

Cancer and HIV: The Molecular Mechanisms of the Deadly Duo


{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. AIDS-Defining Malignancies
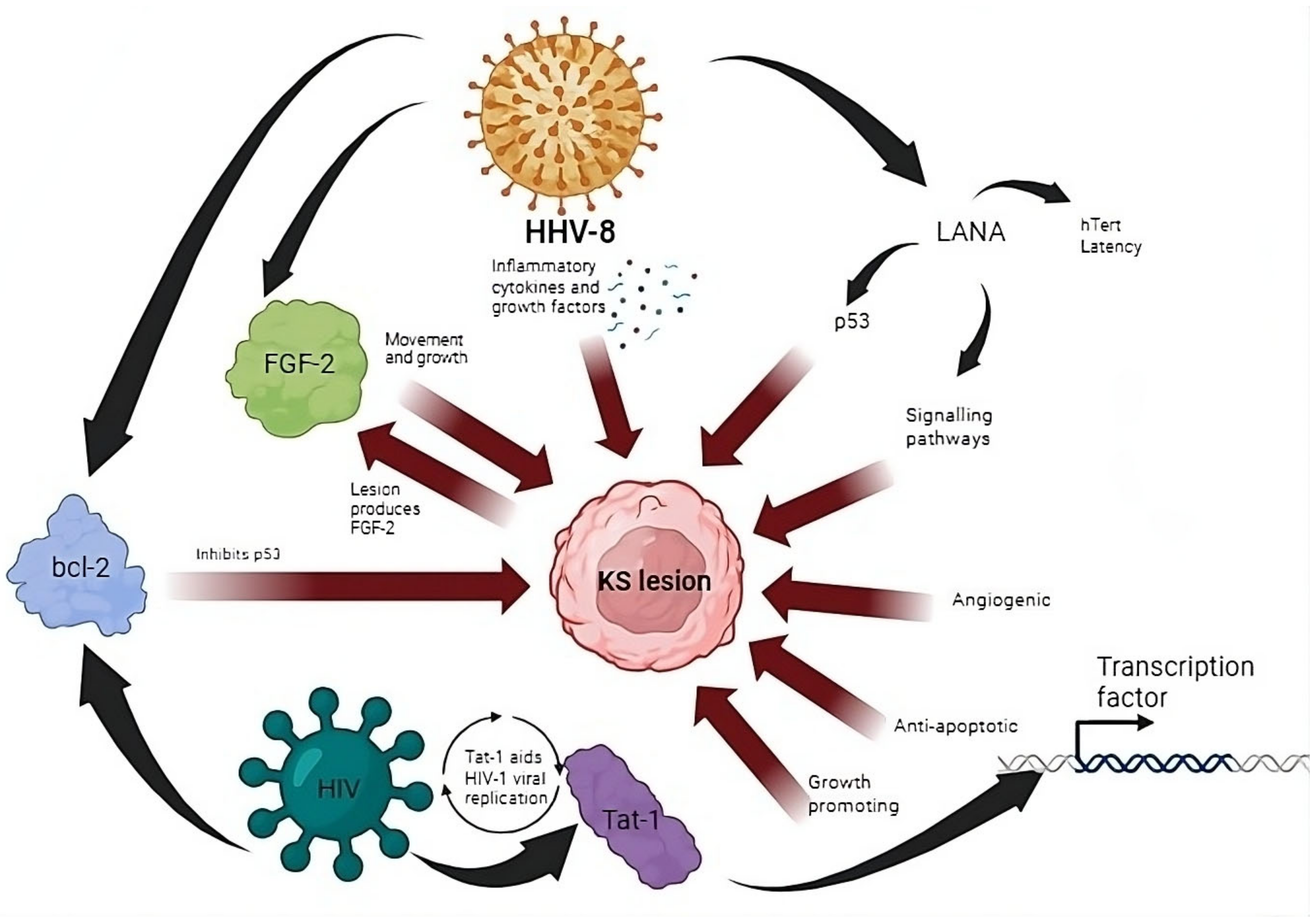
2.1. Inflammatory Cytokines and Signaling in the Formation of KS
2.2. Oncogenic Viral Integration into Telomeres
2.3. Immune Dysregulation and Immune Evasion in KS Development
2.4. An Opportunity for Therapeutic Targeting
2.5. Cervical Cancer Formation Due to Co-Infection with HIV-1 and Multiple HPV Subtypes
2.6. HPV Inhibition of Tumor Suppressor p53 as a Promising Therapeutic Strategy
2.7. Burkitt Lymphoma (BL) Caused by EBV Dysregulation of Protooncogene MYC
2.8. NHL Development Due to Immunosuppression
2.9. Role of PTEN in BL Formation
2.10. Immunomodulatory Approach Targeting EBV-Related Cancers
2.11. Overall View of ADMs in Africa
3. Post-ART Rise in Non-ADMs
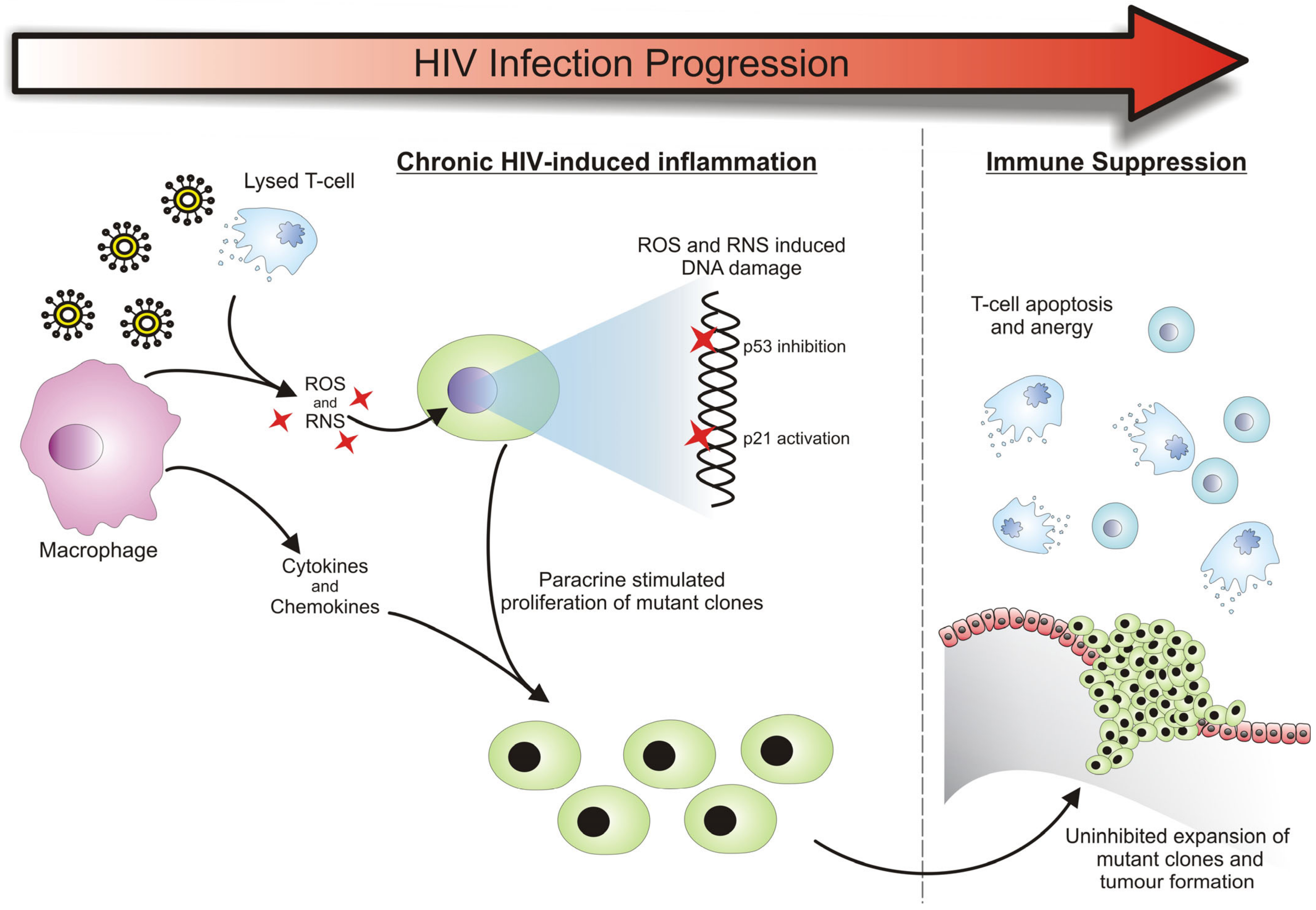
3.1. HIV-Related Chronic Inflammation in the Development of Cancers
3.2. Cancer Susceptibility Due to a Deregulated Immune System
3.3. Cancer Formation Due to HIV Provirus Integration
3.4. Role of HIV-1 Tat-1 Protein in Cancer Development
3.5. HIV-1 Nef Protein Involvement in Cancer
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Frisch, M.; Biggar, R.J.; Engels, E.A.; Goedert, J.J. Association of cancer with AIDS-related immunosuppression in adults. JAMA J. Am. Med. Assoc. 2001, 285, 1736–1745. [Google Scholar] [CrossRef] [PubMed]
- Nordell, A.D.; Okulicz, J.F.; Palfreeman, A.; Horban, A.; Kedem, E.; Neuhaus, J.; Jacobs, D.R., Jr.; Duprez, D.A.; Neaton, J.D. Inflammation Related Morbidity and Mortality Among HIV-Positive Adults: How Extensive Is It? J. Acquir. Immune Defic. Syndr. 2018, 77, 1. [Google Scholar]
- Robbins, H.A.; Pfeiffer, R.M.; Shiels, M.S.; Li, J.; Hall, H.I.; Engels, E.A. Excess cancers among HIV-infected people in the United States. J. Natl. Cancer Inst. 2015, 107, dju503. [Google Scholar] [CrossRef] [PubMed]
- Shmakova, A.; Germini, D.; Vassetzky, Y. HIV-1, HAART and cancer: A complex relationship. Int. J. Cancer. J. Int. Cancer 2020, 146, 2666–2679. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. 1993 revised classification system for HIV infection and expanded surveillance case definition for AIDS among adolescents and adults. MMWR. Recomm. Rep. Morb. Mortal. Wkly. Rep. Recomm. Rep. 1992, 41, 1–19. [Google Scholar]
- Park, L.S.; Tate, J.P.; Sigel, K.; Rimland, D.; Crothers, K.; Gibert, C.; Rodriguez-Barradas, M.C.; Goetz, M.B.; Bedimo, R.J.; Brown, S.T.; et al. Time trends in cancer incidence in persons living with HIV/AIDS in the antiretroviral therapy era: 1997–2012. AIDS 2016, 30, 1795–1806. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, S.; Lise, M.; Clifford, G.M.; Rickenbach, M.; Levi, F.; Maspoli, M.; Bouchardy, C.; Dehler, S.; Jundt, G.; Ess, S.; et al. Changing patterns of cancer incidence in the early- and late-HAART periods: The Swiss HIV Cohort Study. Br. J. Cancer 2010, 103, 416–422. [Google Scholar] [CrossRef]
- Powles, T.; Robinson, D.; Stebbing, J.; Shamash, J.; Nelson, M.; Gazzard, B.; Mandelia, S.; Moller, H.; Bower, M. Highly active antiretroviral therapy and the incidence of non-AIDS-defining cancers in people with HIV infection. J. Clin. Oncol. 2009, 27, 884–890. [Google Scholar] [CrossRef]
- Coghill, A.E.; Shiels, M.S.; Suneja, G.; Engels, E.A. Elevated Cancer-Specific Mortality Among HIV-Infected Patients in the United States. J. Clin. Oncol. 2015, 33, 2376–2383. [Google Scholar] [CrossRef]
- Engels, E.A.; Yanik, E.L.; Wheeler, W.; Gill, M.J.; Shiels, M.S.; Dubrow, R.; Althoff, K.N.; Silverberg, M.J.; Brooks, J.T.; Kitahata, M.M.; et al. Cancer-Attributable Mortality Among People with Treated Human Immunodeficiency Virus Infection in North America. Clin. Infect. Dis. 2017, 65, 636–643. [Google Scholar] [CrossRef]
- Goehringer, F.; Bonnet, F.; Salmon, D.; Cacoub, P.; Paye, A.; Chene, G.; Morlat, P.; May, T.; Grp, A.E.M.S. Causes of Death in HIV-Infected Individuals with Immunovirologic Success in a National Prospective Survey. Aids Res. Hum. Retroviruses 2017, 33, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Marima, R.; Hull, R.; Lolas, G.; Syrigos, K.N.; Kgoebane-Maseko, M.; Kaufmann, A.M.; Dlamini, Z. The Catastrophic HPV/HIV Dual Viral Oncogenomics in Concert with Dysregulated Alternative Splicing in Cervical Cancer. Int. J. Mol. Sci. 2021, 22, 115. [Google Scholar] [CrossRef]
- Lekoane, K.M.B.; Kuupiel, D.; Mashamba-Thompson, T.P.; Ginindza, T.G. The interplay of HIV and human papillomavirus-related cancers in sub-Saharan Africa: Scoping review. Syst. Rev. 2020, 9, 88. [Google Scholar] [CrossRef] [PubMed]
- Portilla-Tamarit, J.; Reus, S.; Portilla, I.; Fuster Ruiz-de-Apodaca, M.J.; Portilla, J. Impact of Advanced HIV Disease on Quality of Life and Mortality in the Era of Combined Antiretroviral Treatment. J. Clin. Med. 2021, 10, 716. [Google Scholar] [CrossRef] [PubMed]
- Nachega, J.B.; Parienti, J.J.; Uthman, O.A.; Gross, R.; Dowdy, D.W.; Sax, P.E.; Gallant, J.E.; Mugavero, M.J.; Mills, E.J.; Giordano, T.P. Lower pill burden and once-daily antiretroviral treatment regimens for HIV infection: A meta-analysis of randomized controlled trials. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2014, 58, 1297–1307. [Google Scholar] [CrossRef] [PubMed]
- Eyawo, O.; Franco-Villalobos, C.; Hull, M.W.; Nohpal, A.; Samji, H.; Sereda, P.; Lima, V.D.; Shoveller, J.; Moore, D.; Montaner, J.S.; et al. Changes in mortality rates and causes of death in a population-based cohort of persons living with and without HIV from 1996 to 2012. BMC Infect. Dis. 2017, 17, 174. [Google Scholar] [CrossRef] [PubMed]
- Shiels, M.S.; Engels, E.A. Evolving epidemiology of HIV-associated malignancies. Curr. Opin. HIV AIDS 2017, 12, 6–11. [Google Scholar] [CrossRef]
- Yang, H.Y.; Beymer, M.R.; Suen, S.C. Chronic Disease Onset Among People Living with HIV and AIDS in a Large Private Insurance Claims Dataset. Sci. Rep. 2019, 9, 18514. [Google Scholar] [CrossRef]
- Yuan, T.; Hu, Y.; Zhou, X.; Yang, L.; Wang, H.; Li, L.; Wang, J.; Qian, H.Z.; Clifford, G.M.; Zou, H. Incidence and mortality of non-AIDS-defining cancers among people living with HIV: A systematic review and meta-analysis. EClinicalMedicine 2022, 52, 101613. [Google Scholar] [CrossRef]
- Pontali, E.; Cenderello, G.; Cassola, G.; Torresin, A. Specific issues in the management of hepatocellular carcinoma in hepatitis B virus/hepatitis C virus-human immunodeficiency virus co-infected patients. Int. STD Res. Rev. 2015, 3, 69–83. [Google Scholar] [CrossRef]
- Shiferaw, E.; Alemayehu, T.; Hailu, D.; Teka, G. Non-aids defining and aids defining malignancies and determi-nants among children and adolescents living with HIV in Addis Ababa, Ethiopia. Ethiop. Med. J. 2021, 59, 217–222. [Google Scholar]
- Fu, L.; Tian, T.; Wang, B.; Lu, Z.; Gao, Y.; Sun, Y.; Lin, Y.-F.; Zhang, W.; Li, Y.; Zou, H. Global patterns and trends in Kaposi sarcoma incidence: A population-based study. Lancet Glob. Health 2023, 11, e1566–e1575. [Google Scholar] [CrossRef] [PubMed]
- Ensoli, B.; Sgadari, C.; Barillari, G.; Sirianni, M.C.; Sturzl, M.; Monini, P. Biology of Kaposi’s sarcoma. Eur. J. Cancer 2001, 37, 1251–1269. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E.; Damania, B.; Krown, S.E.; Martin, J.; Bower, M.; Whitby, D. Kaposi sarcoma. Nat. Rev. Dis. Primers 2019, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Karabajakian, A.; Ray-Coquard, I.; Blay, J.Y. Molecular Mechanisms of Kaposi Sarcoma Development. Cancers 2022, 14, 1869. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.S.; Chang, Y. Kaposi’s sarcoma-associated herpesvirus immunoevasion and tumorigenesis: Two sides of the same coin? Annu. Rev. Microbiol. 2003, 57, 609–639. [Google Scholar] [CrossRef] [PubMed]
- Tagawa, T.; Serquina, A.; Kook, I.; Ziegelbauer, J. Viral non-coding RNAs: Stealth strategies in the tug-of-war between humans and herpesviruses. Semin. Cell Dev. Biol. 2021, 111, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.R.; Brulois, K.; Wong, L.; Jung, J.U. Modulation of Immune System by Kaposi’s Sarcoma-Associated Herpesvirus: Lessons from Viral Evasion Strategies. Front. Microbiol. 2012, 3, 44. [Google Scholar] [CrossRef]
- Salimi-Jeda, A.; Badrzadeh, F.; Esghaei, M.; Abdoli, A. The role of telomerase and viruses interaction in cancer development, and telomerase-dependent therapeutic approaches. Cancer Treat. Res. Commun. 2021, 27, 100323. [Google Scholar] [CrossRef]
- Kamranvar, S.A.; Masucci, M.G.J.V. Regulation of telomere homeostasis during Epstein-Barr virus infection and immortalization. Viruses 2017, 9, 217. [Google Scholar] [CrossRef]
- Yuan, X.; Larsson, C.; Xu, D. Mechanisms underlying the activation of TERT transcription and telomerase activity in human cancer: Old actors and new players. Oncogene 2019, 38, 6172–6183. [Google Scholar] [CrossRef]
- Shamay, M.; Liu, J.; Li, R.; Liao, G.; Shen, L.; Greenway, M.; Hu, S.; Zhu, J.; Xie, Z.; Ambinder, R.F.; et al. A protein array screen for Kaposi’s sarcoma-associated herpesvirus LANA interactors links LANA to TIP60, PP2A activity, and telomere shortening. J. Virol. 2012, 86, 5179–5191. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.C.; Borah, S.; Robertson, E.S. Latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus up-regulates transcription of human telomerase reverse transcriptase promoter through interaction with transcription factor Sp1. J. Virol. 2004, 78, 10348–10359. [Google Scholar] [CrossRef] [PubMed]
- Broussard, G.; Damania, B. KSHV: Immune Modulation and Immunotherapy. Front. Immunol. 2019, 10, 3084. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dalla Pria, A.; Pinato, D.J.; Bracchi, M.; Bower, M. Recent advances in HIV-associated Kaposi sarcoma. F1000Research 2019, 8, 970. [Google Scholar] [CrossRef] [PubMed]
- Shankar, P.; Russo, M.; Harnisch, B.; Patterson, M.; Skolnik, P.; Lieberman, J. Impaired function of circulating HIV-specific CD8(+) T cells in chronic human immunodeficiency virus infection. Blood 2000, 96, 3094–3101. [Google Scholar] [CrossRef] [PubMed]
- Dupin, N. Update on oncogenesis and therapy for Kaposi sarcoma. Curr. Opin. Oncol. 2020, 32, 122–128. [Google Scholar] [CrossRef]
- Galanina, N.; Goodman, A.M.; Cohen, P.R.; Frampton, G.M.; Kurzrock, R. Successful Treatment of HIV-Associated Kaposi Sarcoma with Immune Checkpoint Blockade. Cancer Immunol. Res. 2018, 6, 1129–1135. [Google Scholar] [CrossRef]
- Trautmann, L.; Janbazian, L.; Chomont, N.; Said, E.A.; Gimmig, S.; Bessette, B.; Boulassel, M.R.; Delwart, E.; Sepulveda, H.; Balderas, R.S.; et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat. Med. 2006, 12, 1198–1202. [Google Scholar] [CrossRef]
- Host, K.M.; Jacobs, S.R.; West, J.A.; Zhang, Z.; Costantini, L.M.; Stopford, C.M.; Dittmer, D.P.; Damania, B. Kaposi’s Sarcoma-Associated Herpesvirus Increases PD-L1 and Proinflammatory Cytokine Expression in Human Monocytes. mBio 2017, 8, 10-1128. [Google Scholar] [CrossRef]
- Delyon, J.; Rabate, C.; Euvrard, S.; Harwood, C.A.; Proby, C.; Gulec, A.T.; Seckin, D.; Del Marmol, V.; Bouwes-Bavinck, J.N.; Ferrandiz-Pulido, C.; et al. Management of Kaposi sarcoma after solid organ transplantation: A European retrospective study. J. Am. Acad. Dermatol. 2019, 81, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Polizzotto, M.N.; Uldrick, T.S.; Wyvill, K.M.; Aleman, K.; Peer, C.J.; Bevans, M.; Sereti, I.; Maldarelli, F.; Whitby, D.; Marshall, V.; et al. Pomalidomide for Symptomatic Kaposi’s Sarcoma in People with and without HIV Infection: A Phase I/II Study. J. Clin. Oncol. 2016, 34, 4125–4131. [Google Scholar] [CrossRef] [PubMed]
- Pourcher, V.; Desnoyer, A.; Assoumou, L.; Lebbe, C.; Curjol, A.; Marcelin, A.G.; Cardon, F.; Gibowski, S.; Salmon, D.; Chennebault, J.M.; et al. Phase II Trial of Lenalidomide in HIV-Infected Patients with Previously Treated Kaposi’s Sarcoma: Results of the ANRS 154 Lenakap Trial. AIDS Res. Hum. Retroviruses 2017, 33, 1–10. [Google Scholar] [CrossRef]
- Uldrick, T.S.; Goncalves, P.H.; Wyvill, K.M.; Peer, C.J.; Bernstein, W.; Aleman, K.; Polizzotto, M.N.; Venzon, D.; Steinberg, S.M.; Marshall, V.; et al. A Phase Ib Study of Sorafenib (BAY 43-9006) in Patients with Kaposi Sarcoma. Oncologist 2017, 22, e505–e549. [Google Scholar] [CrossRef] [PubMed]
- Koon, H.B.; Bubley, G.J.; Pantanowitz, L.; Masiello, D.; Smith, B.; Crosby, K.; Proper, J.; Weeden, W.; Miller, T.E.; Chatis, P.; et al. Imatinib-induced regression of AIDS-related Kaposi’s sarcoma. J. Clin. Oncol. 2005, 23, 982–989. [Google Scholar] [CrossRef] [PubMed]
- Krown, S.E.; Roy, D.; Lee, J.Y.; Dezube, B.J.; Reid, E.G.; Venkataramanan, R.; Han, K.; Cesarman, E.; Dittmer, D.P. Rapamycin with Antiretroviral Therapy in AIDS-Associated Kaposi Sarcoma: An AIDS Malignancy Consortium Study. JAIDS J. Acquir. Immune Defic. Syndr. 2012, 59, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Arbyn, M.; Weiderpass, E.; Bruni, L.; de Sanjose, S.; Saraiya, M.; Ferlay, J.; Bray, F. Estimates of incidence and mortality of cervical cancer in 2018: A worldwide analysis. Lancet Glob. Health 2020, 8, e191–e203. [Google Scholar] [CrossRef]
- He, W.Q.; Li, C. Recent global burden of cervical cancer incidence and mortality, predictors, and temporal trends. Gynecol. Oncol. 2021, 163, 583–592. [Google Scholar] [CrossRef]
- Stelzle, D.; Tanaka, L.F.; Lee, K.K.; Ibrahim Khalil, A.; Baussano, I.; Shah, A.S.V.; McAllister, D.A.; Gottlieb, S.L.; Klug, S.J.; Winkler, A.S.; et al. Estimates of the global burden of cervical cancer associated with HIV. Lancet Glob. Health 2021, 9, e161–e169. [Google Scholar] [CrossRef]
- Ibrahim Khalil, A.; Mpunga, T.; Wei, F.; Baussano, I.; de Martel, C.; Bray, F.; Stelzle, D.; Dryden-Peterson, S.; Jaquet, A.; Horner, M.J.; et al. Age-specific burden of cervical cancer associated with HIV: A global analysis with a focus on sub-Saharan Africa. Int. J. Cancer. J. Int. Cancer 2022, 150, 761–772. [Google Scholar] [CrossRef]
- Zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Hull, R.; Mbele, M.; Makhafola, T.; Hicks, C.; Wang, S.M.; Reis, R.M.; Mehrotra, R.; Mkhize-Kwitshana, Z.; Kibiki, G.; Bates, D.O.; et al. Cervical cancer in low and middle-income countries. Oncol. Lett. 2020, 20, 2058–2074. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.K.; Aimagambetova, G.; Ukybassova, T.; Kongrtay, K.; Azizan, A. Human Papillomavirus Infection and Cervical Cancer: Epidemiology, Screening, and Vaccination-Review of Current Perspectives. J. Oncol. 2019, 2019, 3257939. [Google Scholar] [CrossRef] [PubMed]
- Lacey, C.J. HPV vaccination in HIV infection. Papillomavirus Res. 2019, 8, 100174. [Google Scholar] [CrossRef] [PubMed]
- Spinillo, A.; Dominoni, M.; Boschi, A.C.; Sosso, C.; Fiandrino, G.; Cesari, S.; Gardella, B. Clinical Significance of the Interaction between Human Papillomavirus (HPV) Type 16 and Other High-Risk Human Papillomaviruses in Women with Cervical Intraepithelial Neoplasia (CIN) and Invasive Cervical Cancer. J. Oncol. 2020, 2020, 6508180. [Google Scholar] [CrossRef] [PubMed]
- Mapanga, W.; Singh, E.; Feresu, S.A.; Girdler-Brown, B. Treatment of pre- and confirmed cervical cancer in HIV-seropositive women from developing countries: A systematic review. Syst. Rev. 2020, 9, 79. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Sharma, M.; Tan, N.; Barnabas, R.V. HIV-positive women have higher risk of human papilloma virus infection, precancerous lesions, and cervical cancer. AIDS 2018, 32, 795–808. [Google Scholar] [CrossRef] [PubMed]
- Maksimovic-Ivanic, D.; Fagone, P.; McCubrey, J.; Bendtzen, K.; Mijatovic, S.; Nicoletti, F. HIV-protease inhibitors for the treatment of cancer: Repositioning HIV protease inhibitors while developing more potent NO-hybridized derivatives? Int. J. Cancer. J. Int. Cancer 2017, 140, 1713–1726. [Google Scholar] [CrossRef]
- Park, S.; Auyeung, A.; Lee, D.L.; Lambert, P.F.; Carchman, E.H.; Sherer, N.M. HIV-1 Protease Inhibitors Slow HPV16-Driven Cell Proliferation through Targeted Depletion of Viral E6 and E7 Oncoproteins. Cancers 2021, 13, 949. [Google Scholar] [CrossRef]
- Hietanen, S.; Lain, S.; Krausz, E.; Blattner, C.; Lane, D.P. Activation of p53 in cervical carcinoma cells by small molecules. Proc. Natl. Acad. Sci. USA 2000, 97, 8501–8506. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Zapien, D.; Ruiz, F.X.; Poirson, J.; Mitschler, A.; Ramirez, J.; Forster, A.; Cousido-Siah, A.; Masson, M.; Vande Pol, S.; Podjarny, A.; et al. Structure of the E6/E6AP/p53 complex required for HPV-mediated degradation of p53. Nature 2016, 529, 541–545. [Google Scholar] [CrossRef]
- Makgoo, L.; Mosebi, S.; Mbita, Z. Molecular Mechanisms of HIV Protease Inhibitors Against HPV-Associated Cervical Cancer: Restoration of TP53 Tumour Suppressor Activities. Front. Mol. Biosci. 2022, 9, 875208. [Google Scholar] [CrossRef] [PubMed]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Hoya, A.; Soto-Cruz, I. Role of the JAK/STAT Pathway in Cervical Cancer: Its Relationship with HPV E6/E7 Oncoproteins. Cells 2020, 9, 2297. [Google Scholar] [CrossRef] [PubMed]
- Porras, G.O.R.; Nogueda, J.C.; Chacón, A.P. Chemotherapy and molecular therapy in cervical cancer. Rep. Pract. Oncol. Radiother. 2018, 23, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Zhou, J.; Fu, M.; Liang, L.; Deng, Q.; Liu, X.; Liu, F. Efficacy of recombinant human adenovirus-p53 combined with chemotherapy for locally advanced cervical cancer: A clinical trial. Oncol. Lett. 2017, 13, 3676–3680. [Google Scholar] [CrossRef]
- Zhao, X.; Sun, W.; Ren, Y.; Lu, Z. Therapeutic potential of p53 reactivation in cervical cancer. Crit. Rev. Oncol. Hematol. 2021, 157, 103182. [Google Scholar] [CrossRef]
- Molyneux, E.M.; Rochford, R.; Griffin, B.; Newton, R.; Jackson, G.; Menon, G.; Harrison, C.J.; Israels, T.; Bailey, S. Burkitt’s lymphoma. Lancet 2012, 379, 1234–1244. [Google Scholar] [CrossRef]
- Hubel, K. The Changing Landscape of Lymphoma Associated with HIV Infection. Curr. Oncol. Rep. 2020, 22, 111. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef]
- Gopal, S.; Gross, T.G. How I treat Burkitt lymphoma in children, adolescents, and young adults in sub-Saharan Africa. Blood 2018, 132, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Gastwirt, J.P.; Roschewski, M. Management of adults with Burkitt lymphoma. Clin. Adv. Hematol. Oncol. 2018, 16, 812–822. [Google Scholar] [PubMed]
- Crombie, J.; LaCasce, A. The treatment of Burkitt lymphoma in adults. Blood 2021, 137, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.A.; Allday, M.J.; Crawford, D.H. Ebstein-Barr Virus Associated Lymphoproliferative Disorders in Immunocompromised Individuals; George, F., Vande Woude, G.K., Eds.; Academic Press Inc.: Cambridge, MA, USA, 1991; Volume 57, p. 455. [Google Scholar]
- Shindiapina, P.; Ahmed, E.H.; Mozhenkova, A.; Abebe, T.; Baiocchi, R.A. Immunology of EBV-Related Lymphoproliferative Disease in HIV-Positive Individuals. Front. Oncol. 2020, 10, 1723. [Google Scholar] [CrossRef]
- Atallah-Yunes, S.A.; Murphy, D.J.; Noy, A. HIV-associated Burkitt lymphoma. Lancet Haematol. 2020, 7, e594–e600. [Google Scholar] [CrossRef] [PubMed]
- Orem, J.; Mbidde, E.K.; Lambert, B.; de Sanjose, S.; Weiderpass, E. Burkitt’s lymphoma in Africa, a review of the epidemiology and etiology. Afr. Health Sci. 2007, 7, 166–175. [Google Scholar] [PubMed]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2017; Volume 2. [Google Scholar]
- Marques-Piubelli, M.L.; Salas, Y.I.; Pachas, C.; Becker-Hecker, R.; Vega, F.; Miranda, R.N. Epstein-Barr virus-associated B-cell lymphoproliferative disorders and lymphomas: A review. Pathology 2020, 52, 40–52. [Google Scholar] [CrossRef]
- Carbone, A.; Volpi, C.C.; Gualeni, A.V.; Gloghini, A. Epstein-Barr virus associated lymphomas in people with HIV. Curr. Opin. HIV AIDS 2017, 12, 39–46. [Google Scholar] [CrossRef]
- Thacker, N.; Abla, O. Epidemiology of non-hodgkin lymphomas in childhood and adolescence. In Non-Hodgkin’s Lymphoma in Childhood and Adolescence; Springer: Berlin/Heidelberg, Germany, 2019; pp. 15–22. [Google Scholar]
- Noy, A. HIV Lymphoma and Burkitts Lymphoma. Cancer J. 2020, 26, 260–268. [Google Scholar] [CrossRef]
- Collaboration of Observational HIV Epidemiological Research Europe (COHERE) Study Group; Bohlius, J.; Schmidlin, K.; Costagliola, D.; Fatkenheuer, G.; May, M.; Caro-Murillo, A.M.; Mocroft, A.; Bonnet, F.; Clifford, G.; et al. Incidence and risk factors of HIV-related non-Hodgkin’s lymphoma in the era of combination antiretroviral therapy: A European multicohort study. Antivir. Ther. 2009, 14, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.; Xu, L.; Abrams, D.; Leyden, W.; Horberg, M.; Towner, W.; Klein, D.; Tang, B.; Silverberg, M.J.A. Survival of non-Hodgkin lymphoma patients with and without HIV infection in the era of combined antiretroviral therapy. AIDS 2010, 24, 1765–1770. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, F.; Jouvencel, A.C.; Parrens, M.; Leon, M.J.; Cotto, E.; Garrigue, I.; Morlat, P.; Beylot, J.; Fleury, H.; Lafon, M.E. A longitudinal and prospective study of Epstein-Barr virus load in AIDS-related non-Hodgkin lymphoma. J. Clin. Virol. Off. Publ. Pan Am. Soc. Clin. Virol. 2006, 36, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Noy, A. HIV-associated lymphoma including Burkitt in the general population. Cancer J. 2020, 26, 260. [Google Scholar] [CrossRef] [PubMed]
- Meister, A.; Hentrich, M.; Wyen, C.; Hubel, K. Malignant lymphoma in the HIV-positive patient. Eur. J. Haematol. 2018, 101, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Xu, Y.; Xin, P.; Zheng, Y.; Zhu, X. Role and mechanism of PTEN in Burkitt’s lymphoma. Oncol. Rep. 2020, 43, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.I. Vaccine Development for Epstein-Barr Virus. Adv. Exp. Med. Biol. 2018, 1045, 477–493. [Google Scholar] [CrossRef] [PubMed]
- Houghtelin, A.; Bollard, C.M. Virus-Specific T Cells for the Immunocompromised Patient. Front. Immunol. 2017, 8, 1272. [Google Scholar] [CrossRef]
- Han, C.; Singh, R.; Kim, S.-H.; Choi, B.K.; Kwon, B.S. Suppression of EBV-Induced LCLs Using CAR T Cells Redirected against HLA-DR; American Society of Clinical Oncology: Alexandria, VA, USA, 2017. [Google Scholar]
- Shen, R.R.; Pham, C.D.; Wu, M.M.; Foubert, P.; Spindler, T.J.; Munson, D.J.; Aftab, B.T. Functional demonstration of CD19 chimeric antigen receptor (CAR) engineered Epstein-Barr virus (EBV) specific T cells: An off-the-shelf, allogeneic CAR T-cell immunotherapy platform. Cell 2019, 45, 2. [Google Scholar]
- Broussard, G.; Damania, B. Regulation of KSHV Latency and Lytic Reactivation. Viruses 2020, 12, 1034. [Google Scholar] [CrossRef]
- Aneja, K.K.; Yuan, Y. Reactivation and Lytic Replication of Kaposi’s Sarcoma-Associated Herpesvirus: An Update. Front. Microbiol. 2017, 8, 613. [Google Scholar] [CrossRef] [PubMed]
- Reid, E.G.; Looney, D.; Maldarelli, F.; Noy, A.; Henry, D.; Aboulafia, D.; Ramos, J.C.; Sparano, J.; Ambinder, R.F.; Lee, J.; et al. Safety and efficacy of an oncolytic viral strategy using bortezomib with ICE/R in relapsed/refractory HIV-positive lymphomas. Blood Adv. 2018, 2, 3618–3626. [Google Scholar] [CrossRef] [PubMed]
- Hartlage, A.S.; Liu, T.; Patton, J.T.; Garman, S.L.; Zhang, X.; Kurt, H.; Lozanski, G.; Lustberg, M.E.; Caligiuri, M.A.; Baiocchi, R.A. The Epstein–Barr Virus lytic protein BZLF1 as a candidate target antigen for vaccine development. Cancer Immunol. Res. 2015, 3, 787–794. [Google Scholar] [CrossRef]
- Burns, D.M.; Chaganti, S. Epstein-Barr virus-associated lymphoproliferative disorders in immunosuppressed patients. Hum. Pathol. 2021, 38, 1293–1304. [Google Scholar] [CrossRef]
- Ho, J.W.Y.; Li, L.; Wong, K.Y.; Srivastava, G.; Tao, Q. Comprehensive Profiling of EBV Gene Expression and Promoter Methylation Reveals Latency II Viral Infection and Sporadic Abortive Lytic Activation in Peripheral T-Cell Lymphomas. Viruses 2023, 15, 423. [Google Scholar] [CrossRef]
- Saha, A.; Robertson, E.S. Mechanisms of B-Cell Oncogenesis Induced by Epstein-Barr Virus. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.L.; Lakomy, D.S.; Chiao, E.Y.; Strother, R.M.; Wirth, M.; Cesarman, E.; Borok, M.; Busakhala, N.; Chibwesha, C.J.; Chinula, L.; et al. Clinical Trials for Treatment and Prevention of HIV-Associated Malignancies in Sub-Saharan Africa: Building Capacity and Overcoming Barriers. JCO Glob. Oncol. 2020, 6, 1134–1146. [Google Scholar] [CrossRef]
- Chen, W.C.; Singh, E.; Muchengeti, M.; Bradshaw, D.; Mathew, C.G.; Babb de Villiers, C.; Lewis, C.M.; Waterboer, T.; Newton, R.; Sitas, F. Johannesburg Cancer Study (JCS): Contribution to knowledge and opportunities arising from 20 years of data collection in an African setting. Cancer Epidemiol. 2020, 65, 101701. [Google Scholar] [CrossRef]
- Dlamini, Z.; Mbele, M.; Makhafola, T.J.; Hull, R.; Marima, R. HIV-Associated Cancer Biomarkers: A Requirement for Early Diagnosis. Int. J. Mol. Sci. 2021, 22, 8127. [Google Scholar] [CrossRef]
- Montano, M.A.; Mtisi, T.; Ndlovu, N.; Borok, M.; Bula, A.; Joffe, M.; Ignacio, R.B.; Chagomerana, M.B. Characterizing HIV status documentation among cancer patients at regional cancer centers in Malawi, Zimbabwe, and South Africa. medRxiv 2023. [Google Scholar] [CrossRef]
- Rapiti, N.; Abdelatif, N.; Rapiti, A.; Moosa, M.Y. Patient characteristics and outcome of CD20-positive HIV-associated lymphoma: A single-center KwaZulu-Natal, South African hospital 12-year retrospective review. J. Egypt. Natl. Canc. Inst. 2022, 34, 32. [Google Scholar] [CrossRef] [PubMed]
- Vogt, S.L.; Patel, M.; Lakha, A.; Philip, V.; Omar, T.; Ashmore, P.; Pather, S.; Haley, L.M.; Zheng, G.; Stone, J.; et al. Feasibility of Cell-Free DNA Collection and Clonal Immunoglobulin Sequencing in South African Patients with HIV-Associated Lymphoma. JCO Glob. Oncol. 2021, 7, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Horner, M.J.; Shiels, M.S.; Pfeiffer, R.M.; Engels, E.A. Deaths Attributable to Cancer in the US Human Immunodeficiency Virus Population During 2001–2015. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2021, 72, e224–e231. [Google Scholar] [CrossRef] [PubMed]
- Cobucci, R.N.; Lima, P.H.; de Souza, P.C.; Costa, V.V.; Cornetta Mda, C.; Fernandes, J.V.; Goncalves, A.K. Assessing the impact of HAART on the incidence of defining and non-defining AIDS cancers among patients with HIV/AIDS: A systematic review. J. Infect. Public Health 2015, 8, 1–10. [Google Scholar] [CrossRef]
- Spano, J.-P.; Costagliola, D.; Katlama, C.; Mounier, N.; Oksenhendler, E.; Khayat, D. AIDS-related malignancies: State of the art and therapeutic challenges. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 4834–4842. [Google Scholar] [CrossRef] [PubMed]
- Engels, E. Non-AIDS-defining malignancies in HIV-infected persons: Etiologic puzzles, epidemiologic perils, prevention opportunities. AIDS 2009, 23, 875–885. [Google Scholar] [CrossRef]
- Ceccarelli, M.; Venanzi Rullo, E.; Marino, M.A.; d’Aleo, F.; Pellicano, G.F.; D’Andrea, F.; Marino, A.; Cacopardo, B.; Celesia, B.M.; La Rocca, G.; et al. Non-AIDS defining cancers: A comprehensive update on diagnosis and management. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 3849–3875. [Google Scholar] [CrossRef]
- Chiao, E.Y.; Coghill, A.; Kizub, D.; Fink, V.; Ndlovu, N.; Mazul, A.; Sigel, K. The effect of non-AIDS-defining cancers on people living with HIV. Lancet Oncol. 2021, 22, e240–e253. [Google Scholar] [CrossRef]
- Corrigan, K.L.; Wall, K.C.; Bartlett, J.A.; Suneja, G. Cancer disparities in people with HIV: A systematic review of screening for non-AIDS-defining malignancies. Cancer 2019, 125, 843–853. [Google Scholar] [CrossRef]
- Franzetti, M.; Ricci, E.; Bonfanti, P. The Pattern of Non-AIDS-defining Cancers in the HIV Population: Epidemiology, Risk Factors and Prognosis. A Review. Curr. HIV Res. 2019, 17, 1–12. [Google Scholar] [CrossRef]
- Suneja, G.; Boyer, M.; Yehia, B.R.; Shiels, M.S.; Engels, E.A.; Bekelman, J.E.; Long, J.A. Cancer Treatment in Patients with HIV Infection and Non-AIDS-Defining Cancers: A Survey of US Oncologists. J. Oncol. Pr. 2015, 11, e380–e387. [Google Scholar] [CrossRef]
- Reid, E.; Suneja, G.; Ambinder, R.F.; Ard, K.; Baiocchi, R.; Barta, S.K.; Carchman, E.; Cohen, A.; Gupta, N.; Johung, K.L.; et al. Cancer in People Living with HIV, Version 1.2018, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2018, 16, 986–1017. [Google Scholar] [CrossRef] [PubMed]
- Sigel, K.; Dubrow, R.; Silverberg, M.; Crothers, K.; Braithwaite, S.; Justice, A. Cancer screening in patients infected with HIV. Curr. HIV/AIDS Rep. 2011, 8, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Baniyash, M. Chronic inflammation, immunosuppression and cancer: New insights and outlook. Semin. Cancer Biol. 2006, 16, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Breen, E.; Hussain, S.; Magpantay, L.; Jacobson, L.; Detels, R.; Rabkin, C.; Kaslow, R.; Variakojis, D.; Bream, J.; Rinaldo, C.; et al. B-cell stimulatory cytokines and markers of immune activation are elevated several years prior to the diagnosis of systemic AIDS-associated non-Hodgkin B-cell lymphoma. Cancer Epidemiol. Biomark. Prev. 2011, 20, 1303–1314. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Valuev-Elliston, V.T.; Ivanova, O.N.; Kochetkov, S.N.; Starodubova, E.S.; Bartosch, B.; Isaguliants, M.G. Oxidative Stress during HIV Infection: Mechanisms and Consequences. Oxid. Med. Cell. Longev. 2016, 2016, 8910396. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, O.M.; Mohammed, M.T. Oxidative stress: The role of reactive oxygen species (ROS) and antioxidants in human diseases. Plant Arch. 2020, 20, 4089–4095. [Google Scholar]
- Zahra, K.F.; Lefter, R.; Ali, A.; Abdellah, E.C.; Trus, C.; Ciobica, A.; Timofte, D. The Involvement of the Oxidative Stress Status in Cancer Pathology: A Double View on the Role of the Antioxidants. Oxid. Med. Cell. Longev. 2021, 2021, 9965916. [Google Scholar] [CrossRef]
- Calmels, S.; Hainaut, P.; Ohshima, H. Nitric oxide induces conformational and functional modifications of wild-type p53 tumor suppressor protein. Cancer Res. 1997, 57, 3365–3369. [Google Scholar]
- Ohshima, H.; Tatemichi, M.; Sawa, T. Chemical basis of inflammation-induced carcinogenesis. Arch. Biochem. Biophys. 2003, 417, 3–11. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Baby, D.; Rajguru, J.P.; Patil, P.B.; Thakkannavar, S.S.; Pujari, V.B. Inflammation and cancer. Ann. Afr. Med. 2019, 18, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Sara De, B.; Marcello, P.; Milena, N.; Lara, G.; Linda, B.; Serena, M.; Cristina, M.; Andrea, C. HIV-1 Infection and the Aging of the Immune System: Facts, Similarities and Perspectives. J. Exp. Clin. Med. 2011, 3, 143–150. [Google Scholar] [CrossRef]
- Babu, H.; Ambikan, A.T.; Gabriel, E.E.; Svensson Akusjarvi, S.; Palaniappan, A.N.; Sundaraj, V.; Mupanni, N.R.; Sperk, M.; Cheedarla, N.; Sridhar, R.; et al. Systemic Inflammation and the Increased Risk of Inflamm-Aging and Age-Associated Diseases in People Living with HIV on Long Term Suppressive Antiretroviral Therapy. Front. Immunol. 2019, 10, 1965. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S. HIV infection, inflammation, immunosenescence, and aging. Annu. Rev. Med. 2011, 62, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.W.; Swann, J.B.; Koebel, C.M.; Schreiber, R.D.; Smyth, M.J. Immune-mediated dormancy: An equilibrium with cancer. J. Leukoc. Biol. 2008, 84, 988–993. [Google Scholar] [CrossRef] [PubMed]
- Vajdic, C.M.; van Leeuwen, M.T. Cancer incidence and risk factors after solid organ transplantation. Int. J. Cancer 2009, 125, 1747–1754. [Google Scholar] [CrossRef] [PubMed]
- Killebrew, D.; Shiramizu, B. Pathogenesis of HIV-associated non-Hodgkin lymphoma. Curr. HIV Res. 2004, 2, 215–221. [Google Scholar] [CrossRef]
- Linden, N.; Jones, R.B. Potential multi-modal effects of provirus integration on HIV-1 persistence: Lessons from other viruses. Trends Immunol. 2022, 43, 617–629. [Google Scholar] [CrossRef]
- Shiramizu, B.; Herndier, B.; McGrath, M. Identification of a common clonal human immunodeficiency virus integration site in human immunodeficiency virus-associated lymphomas. Cancer Res. 1994, 54, 2069–2072. [Google Scholar]
- Mack, K.D. HIV insertions within and proximal to host cell genes are a common finding in tissues containing high levels of HIV DNA and macrophage associated p24 antigen expression. J. Acquir. Immune Defic. Syndr. 2003, 33, 308–320. [Google Scholar] [CrossRef] [PubMed]
- Batzer, M.A.; Deininger, P.L. Alu repeats and human genomic diversity. Nat. Rev. Genet. 2002, 3, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Arendt, C.; Littman, D. HIV: Master of the host cell. Genome Biol. 2001, 2, 1. [Google Scholar] [CrossRef] [PubMed]
- Isaguliants, M.; Bayurova, E.; Avdoshina, D.; Kondrashova, A.; Chiodi, F.; Palefsky, J.M. Oncogenic Effects of HIV-1 Proteins, Mechanisms Behind. Cancers 2021, 13, 305. [Google Scholar] [CrossRef] [PubMed]
- Ameya, G.; Birri, D.J. The molecular mechanisms of virus-induced human cancers. Microb. Pathog. 2023, 183, 106292. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Li, W.; Xia, L.; Zou, R.; Chen, X.; Zou, W. Malignancies in people living with HIV. AIDS Rev. 2022, 24, 69–78. [Google Scholar] [CrossRef]
- Clark, E.; Santiago, F.; Deng, L.; Chong, S.; de La Fuente, C.; Wang, L.; Fu, P.; Stein, D.; Denny, T.; Lanka, V.; et al. Loss of G(1)/S checkpoint in human immunodeficiency virus type 1-infected cells is associated with a lack of cyclin-dependent kinase inhibitor p21/Waf1. J. Virol. 2000, 74, 5040–5052. [Google Scholar] [CrossRef]
- Fitzpatrick, M.; Brooks, J.T.; Kaplan, J.E. Epidemiology of HIV-Associated Lung Disease in the United States. Semin. Respir. Crit. Care Med. 2016, 37, 181–198. [Google Scholar] [CrossRef]
- Hessol, N.A.; Martinez-Maza, O.; Levine, A.M.; Morris, A.; Margolick, J.B.; Cohen, M.H.; Jacobson, L.P.; Seaberg, E.C. Lung cancer incidence and survival among HIV-infected and uninfected women and men. AIDS 2015, 29, 1183–1193. [Google Scholar] [CrossRef]
- Marecki, J.C.; Cool, C.D.; Parr, J.E.; Beckey, V.E.; Luciw, P.A.; Tarantal, A.F.; Carville, A.; Shannon, R.P.; Cota-Gomez, A.; Tuder, R.M.; et al. HIV-1 Nef is associated with complex pulmonary vascular lesions in SHIV-nef-infected macaques. Am. J. Respir. Crit. Care Med. 2006, 174, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Landi, A.; Iannucci, V.; Nuffel, A.V.; Meuwissen, P.; Verhasselt, B. One protein to rule them all: Modulation of cell surface receptors and molecules by HIV Nef. Curr. HIV Res. 2011, 9, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Guo, Y.; Yao, S.; Yan, Q.; Xue, M.; Hao, T.; Zhou, F.; Zhu, J.; Qin, D.; Lu, C. Synergy between Kaposi’s sarcoma-associated herpesvirus (KSHV) vIL-6 and HIV-1 Nef protein in promotion of angiogenesis and oncogenesis: Role of the AKT signaling pathway. Oncogene 2014, 33, 1986–1996. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Witte, V.; Laffert, B.; Blume, K.; Stromer, E.; Trapp, S.; d’Aloja, P.; Schurmann, A.; Baur, A.S. HIV-1 Nef associated PAK and PI3-kinases stimulate Akt-independent Bad-phosphorylation to induce anti-apoptotic signals. Nat. Med. 2001, 7, 1217–1224. [Google Scholar] [CrossRef] [PubMed]
- Geleziunas, R.; Xu, W.; Takeda, K.; Ichijo, H.; Greene, W.C. HIV-1 Nef inhibits ASK1-dependent death signalling providing a potential mechanism for protecting the infected host cell. Nature 2001, 410, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Santerre, M.; Chatila, W.; Wang, Y.; Mukerjee, R.; Sawaya, B.E. HIV-1 Nef promotes cell proliferation and microRNA dysregulation in lung cells. Cell Cycle 2019, 18, 130–142. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Omar, A.; Marques, N.; Crawford, N. Cancer and HIV: The Molecular Mechanisms of the Deadly Duo. Cancers 2024, 16, 546. https://doi.org/10.3390/cancers16030546
Omar A, Marques N, Crawford N. Cancer and HIV: The Molecular Mechanisms of the Deadly Duo. Cancers. 2024; 16(3):546. https://doi.org/10.3390/cancers16030546
Chicago/Turabian StyleOmar, Aadilah, Natasia Marques, and Nicole Crawford. 2024. "Cancer and HIV: The Molecular Mechanisms of the Deadly Duo" Cancers 16, no. 3: 546. https://doi.org/10.3390/cancers16030546
APA StyleOmar, A., Marques, N., & Crawford, N. (2024). Cancer and HIV: The Molecular Mechanisms of the Deadly Duo. Cancers, 16(3), 546. https://doi.org/10.3390/cancers16030546