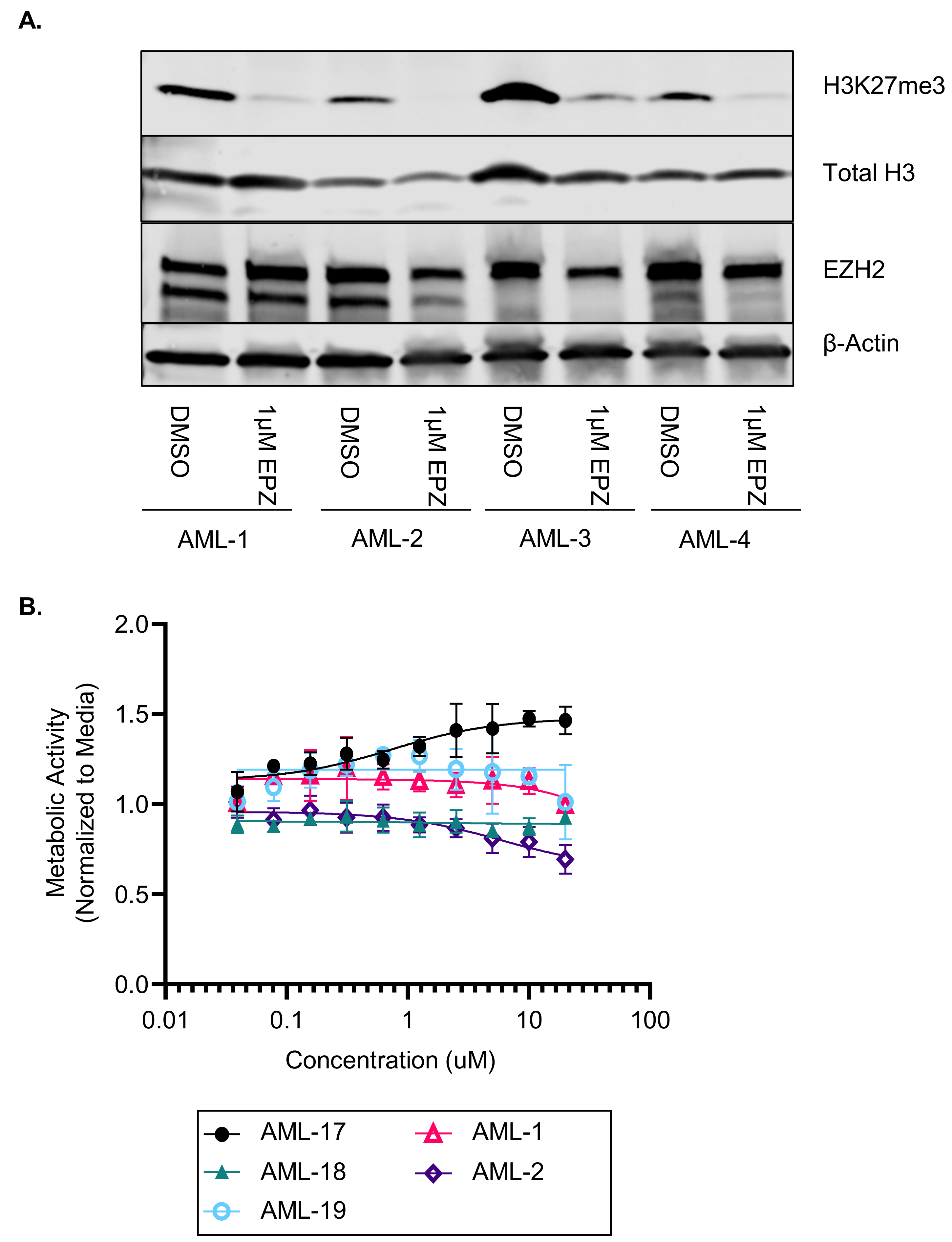
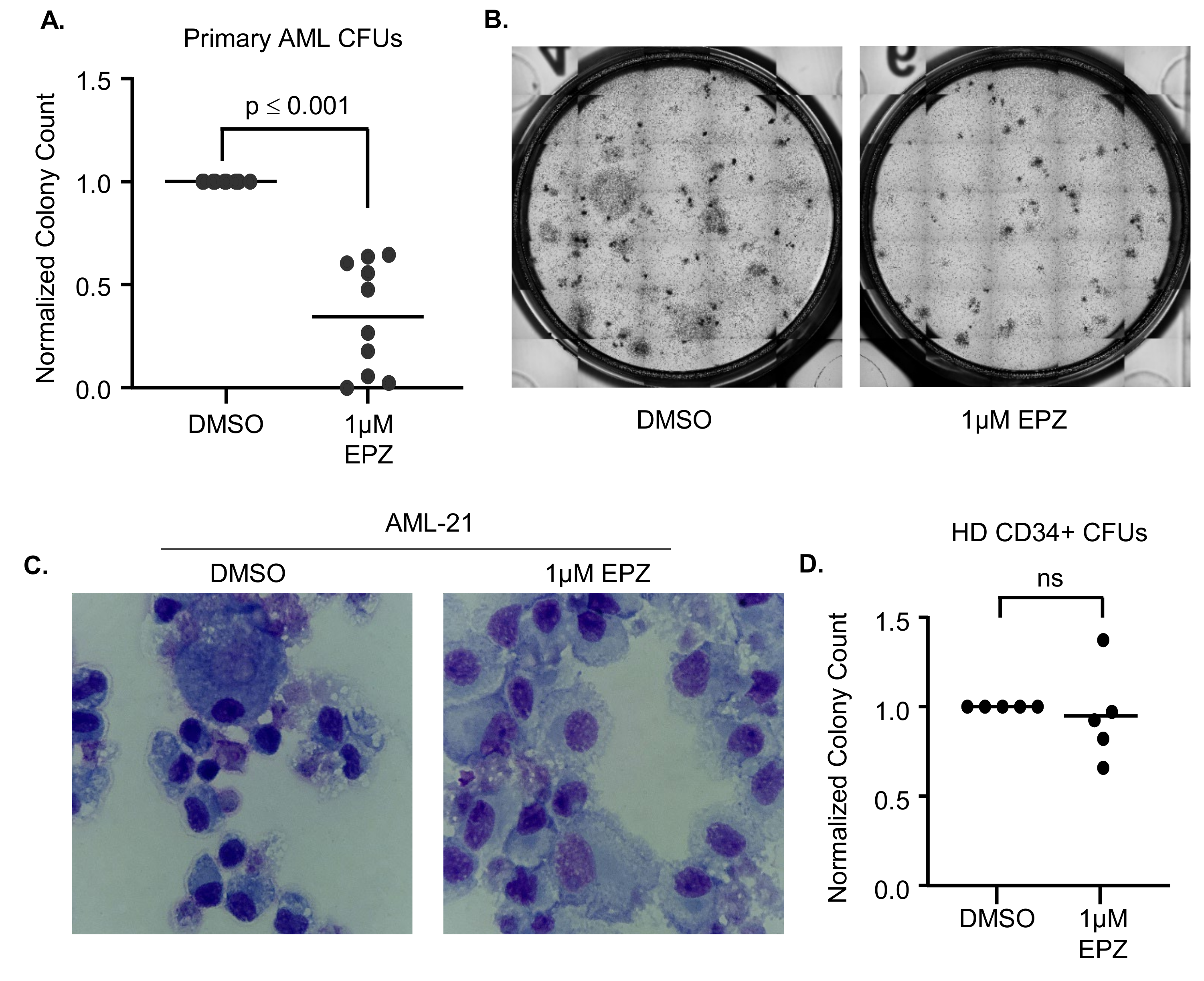
3.2. EZH2 Inhibition Induces Differentiation in Primary AML Samples
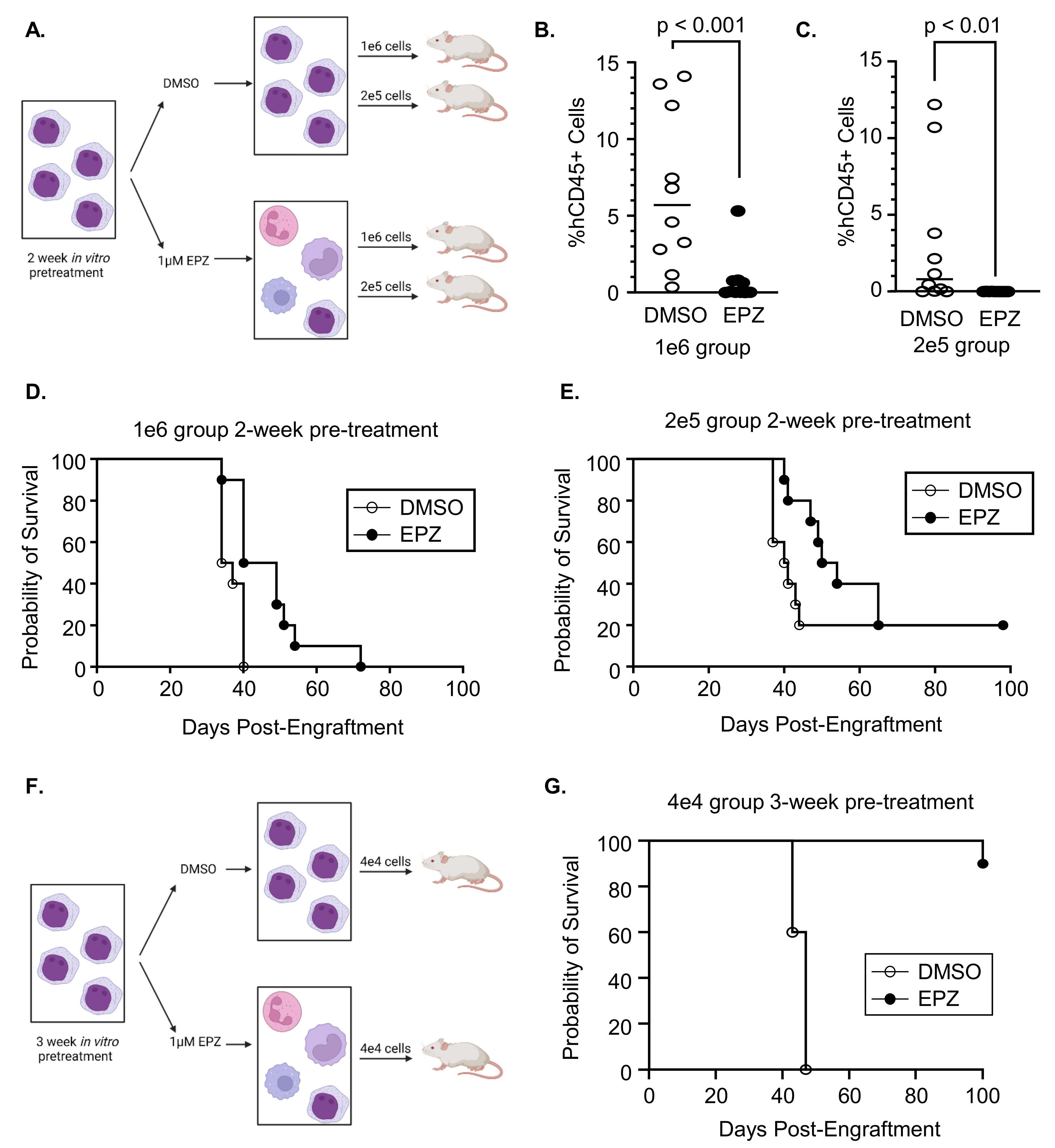
3.3. Inhibition of EZH2 Prior to Engraftment Delays Expansion of Primary AML Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Kouchkovsky, I.; Abdul-Hay, M. Acute myeloid leukemia: A comprehensive review and 2016 update. Blood Cancer J. 2016, 6, e441. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; Kadia, T.; DiNardo, C.; Daver, N.; Borthakur, G.; Jabbour, E.; Garcia-Manero, G.; Konopleva, M.; Ravandi, F. Acute myeloid leukemia: Current progress and future directions. Blood Cancer J. 2021, 11, 41. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, H.F.; Sun, Z.; Yao, X.; Litzow, M.R.; Luger, S.M.; Paietta, E.M.; Racevskis, J.; Dewald, G.W.; Ketterling, R.P.; Bennett, J.M.; et al. Anthracycline dose intensification in acute myeloid leukemia. N. Engl. J. Med. 2009, 361, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Lowenberg, B.; Ossenkoppele, G.J.; van Putten, W.; Schouten, H.C.; Graux, C.; Ferrant, A.; Sonneveld, P.; Maertens, J.; Jongen-Lavrencic, M.; von Lilienfeld-Toal, M.; et al. High-dose daunorubicin in older patients with acute myeloid leukemia. N. Engl. J. Med. 2009, 361, 1235–1248. [Google Scholar] [CrossRef] [PubMed]
- McMahon, C.M.; Canaani, J.; Rea, B.; Sargent, R.L.; Qualtieri, J.N.; Watt, C.D.; Morrissette, J.J.D.; Carroll, M.; Perl, A.E. Gilteritinib induces differentiation in relapsed and refractory FLT3-mutated acute myeloid leukemia. Blood Adv. 2019, 3, 1581–1585. [Google Scholar] [CrossRef]
- Norsworthy, K.J.; Mulkey, F.; Scott, E.C.; Ward, A.F.; Przepiorka, D.; Charlab, R.; Dorff, S.E.; Deisseroth, A.; Kazandjian, D.; Sridhara, R.; et al. Differentiation Syndrome with Ivosidenib and Enasidenib Treatment in Patients with Relapsed or Refractory IDH-Mutated AML: A U.S. Food and Drug Administration Systematic Analysis. Clin. Cancer Res. 2020, 26, 4280–4288. [Google Scholar] [CrossRef] [PubMed]
- Fathi, A.T.; DiNardo, C.D.; Kline, I.; Kenvin, L.; Gupta, I.; Attar, E.C.; Stein, E.M.; de Botton, S.; Investigators, A.C.S. Differentiation Syndrome Associated With Enasidenib, a Selective Inhibitor of Mutant Isocitrate Dehydrogenase 2: Analysis of a Phase 1/2 Study. JAMA Oncol. 2018, 4, 1106–1110. [Google Scholar] [CrossRef] [PubMed]
- McMahon, C.M.; Ferng, T.; Canaani, J.; Wang, E.S.; Morrissette, J.J.D.; Eastburn, D.J.; Pellegrino, M.; Durruthy-Durruthy, R.; Watt, C.D.; Asthana, S.; et al. Clonal Selection with RAS Pathway Activation Mediates Secondary Clinical Resistance to Selective FLT3 Inhibition in Acute Myeloid Leukemia. Cancer Discov. 2019, 9, 1050–1063. [Google Scholar] [CrossRef]
- Intlekofer, A.M.; Shih, A.H.; Wang, B.; Nazir, A.; Rustenburg, A.S.; Albanese, S.K.; Patel, M.; Famulare, C.; Correa, F.M.; Takemoto, N.; et al. Acquired resistance to IDH inhibition through trans or cis dimer-interface mutations. Nature 2018, 559, 125–129. [Google Scholar] [CrossRef]
- Quek, L.; David, M.D.; Kennedy, A.; Metzner, M.; Amatangelo, M.; Shih, A.; Stoilova, B.; Quivoron, C.; Heiblig, M.; Willekens, C.; et al. Clonal heterogeneity of acute myeloid leukemia treated with the IDH2 inhibitor enasidenib. Nat. Med. 2018, 24, 1167–1177. [Google Scholar] [CrossRef]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Yap, D.B.; Chu, J.; Berg, T.; Schapira, M.; Cheng, S.W.; Moradian, A.; Morin, R.D.; Mungall, A.J.; Meissner, B.; Boyle, M.; et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood 2011, 117, 2451–2459. [Google Scholar] [CrossRef] [PubMed]
- Knutson, S.K.; Kawano, S.; Minoshima, Y.; Warholic, N.M.; Huang, K.C.; Xiao, Y.; Kadowaki, T.; Uesugi, M.; Kuznetsov, G.; Kumar, N.; et al. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol. Cancer Ther. 2014, 13, 842–854. [Google Scholar] [CrossRef] [PubMed]
- Morschhauser, F.; Tilly, H.; Chaidos, A.; McKay, P.; Phillips, T.; Assouline, S.; Batlevi, C.L.; Campbell, P.; Ribrag, V.; Damaj, G.L.; et al. Tazemetostat for patients with relapsed or refractory follicular lymphoma: An open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol. 2020, 21, 1433–1442. [Google Scholar] [CrossRef] [PubMed]
- Ernst, T.; Chase, A.J.; Score, J.; Hidalgo-Curtis, C.E.; Bryant, C.; Jones, A.V.; Waghorn, K.; Zoi, K.; Ross, F.M.; Reiter, A.; et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat. Genet. 2010, 42, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Kempf, J.M.; Weser, S.; Bartoschek, M.D.; Metzeler, K.H.; Vick, B.; Herold, T.; Volse, K.; Mattes, R.; Scholz, M.; Wange, L.E.; et al. Loss-of-function mutations in the histone methyltransferase EZH2 promote chemotherapy resistance in AML. Sci. Rep. 2021, 11, 5838. [Google Scholar] [CrossRef] [PubMed]
- Stomper, J.; Meier, R.; Ma, T.; Pfeifer, D.; Ihorst, G.; Blagitko-Dorfs, N.; Greve, G.; Zimmer, D.; Platzbecker, U.; Hagemeijer, A.; et al. Integrative study of EZH2 mutational status, copy number, protein expression and H3K27 trimethylation in AML/MDS patients. Clin. Epigenet. 2021, 13, 77. [Google Scholar] [CrossRef]
- Basheer, F.; Giotopoulos, G.; Meduri, E.; Yun, H.; Mazan, M.; Sasca, D.; Gallipoli, P.; Marando, L.; Gozdecka, M.; Asby, R.; et al. Contrasting requirements during disease evolution identify EZH2 as a therapeutic target in AML. J. Exp. Med. 2019, 216, 966–981. [Google Scholar] [CrossRef]
- Campbell, J.E.; Kuntz, K.W.; Knutson, S.K.; Warholic, N.M.; Keilhack, H.; Wigle, T.J.; Raimondi, A.; Klaus, C.R.; Rioux, N.; Yokoi, A.; et al. EPZ011989, A Potent, Orally-Available EZH2 Inhibitor with Robust in Vivo Activity. ACS Med. Chem. Lett. 2015, 6, 491–495. [Google Scholar] [CrossRef]
- Eisenmann, E.D.; Stromatt, J.C.; Fobare, S.; Huang, K.M.; Buelow, D.R.; Orwick, S.; Jeon, J.Y.; Weber, R.H.; Larsen, B.; Mims, A.S.; et al. TP-0903 Is Active in Preclinical Models of Acute Myeloid Leukemia with TP53 Mutation/Deletion. Cancers 2022, 15, 29. [Google Scholar] [CrossRef]
- Chen, Y.; Jacamo, R.; Konopleva, M.; Garzon, R.; Croce, C.; Andreeff, M. CXCR4 downregulation of let-7a drives chemoresistance in acute myeloid leukemia. J. Clin. Investig. 2013, 123, 2395–2407. [Google Scholar] [CrossRef]
- Stacchiotti, S.; Zuco, V.; Tortoreto, M.; Cominetti, D.; Frezza, A.M.; Percio, S.; Indio, V.; Barisella, M.; Monti, V.; Brich, S.; et al. Comparative Assessment of Antitumor Effects and Autophagy Induction as a Resistance Mechanism by Cytotoxics and EZH2 Inhibition in INI1-Negative Epithelioid Sarcoma Patient-Derived Xenograft. Cancers 2019, 11, 1015. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, F.E.; Cashman, J.D.; Hogge, D.E.; Humphries, R.K.; Eaves, C.J. NOD/SCID mice engineered to express human IL-3, GM-CSF and Steel factor constitutively mobilize engrafted human progenitors and compromise human stem cell regeneration. Leukemia 2004, 18, 341–347. [Google Scholar] [CrossRef]
- Thompson, H.W.; Mera, R.; Prasad, C. The Analysis of Variance (ANOVA). Nutr. Neurosci. 1999, 2, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Kleinbaum, D.G.; Klein, M. A Self-Learning Text. In Survival Analysis, 3rd ed.; Springer: New York, NY, USA, 2012. [Google Scholar]
- SAS. Statistical Analysis System, SAS Release 9.1 for Windows; SAS Institute Inc.: Cary, NC, USA, 2003. [Google Scholar]
- He, S.; Liu, Y.; Meng, L.; Sun, H.; Wang, Y.; Ji, Y.; Purushe, J.; Chen, P.; Li, C.; Madzo, J.; et al. Ezh2 phosphorylation state determines its capacity to maintain CD8+ T memory precursors for antitumor immunity. Nat. Commun. 2017, 8, 2125. [Google Scholar] [CrossRef] [PubMed]
- Sung, P.J.; Selvam, M.; Riedel, S.S.; Xie, H.M.; Bryant, K.; Manning, B.; Wertheim, G.B.; Kulej, K.; Pham, L.; Bowman, R.L.; et al. FLT3 tyrosine kinase inhibition modulates PRC2 and promotes differentiation in acute myeloid leukemia. Leukemia 2024. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Chen, X.; Zhang, Y.; Ren, F.; Ma, Y. MEK/ERK and PI3K/AKT pathway inhibitors affect the transformation of myelodysplastic syndrome into acute myeloid leukemia via H3K27me3 methylases and de-methylases. Int. J. Oncol. 2023, 63, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Alqazzaz, M.A.; Luciani, G.M.; Vu, V.; Machado, R.A.C.; Szewczyk, M.M.; Adamson, E.C.; Cheon, S.; Li, F.; Arrowsmith, C.H.; Minden, M.D.; et al. Epigenetic vulnerabilities of leukemia harboring inactivating EZH2 mutations. Exp. Hematol. 2023, 104135. [Google Scholar] [CrossRef]
- Kaundal, B.; Srivastava, A.K.; Dev, A.; Mohanbhai, S.J.; Karmakar, S.; Roy Choudhury, S. Nanoformulation of EPZ011989 Attenuates EZH2-c-Myb Epigenetic Interaction by Proteasomal Degradation in Acute Myeloid Leukemia. Mol. Pharm. 2020, 17, 604–621. [Google Scholar] [CrossRef]
- Porazzi, P.; Petruk, S.; Pagliaroli, L.; De Dominici, M.; Deming, D., 2nd; Puccetti, M.V.; Kushinsky, S.; Kumar, G.; Minieri, V.; Barbieri, E.; et al. Targeting Chemotherapy to Decondensed H3K27me3-Marked Chromatin of AML Cells Enhances Leukemia Suppression. Cancer Res. 2022, 82, 458–471. [Google Scholar] [CrossRef]
- Wen, S.; Wang, J.; Liu, P.; Li, Y.; Lu, W.; Hu, Y.; Liu, J.; He, Z.; Huang, P. Novel combination of histone methylation modulators with therapeutic synergy against acute myeloid leukemia in vitro and in vivo. Cancer Lett. 2018, 413, 35–45. [Google Scholar] [CrossRef]
- Aldana, J.; Gardner, M.L.; Freitas, M.A. Integrative Multi-Omics Analysis of Oncogenic EZH2 Mutants: From Epigenetic Reprogramming to Molecular Signatures. Int. J. Mol. Sci. 2023, 24, 11378. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fobare, S.; Elgamal, O.A.; Wunderlich, M.; Stahl, E.; Mehmood, A.; Furby, C.; Lerma, J.R.; Sesterhenn, T.M.; Pan, J.; Rai, J.; et al. Inhibition of Enhancer of Zeste Homolog 2 Induces Blast Differentiation, Impairs Engraftment and Prolongs Survival in Murine Models of Acute Myeloid Leukemia. Cancers 2024, 16, 569. https://doi.org/10.3390/cancers16030569
Fobare S, Elgamal OA, Wunderlich M, Stahl E, Mehmood A, Furby C, Lerma JR, Sesterhenn TM, Pan J, Rai J, et al. Inhibition of Enhancer of Zeste Homolog 2 Induces Blast Differentiation, Impairs Engraftment and Prolongs Survival in Murine Models of Acute Myeloid Leukemia. Cancers. 2024; 16(3):569. https://doi.org/10.3390/cancers16030569
Chicago/Turabian StyleFobare, Sydney, Ola A. Elgamal, Mark Wunderlich, Emily Stahl, Abeera Mehmood, Casie Furby, James R. Lerma, Thomas M. Sesterhenn, Jianmin Pan, Jayesh Rai, and et al. 2024. "Inhibition of Enhancer of Zeste Homolog 2 Induces Blast Differentiation, Impairs Engraftment and Prolongs Survival in Murine Models of Acute Myeloid Leukemia" Cancers 16, no. 3: 569. https://doi.org/10.3390/cancers16030569
APA StyleFobare, S., Elgamal, O. A., Wunderlich, M., Stahl, E., Mehmood, A., Furby, C., Lerma, J. R., Sesterhenn, T. M., Pan, J., Rai, J., Johnstone, M. E., Abdul-Aziz, A., Johnson, M. L., Rai, S. N., Byrd, J. C., & Hertlein, E. (2024). Inhibition of Enhancer of Zeste Homolog 2 Induces Blast Differentiation, Impairs Engraftment and Prolongs Survival in Murine Models of Acute Myeloid Leukemia. Cancers, 16(3), 569. https://doi.org/10.3390/cancers16030569