
Antibody-Based Immunotherapies for the Treatment of Hematologic Malignancies


Simple Summary
Abstract
1. Introduction
2. Immunotherapeutics in Hematology Malignancies
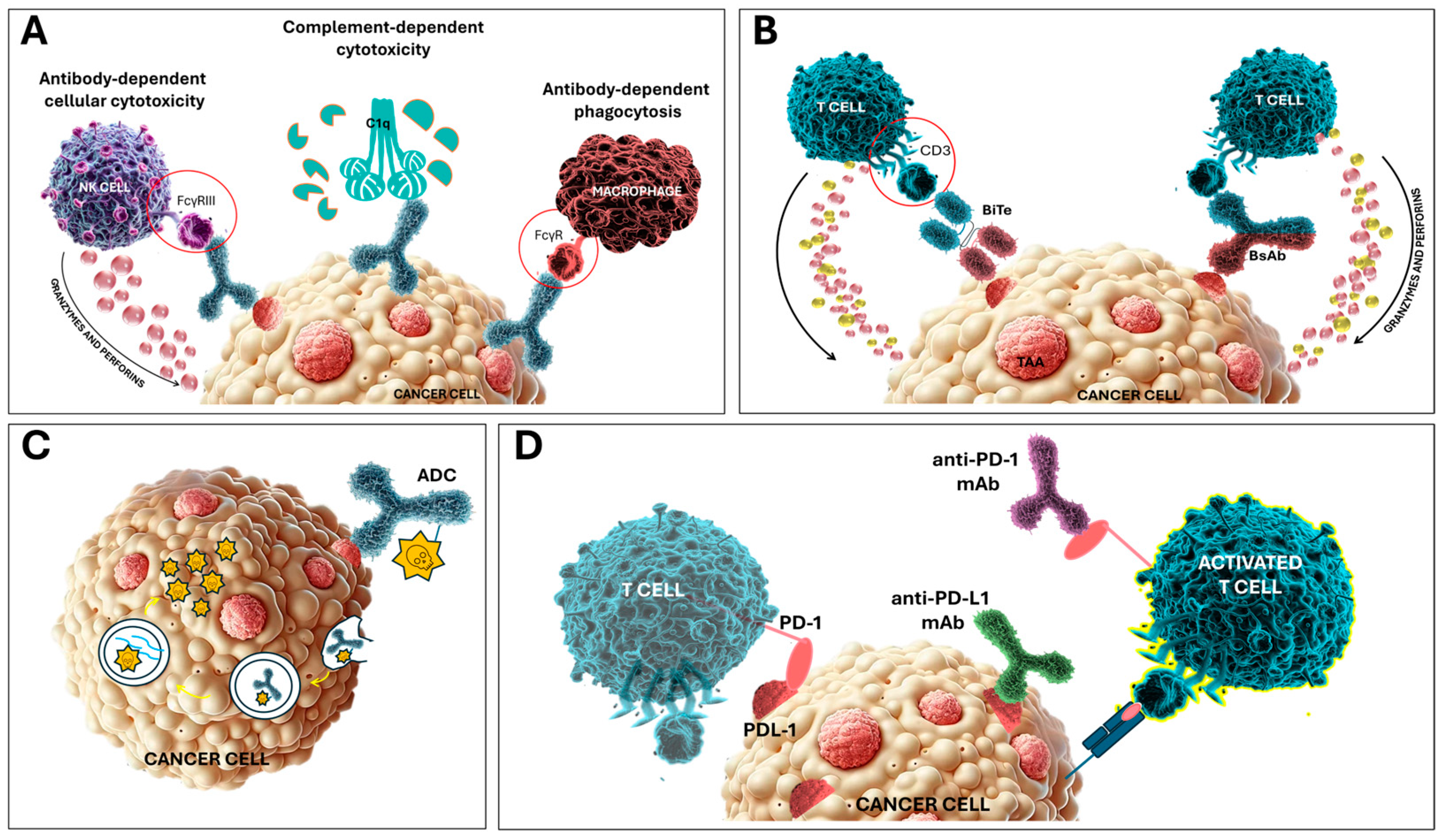
2.1. Monoclonal Antibodies
2.2. Bispecific Antibodies and T-Cell Engagers
2.3. Antibody–Drug Conjugates
2.4. Immune Checkpoint Inhibitors
3. The Role of the Tumor Microenvironment in Regulating Responses to Antibody-Based Therapies in Hematological Malignancies
4. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Couzin-Frankel, J. Cancer Immunotherapy. Science 2013, 342, 1432–1433. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.-W.; Chang, J.W.-C. Immune Checkpoint Inhibitors Win the 2018 Nobel Prize. Biomed. J. 2019, 42, 299–306. [Google Scholar] [CrossRef]
- Coley, W.B. The Treatment of Malignant Tumors by Repeated Inoculations of Erysipelas. With a Report of Ten Original Cases. 1893. Clin. Orthop. Relat. Res. 1991, 3–11. [Google Scholar]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The Three Es of Cancer Immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef] [PubMed]
- Weiden, P.L.; Flournoy, N.; Thomas, E.D.; Prentice, R.; Fefer, A.; Buckner, C.D.; Storb, R. Antileukemic Effect of Graft-versus-Host Disease in Human Recipients of Allogeneic-Marrow Grafts. N. Engl. J. Med. 1979, 300, 1068–1073. [Google Scholar] [CrossRef] [PubMed]
- Maurer, K.; Antin, J.H. The Graft versus Leukemia Effect: Donor Lymphocyte Infusions and Cellular Therapy. Front. Immunol. 2024, 15, 1328858. [Google Scholar] [CrossRef]
- Strohl, W.R. Current Progress in Innovative Engineered Antibodies. Protein Cell 2018, 9, 86–120. [Google Scholar] [CrossRef] [PubMed]
- Sifniotis, V.; Cruz, E.; Eroglu, B.; Kayser, V. Current Advancements in Addressing Key Challenges of Therapeutic Antibody Design, Manufacture, and Formulation. Antibodies 2019, 8, 36. [Google Scholar] [CrossRef]
- Kaplon, H.; Crescioli, S.; Chenoweth, A.; Visweswaraiah, J.; Reichert, J.M. Antibodies to Watch in 2023. MAbs 2023, 15, 2153410. [Google Scholar] [CrossRef] [PubMed]
- Crescioli, S.; Kaplon, H.; Chenoweth, A.; Wang, L.; Visweswaraiah, J.; Reichert, J.M. Antibodies to Watch in 2024. MAbs 2024, 16, 2297450. [Google Scholar] [CrossRef]
- Grilo, A.L.; Mantalaris, A. The Increasingly Human and Profitable Monoclonal Antibody Market. Trends Biotechnol. 2019, 37, 9–16. [Google Scholar] [CrossRef]
- Kinch, M.S.; Kraft, Z.; Schwartz, T. Monoclonal Antibodies: Trends in Therapeutic Success and Commercial Focus. Drug Discov. Today 2023, 28, 103415. [Google Scholar] [CrossRef] [PubMed]
- Köhler, G.; Milstein, C. Continuous Cultures of Fused Cells Secreting Antibody of Predefined Specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Harding, F.A.; Stickler, M.M.; Razo, J.; DuBridge, R.B. The Immunogenicity of Humanized and Fully Human Antibodies: Residual Immunogenicity Resides in the CDR Regions. MAbs 2010, 2, 256–265. [Google Scholar] [CrossRef]
- Weiner, G.J. Building Better Monoclonal Antibody-Based Therapeutics. Nat. Rev. Cancer 2015, 15, 361–370. [Google Scholar] [CrossRef]
- Almagro, J.C.; Daniels-Wells, T.R.; Perez-Tapia, S.M.; Penichet, M.L. Progress and Challenges in the Design and Clinical Development of Antibodies for Cancer Therapy. Front. Immunol. 2018, 8, 1751. [Google Scholar] [CrossRef]
- Almagro, J.C.; Pedraza-Escalona, M.; Arrieta, H.I.; Pérez-Tapia, S.M. Phage Display Libraries for Antibody Therapeutic Discovery and Development. Antibodies 2019, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Najfeld, V. Conventional and Molecular Cytogenomic Basis of Hematologic Malignancies. In Hematology: Basic Principles and Practice; Elsevier: Amsterdam, The Netherlands, 2018; pp. 774–848. [Google Scholar] [CrossRef]
- Hartmann, L.; Metzeler, K.H. Clonal Hematopoiesis and Preleukemia-Genetics, Biology, and Clinical Implications. Genes Chromosomes Cancer 2019, 58, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Niroula, A.; Sekar, A.; Murakami, M.A.; Trinder, M.; Agrawal, M.; Wong, W.J.; Bick, A.G.; Uddin, M.M.; Gibson, C.J.; Griffin, G.K.; et al. Distinction of Lymphoid and Myeloid Clonal Hematopoiesis. Nat. Med. 2021, 27, 1921–1927. [Google Scholar] [CrossRef]
- Filipek-Gorzała, J.; Kwiecińska, P.; Szade, A.; Szade, K. The Dark Side of Stemness—The Role of Hematopoietic Stem Cells in Development of Blood Malignancies. Front. Oncol. 2024, 14, 1308709. [Google Scholar] [CrossRef]
- Hu, D.; Shilatifard, A. Epigenetics of Hematopoiesis and Hematological Malignancies. Genes Dev. 2016, 30, 2021–2041. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.; Xiao, W.; Abdel-Wahab, O. Diagnosis and Classification of Hematologic Malignancies on the Basis of Genetics. Blood 2017, 130, 410–423. [Google Scholar] [CrossRef]
- Jaffe, E.S.; Harris, N.L.; Stein, H.; Vardiman, J.W. (Eds.) Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues; Iarc Who Classification of Tumours Series; IARC Press: Lyon, France, 2001; ISBN 9789283224112. [Google Scholar]
- Salama, M.E.; Hoffman, R. Progress in the Classification of Hematopoietic and Lymphoid Neoplasms: Clinical Implications. In Hematology: Basic Principles and Practice; Elsevier: Amsterdam, The Netherlands, 2017; pp. 763–773. ISBN 9780323357623. [Google Scholar]
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global Cancer Statistics 2022: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Pulte, D.; Jansen, L.; Brenner, H. Changes in Long Term Survival after Diagnosis with Common Hematologic Malignancies in the Early 21st Century. Blood Cancer J. 2020, 10, 56. [Google Scholar] [CrossRef]
- Maloney, D.G.; Grillo-López, A.J.; White, C.A.; Bodkin, D.; Schilder, R.J.; Neidhart, J.A.; Janakiraman, N.; Foon, K.A.; Liles, T.-M.; Dallaire, B.K.; et al. IDEC-C2B8 (Rituximab) Anti-CD20 Monoclonal Antibody Therapy in Patients With Relapsed Low-Grade Non-Hodgkin’s Lymphoma. Blood 1997, 90, 2188–2195. [Google Scholar] [CrossRef]
- Weiner, G.J. Rituximab: Mechanism of Action. Semin. Hematol. 2010, 47, 115–123. [Google Scholar] [CrossRef]
- Nadler, L.M.; Ritz, J.; Hardy, R.; Pesando, J.M.; Schlossman, S.F.; Stashenko, P. A Unique Cell Surface Antigen Identifying Lymphoid Malignancies of B Cell Origin. J. Clin. Investig. 1981, 67, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Johnson, P.; Glennie, M. The Mechanisms of Action of Rituximab in the Elimination of Tumor Cells. Semin. Oncol. 2003, 30, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.R.; Cohen, P.L. Living Life without B Cells: Is Repeated B-Cell Depletion a Safe and Effective Long-Term Treatment Plan for Rheumatoid Arthritis? Int. J. Clin. Rheumtol. 2012, 7, 159. [Google Scholar] [CrossRef]
- Athni, T.S.; Barmettler, S. Hypogammaglobulinemia, Late-Onset Neutropenia, and Infections Following Rituximab. Ann. Allergy. Asthma Immunol. 2023, 130, 699. [Google Scholar] [CrossRef]
- Salles, G.; Barrett, M.; Foà, R.; Maurer, J.; O’Brien, S.; Valente, N.; Wenger, M.; Maloney, D.G. Rituximab in B-Cell Hematologic Malignancies: A Review of 20 Years of Clinical Experience. Adv. Ther. 2017, 34, 2232–2273. [Google Scholar] [CrossRef] [PubMed]
- Pierpont, T.M.; Limper, C.B.; Richards, K.L. Past, Present, and Future of Rituximab-The World’s First Oncology Monoclonal Antibody Therapy. Front. Oncol. 2018, 8, 163. [Google Scholar] [CrossRef] [PubMed]
- Al Hamed, R.; Bazarbachi, A.H.; Mohty, M. Epstein-Barr Virus-Related Post-Transplant Lymphoproliferative Disease (EBV-PTLD) in the Setting of Allogeneic Stem Cell Transplantation: A Comprehensive Review from Pathogenesis to Forthcoming Treatment Modalities. Bone Marrow Transplant. 2020, 55, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Marjańska, A.; Pogorzała, M.; Dziedzic, M.; Czyżewski, K.; Richert-Przygońska, M.; Dębski, R.; Bogiel, T. Impact of prophylaxis with rituximab on EBV-related complications after allogeneic hematopoietic cell transplantation in children. Front. Immunol. 2024, 15, 1427637. [Google Scholar] [CrossRef]
- Wierda, W.G.; Kipps, T.J.; Mayer, J.; Stilgenbauer, S.; Williams, C.D.; Hellmann, A.; Robak, T.; Furman, R.R.; Hillmen, P.; Trneny, M.; et al. Ofatumumab as Single-Agent CD20 Immunotherapy in Fludarabine-Refractory Chronic Lymphocytic Leukemia. J. Clin. Oncol. 2010, 28, 1749–1755. [Google Scholar] [CrossRef] [PubMed]
- Hillmen, P.; Robak, T.; Janssens, A.; Babu, K.G.; Kloczko, J.; Grosicki, S.; Doubek, M.; Panagiotidis, P.; Kimby, E.; Schuh, A.; et al. Chlorambucil plus Ofatumumab versus Chlorambucil Alone in Previously Untreated Patients with Chronic Lymphocytic Leukaemia (COMPLEMENT 1): A Randomised, Multicentre, Open-Label Phase 3 Trial. Lancet 2015, 385, 1873–1883. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Blair, H.A. Ofatumumab: A Review in Relapsing Forms of Multiple Sclerosis. Drugs 2022, 82, 55–62. [Google Scholar] [CrossRef]
- Goede, V.; Fischer, K.; Busch, R.; Engelke, A.; Eichhorst, B.; Wendtner, C.M.; Chagorova, T.; de la Serna, J.; Dilhuydy, M.-S.; Illmer, T.; et al. Obinutuzumab plus Chlorambucil in Patients with CLL and Coexisting Conditions. N. Engl. J. Med. 2014, 370, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Sehn, L.H.; Chua, N.; Mayer, J.; Dueck, G.; Trněný, M.; Bouabdallah, K.; Fowler, N.; Delwail, V.; Press, O.; Salles, G.; et al. Obinutuzumab plus Bendamustine versus Bendamustine Monotherapy in Patients with Rituximab-Refractory Indolent Non-Hodgkin Lymphoma (GADOLIN): A Randomised, Controlled, Open-Label, Multicentre, Phase 3 Trial. Lancet. Oncol. 2016, 17, 1081–1093. [Google Scholar] [CrossRef] [PubMed]
- Mössner, E.; Brünker, P.; Moser, S.; Püntener, U.; Schmidt, C.; Herter, S.; Grau, R.; Gerdes, C.; Nopora, A.; Van Puijenbroek, E.; et al. Increasing the Efficacy of CD20 Antibody Therapy through the Engineering of a New Type II Anti-CD20 Antibody with Enhanced Direct and Immune Effector Cell–Mediated B-Cell Cytotoxicity. Blood 2010, 115, 4393. [Google Scholar] [CrossRef]
- Townsend, W.; Hiddemann, W.; Buske, C.; Cartron, G.; Cunningham, D.; Dyer, M.J.S.; Gribben, J.G.; Phillips, E.H.; Dreyling, M.; Seymour, J.F.; et al. Obinutuzumab Versus Rituximab Immunochemotherapy in Previously Untreated INHL: Final Results From the GALLIUM Study. HemaSphere 2023, 7, E919. [Google Scholar] [CrossRef] [PubMed]
- Sehn, L.H.; Martelli, M.; Trněný, M.; Liu, W.; Bolen, C.R.; Knapp, A.; Sahin, D.; Sellam, G.; Vitolo, U.; Sehn, L.H. A Randomized, Open-Label, Phase III Study of Obinutuzumab or Rituximab plus CHOP in Patients with Previously Untreated Diffuse Large B-Cell Lymphoma: Final Analysis of GOYA. J. Hematol. Oncol. 2020, 13, 71. [Google Scholar] [CrossRef] [PubMed]
- Le Gouill, S.; Ghesquières, H.; Oberic, L.; Morschhauser, F.; Tilly, H.; Ribrag, V.; Lamy, T.; Thieblemont, C.; Maisonneuve, H.; Gressin, R.; et al. Obinutuzumab vs Rituximab for Advanced DLBCL: A PET-Guided and Randomized Phase 3 Study by LYSA. Blood 2021, 137, 2307–2320. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.; Kater, A.P.; Sharman, J.P.; Stilgenbauer, S.; Vitolo, U.; Klein, C.; Parreira, J.; Salles, G. Obinutuzumab in the Treatment of B-Cell Malignancies: A Comprehensive Review. Futur. Oncol. 2022, 18, 2943–2966. [Google Scholar] [CrossRef] [PubMed]
- Sarkozy, C.; Callanan, M.; Thieblemont, C.; Obéric, L.; Burroni, B.; Bouabdallah, K.; Damaj, G.; Tessoulin, B.; Ribrag, V.; Houot, R.; et al. Obinutuzumab vs Rituximab for Transplant-Eligible Patients with Mantle Cell Lymphoma. Blood 2024, 144, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Jurczak, W.; Długosz Danecka, M.; Buske, C. Rituximab Biosimilars for Lymphoma in Europe. Expert Opin. Biol. Ther. 2019, 19, 1045–1056. [Google Scholar] [CrossRef]
- Olejarz, W.; Basak, G. Emerging Therapeutic Targets and Drug Resistance Mechanisms in Immunotherapy of Hematological Malignancies. Cancers 2023, 15, 5765. [Google Scholar] [CrossRef]
- Lonial, S.; Dimopoulos, M.; Palumbo, A.; White, D.; Grosicki, S.; Spicka, I.; Walter-Croneck, A.; Moreau, P.; Mateos, M.-V.; Magen, H.; et al. Elotuzumab Therapy for Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2015, 373, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.C.; Kanate, A.S.; Craig, M.; Petros, W.P.; Hazlehurst, L.A. Emerging Combination Therapies for the Management of Multiple Myeloma: The Role of Elotuzumab. Cancer Manag. Res. 2017, 9, 307–314. [Google Scholar] [CrossRef]
- Costa, F.; Palma, B.D.; Giuliani, N. CD38 Expression by Myeloma Cells and Its Role in the Context of Bone Marrow Microenvironment: Modulation by Therapeutic Agents. Cells 2019, 8, 1632. [Google Scholar] [CrossRef]
- McKeage, K. Daratumumab: First Global Approval. Drugs 2016, 76, 275–281. [Google Scholar] [CrossRef]
- Dhillon, S. Isatuximab: First Approval. Drugs 2020, 80, 905–912. [Google Scholar] [CrossRef]
- Frampton, J.E. Isatuximab: A Review of Its Use in Multiple Myeloma. Target. Oncol. 2021, 16, 675–686. [Google Scholar] [CrossRef]
- Facon, T.; Dimopoulos, M.-A.; Leleu, X.P.; Beksac, M.; Pour, L.; Hájek, R.; Liu, Z.; Minarik, J.; Moreau, P.; Romejko-Jarosinska, J.; et al. Isatuximab, Bortezomib, Lenalidomide, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2024, 391. [Google Scholar] [CrossRef]
- Sarkozy, C.; Sehn, L.H. New Drugs for the Management of Relapsed or Refractory Diffuse Large B-Cell Lymphoma. Ann. Lymphoma 2019, 3, 10. [Google Scholar] [CrossRef]
- Hoy, S.M. Tafasitamab: First Approval. Drugs 2020, 80, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Salles, G.; Duell, J.; González Barca, E.; Tournilhac, O.; Jurczak, W.; Liberati, A.M.; Nagy, Z.; Obr, A.; Gaidano, G.; André, M.; et al. Tafasitamab plus Lenalidomide in Relapsed or Refractory Diffuse Large B-Cell Lymphoma (L-MIND): A Multicentre, Prospective, Single-Arm, Phase 2 Study. Lancet. Oncol. 2020, 21, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Duell, J.; Maddocks, K.J.; González-Barca, E.; Jurczak, W.; Liberati, A.M.; de Vos, S.; Nagy, Z.; Obr, A.; Gaidano, G.; Abrisqueta, P.; et al. Long-Term L-MIND Study Outcomes of Tafasitamab from the(MOR208) Phase II plus Lenalidomide in Patients with Relapsed or Refractory Diffuse Large B-Cell Lymphoma. Haematologica 2021, 106, 2417–2426. [Google Scholar] [CrossRef] [PubMed]
- Salles, G.; Długosz-Danecka, M.; Ghesquières, H.; Jurczak, W. Tafasitamab for the Treatment of Relapsed or Refractory Diffuse Large B-Cell Lymphoma. Expert Opin. Biol. Ther. 2021, 21, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Duell, J.; Westin, J. The future of immunotherapy for diffuse large B-cell lymphoma. Int. J. Cancer 2025, 156, 251–261. [Google Scholar] [CrossRef]
- Chames, P.; Baty, D. Bispecific Antibodies for Cancer Therapy: The Light at the End of the Tunnel? MAbs 2009, 1, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Del Bano, J.; Chames, P.; Baty, D.; Kerfelec, B. Taking up Cancer Immunotherapy Challenges: Bispecific Antibodies, the Path Forward? Antibodies 2015, 5, 1. [Google Scholar] [CrossRef]
- Yang, F.; Wen, W.; Qin, W. Bispecific Antibodies as a Development Platform for New Concepts and Treatment Strategies. Int. J. Mol. Sci. 2017, 18, 48. [Google Scholar] [CrossRef]
- Velasquez, M.P.; Bonifant, C.L.; Gottschalk, S. Redirecting T Cells to Hematological Malignancies with Bispecific Antibodies. Blood 2018, 131, 30–38. [Google Scholar] [CrossRef]
- Cech, P.; Skórka, K.; Dziki, L.; Giannopoulos, K. T-Cell Engagers—The Structure and Functional Principle and Application in Hematological Malignancies. Cancers 2024, 16, 1580. [Google Scholar] [CrossRef] [PubMed]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W.H.I. Bispecific Antibodies: A Mechanistic Review of the Pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef]
- Thakur, A.; Huang, M.; Lum, L.G. Bispecific Antibody Based Therapeutics: Strengths and Challenges. Blood Rev. 2018, 32, 339–347. [Google Scholar] [CrossRef]
- Stamova, S.; Koristka, S.; Keil, J.; Arndt, C.; Feldmann, A.; Michalk, I.; Bartsch, H.; Bippes, C.C.; Schmitz, M.; Cartellieri, M.; et al. Cancer Immunotherapy by Retargeting of Immune Effector Cells via Recombinant Bispecific Antibody Constructs. Antibodies 2012, 1, 172–198. [Google Scholar] [CrossRef]
- Blanco, B.; Domínguez-Alonso, C.; Alvarez-Vallina, L. Bispecific Immunomodulatory Antibodies for Cancer Immunotherapy. Clin. Cancer Res. 2021, 27, 5457–5464. [Google Scholar] [CrossRef] [PubMed]
- Huehls, A.M.; Coupet, T.A.; Sentman, C.L. Bispecific T-Cell Engagers for Cancer Immunotherapy. Immunol. Cell Biol. 2015, 93, 290–296. [Google Scholar] [CrossRef]
- Przepiorka, D.; Ko, C.W.; Deisseroth, A.; Yancey, C.L.; Candau-Chacon, R.; Chiu, H.J.; Gehrke, B.J.; Gomez-Broughton, C.; Kane, R.C.; Kirshner, S.; et al. FDA Approval: Blinatumomab. Clin. Cancer Res. 2015, 21, 4035–4039. [Google Scholar] [CrossRef]
- Portell, C.A.; Wenzell, C.M.; Advani, A.S. Clinical and Pharmacologic Aspects of Blinatumomab in the Treatment of B-Cell Acute Lymphoblastic Leukemia. Clin. Pharmacol. Adv. Appl. 2013, 5, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Carter, R.H.; Wang, Y.; Brooks, S. Role of CD19 Signal Transduction in B Cell Biology. Immunol. Res. 2002, 26, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Otero, D.C.; Anzelon, A.N.; Rickert, R.C. CD19 Function in Early and Late B Cell Development: I. Maintenance of Follicular and Marginal Zone B Cells Requires CD19-Dependent Survival Signals. J. Immunol. 2003, 170, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Eibel, H.; Kraus, H.; Sic, H.; Kienzler, A.K.; Rizzi, M. B Cell Biology: An Overview Topical Collection on Basic and Applied Science. Curr. Allergy Asthma Rep. 2014, 14, 434. [Google Scholar] [CrossRef] [PubMed]
- Hammer, O. CD19 as an Attractive Target for Antibody-Based Therapy. MAbs 2012, 4, 571. [Google Scholar] [CrossRef]
- Ali, S.; Moreau, A.; Melchiorri, D.; Camarero, J.; Josephson, F.; Olimpier, O.; Bergh, J.; Karres, D.; Tzogani, K.; Gisselbrecht, C.; et al. Blinatumomab for Acute Lymphoblastic Leukemia: The First Bispecific T-Cell Engager Antibody to Be Approved by the EMA for Minimal Residual Disease. Oncologist 2020, 25, e709–e715. [Google Scholar] [CrossRef] [PubMed]
- Boissel, N.; Chiaretti, S.; Papayannidis, C.; Ribera, J.M.; Bassan, R.; Sokolov, A.N.; Alam, N.; Brescianini, A.; Pezzani, I.; Kreuzbauer, G.; et al. Real-World Use of Blinatumomab in Adult Patients with B-Cell Acute Lymphoblastic Leukemia in Clinical Practice: Results from the NEUF Study. Blood Cancer J. 2023, 13, 2. [Google Scholar] [CrossRef] [PubMed]
- Halford, Z.; Coalter, C.; Gresham, V.; Brown, T. A Systematic Review of Blinatumomab in the Treatment of Acute Lymphoblastic Leukemia: Engaging an Old Problem With New Solutions. Ann. Pharmacother. 2021, 55, 1236–1253. [Google Scholar] [CrossRef] [PubMed]
- van der Sluis, I.M.; de Lorenzo, P.; Kotecha, R.S.; Attarbaschi, A.; Escherich, G.; Nysom, K.; Stary, J.; Ferster, A.; Brethon, B.; Locatelli, F.; et al. Blinatumomab Added to Chemotherapy in Infant Lymphoblastic Leukemia. N. Engl. J. Med. 2023, 388, 1572–1581. [Google Scholar] [CrossRef]
- Sayyed, A.; Chen, C.; Gerbitz, A.; Kim, D.D.H.; Kumar, R.; Lam, W.; Law, A.D.; Lipton, J.H.; Michelis, F.V.; Novitzky-Basso, I.; et al. Pretransplant Blinatumomab Improves Outcomes in B Cell Acute Lymphoblastic Leukemia Patients Who Undergo Allogeneic Hematopoietic Cell Transplantation. Transplant. Cell. Ther. 2024, 30, 520.e1–520.e12. [Google Scholar] [CrossRef]
- Llaurador, G.; Shaver, K.; Wu, M.; Wang, T.; Gillispie, A.; Doherty, E.; Craddock, J.; Read, J.; Yassine, K.; Morales, E.; et al. Blinatumomab Therapy Is Associated with Favorable Outcomes after Allogeneic Hematopoietic Cell Transplantation in Pediatric Patients with B Cell Acute Lymphoblastic Leukemia. Transplant. Cell. Ther. 2024, 30, 217–227. [Google Scholar] [CrossRef]
- Locatelli, F.; Zugmaier, G.; Rizzari, C.; Morris, J.D.; Gruhn, B.; Klingebiel, T.; Parasole, R.; Linderkamp, C.; Flotho, C.; Petit, A.; et al. Effect of Blinatumomab vs Chemotherapy on Event-Free Survival Among Children With High-Risk First-Relapse B-Cell Acute Lymphoblastic Leukemia: A Randomized Clinical Trial. JAMA 2021, 325, 843–854. [Google Scholar] [CrossRef]
- Locatelli, F.; Zugmaier, G.; Rizzari, C.; Morris, J.D.; Gruhn, B.; Klingebiel, T.; Parasole, R.; Linderkamp, C.; Flotho, C.; Petit, A.; et al. Improved Survival and MRD Remission with Blinatumomab vs. Chemotherapy in Children with First High-Risk Relapse B-ALL. Leukemia 2023, 37, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.; Bruno, A.; Rizzari, C.; Brescianini, A.; Von Stackelberg, A.; Linderkamp, C.; Zeng, Y.; Zugmaier, G.; Locatelli, F. Blinatumomab Is Associated with Better Post-Transplant Outcome than Chemotherapy in Children with High-Risk First-Relapse B-Cell Acute Lymphoblastic Leukemia Irrespective of the Conditioning Regimen. Haematologica 2024. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.; Dalle, J.H.; Locatelli, F.; Poetschger, U.; Sedlacek, P.; Buechner, J.; Shaw, P.J.; Staciuk, R.; Ifversen, M.; Pichler, H.; et al. Total Body Irradiation or Chemotherapy Conditioning in Childhood ALL: A Multinational, Randomized, Noninferiority Phase III Study. J. Clin. Oncol. 2021, 39, 295–307. [Google Scholar] [CrossRef]
- Abou Dalle, I.; Dulery, R.; Moukalled, N.; Ricard, L.; Stocker, N.; El-Cheikh, J.; Mohty, M.; Bazarbachi, A. Bi- and Tri-Specific Antibodies in Non-Hodgkin Lymphoma: Current Data and Perspectives. Blood Cancer J. 2024, 14, 23. [Google Scholar] [CrossRef]
- Carpenter, R.O.; Evbuomwan, M.O.; Pittaluga, S.; Rose, J.J.; Raffeld, M.; Yang, S.; Gress, R.E.; Hakim, F.T.; Kochenderfer, J.N. B-Cell Maturation Antigen Is a Promising Target for Adoptive T-Cell Therapy of Multiple Myeloma. Clin. Cancer Res. 2013, 19, 2048–2060. [Google Scholar] [CrossRef] [PubMed]
- Abramson, H.N. B-Cell Maturation Antigen (BCMA) as a Target for New Drug Development in Relapsed and/or Refractory Multiple Myeloma. Int. J. Mol. Sci. 2020, 21, 5192. [Google Scholar] [CrossRef]
- Kang, C. Teclistamab: First Approval. Drugs 2022, 82, 1613. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Garfall, A.L.; van de Donk, N.W.C.J.; Nahi, H.; San-Miguel, J.F.; Oriol, A.; Nooka, A.K.; Martin, T.; Rosinol, L.; Chari, A.; et al. Teclistamab in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2022, 387, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Elranatamab: First Approval. Drugs 2023, 83, 1621–1627. [Google Scholar] [CrossRef]
- Bahlis, N.J.; Costello, C.L.; Raje, N.S.; Levy, M.Y.; Dholaria, B.; Solh, M.; Tomasson, M.H.; Damore, M.A.; Jiang, S.; Basu, C.; et al. Elranatamab in Relapsed or Refractory Multiple Myeloma: The MagnetisMM-1 Phase 1 Trial. Nat. Med. 2023, 29, 2570–2576. [Google Scholar] [CrossRef] [PubMed]
- Lesokhin, A.M.; Tomasson, M.H.; Arnulf, B.; Bahlis, N.J.; Miles Prince, H.; Niesvizky, R.; Rodrίguez-Otero, P.; Martinez-Lopez, J.; Koehne, G.; Touzeau, C.; et al. Elranatamab in Relapsed or Refractory Multiple Myeloma: Phase 2 MagnetisMM-3 Trial Results. Nat. Med. 2023, 29, 2259–2267. [Google Scholar] [CrossRef] [PubMed]
- Tomasson, M.; Iida, S.; Niesvizky, R.; Mohty, M.; Bahlis, N.J.; Martinez-Lopez, J.; Koehne, G.; Rodriguez Otero, P.; Prince, H.M.; Viqueira, A.; et al. Long-Term Efficacy and Safety of Elranatamab Monotherapy in the Phase 2 Magnetismm-3 Trial in Relapsed or Refractory Multiple Myeloma (RRMM). Blood 2023, 142, 3385. [Google Scholar] [CrossRef]
- van de Donk, N.W.C.J.; O’Neill, C.; de Ruijter, M.E.M.; Verkleij, C.P.M.; Zweegman, S. T-Cell Redirecting Bispecific and Trispecific Antibodies in Multiple Myeloma beyond BCMA. Curr. Opin. Oncol. 2023, 35, 601–611. [Google Scholar] [CrossRef]
- Chari, A.; Touzeau, C.; Schinke, C.; Minnema, M.C.; Berdeja, J.; Oriol, A.; Van De Donk, N.W.; Rodriguez Otero, P.; Askari, E.; Mateos, M.-V.; et al. Talquetamab, a G Protein-Coupled Receptor Family C Group 5 Member D x CD3 Bispecific Antibody, in Patients with Relapsed/Refractory Multiple Myeloma (RRMM): Phase 1/2 Results from MonumenTAL-1. Blood 2022, 140, 384–387. [Google Scholar] [CrossRef]
- Jakubowiak, A.J.; Anguille, S.; Karlin, L.; Chari, A.; Schinke, C.; Rasche, L.; San-Miguel, J.; Campagna, M.; Hilder, B.W.; Masterson, T.J.; et al. Updated Results of Talquetamab, a GPRC5D×CD3 Bispecific Antibody, in Patients with Relapsed/Refractory Multiple Myeloma with Prior Exposure to T-Cell Redirecting Therapies: Results of the Phase 1/2 MonumenTAL-1 Study. Blood 2023, 142, 3377. [Google Scholar] [CrossRef]
- Sanchez, L.; Schinke, C.; Krishnan, A.; Berdeja, J.G.; van de Donk, N.W.; Mateos, M.V.; Chari, A.; Parekh, S.; Mouhieddine, T.H.; Jagannath, S.; et al. Clinical Outcomes of Subsequent Therapies in Patients with Relapsed/Refractory Multiple Myeloma Following Talquetamab Treatment: Analyses from the Phase 1/2 MonumenTAL-1 Study. Blood 2023, 142, 2007. [Google Scholar] [CrossRef]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and Challenges for the next Generation of Antibody-Drug Conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef]
- Larson, S.M.; Carrasquillo, J.A.; Cheung, N.K.V.; Press, O.W. Radioimmunotherapy of Human Tumours. Nat. Rev. Cancer 2015, 15, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Baron, J.; Wang, E.S. Gemtuzumab Ozogamicin for the Treatment of Acute Myeloid Leukemia. Expert Rev. Clin. Pharmacol. 2018, 11, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Fostvedt, L.K.; Hibma, J.E.; Masters, J.C.; Vandendries, E.; Ruiz-Garcia, A. Pharmacokinetic/Pharmacodynamic Modeling to Support the Re-approval of Gemtuzumab Ozogamicin. Clin. Pharmacol. Ther. 2019, 106, 1006. [Google Scholar] [CrossRef]
- Nguyen, T.D.; Bordeau, B.M.; Balthasar, J.P. Mechanisms of ADC Toxicity and Strategies to Increase ADC Tolerability. Cancers 2023, 15, 713. [Google Scholar] [CrossRef]
- Birrer, M.J.; Moore, K.N.; Betella, I.; Bates, R.C. Antibody-Drug Conjugate-Based Therapeutics: State of the Science. J. Natl. Cancer Inst. 2019, 111, 538–549. [Google Scholar] [CrossRef]
- Chau, C.H.; Steeg, P.S.; Figg, W.D. Antibody-Drug Conjugates for Cancer. Lancet 2019, 394, 793–804. [Google Scholar] [CrossRef]
- De Claro, R.A.; McGinn, K.; Kwitkowski, V.; Bullock, J.; Khandelwal, A.; Habtemariam, B.; Ouyang, Y.; Saber, H.; Lee, K.; Koti, K.; et al. U.S. Food and Drug Administration Approval Summary: Brentuximab Vedotin for the Treatment of Relapsed Hodgkin Lymphoma or Relapsed Systemic Anaplastic Large-Cell Lymphoma. Clin. Cancer Res. 2012, 18, 5845–5849. [Google Scholar] [CrossRef]
- Connors, J.M.; Jurczak, W.; Straus, D.J.; Ansell, S.M.; Kim, W.S.; Gallamini, A.; Younes, A.; Alekseev, S.; Illés, Á.; Picardi, M.; et al. Brentuximab Vedotin with Chemotherapy for Stage III or IV Hodgkin’s Lymphoma. N. Engl. J. Med. 2018, 378, 331–344. [Google Scholar] [CrossRef]
- Straus, D.J.; Długosz-Danecka, M.; Connors, J.M.; Alekseev, S.; Illés, Á.; Picardi, M.; Lech-Maranda, E.; Feldman, T.; Smolewski, P.; Savage, K.J.; et al. Brentuximab Vedotin with Chemotherapy for Stage III or IV Classical Hodgkin Lymphoma (ECHELON-1): 5-Year Update of an International, Open-Label, Randomised, Phase 3 Trial. Lancet Haematol. 2021, 8, e410–e421. [Google Scholar] [CrossRef]
- Borchmann, P.; Ferdinandus, J.; Schneider, G.; Moccia, A.; Greil, R.; Hertzberg, M.; Schaub, V.; Hüttmann, A.; Keil, F.; Dierlamm, J.; et al. Assessing the Efficacy and Tolerability of PET-Guided BrECADD versus EBEACOPP in Advanced-Stage, Classical Hodgkin Lymphoma (HD21): A Randomised, Multicentre, Parallel, Open-Label, Phase 3 Trial. Lancet 2024, 404, 341–352. [Google Scholar] [CrossRef]
- Lamb, Y.N. Inotuzumab Ozogamicin: First Global Approval. Drugs 2017, 77, 1603–1610. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; DeAngelo, D.J.; Stelljes, M.; Liedtke, M.; Stock, W.; Gökbuget, N.; O’Brien, S.M.; Jabbour, E.; Wang, T.; Liang White, J.; et al. Inotuzumab Ozogamicin versus Standard of Care in Relapsed or Refractory Acute Lymphoblastic Leukemia: Final Report and Long-Term Survival Follow-up from the Randomized, Phase 3 INO-VATE Study. Cancer 2019, 125, 2474–2487. [Google Scholar] [CrossRef] [PubMed]
- Marks, D.I.; Kebriaei, P.; Stelljes, M.; Gökbuget, N.; Kantarjian, H.; Advani, A.S.; Merchant, A.; Stock, W.; Cassaday, R.D.; Wang, T.; et al. Outcomes of Allogeneic Stem Cell Transplantation after Inotuzumab Ozogamicin Treatment for Relapsed or Refractory Acute Lymphoblastic Leukemia. Biol. Blood Marrow Transplant. 2019, 25, 1720–1729. [Google Scholar] [CrossRef]
- Stelmach, P.; Wethmar, K.; Groth, C.; Wenge, D.V.; Albring, J.; Mikesch, J.H.; Schliemann, C.; Reicherts, C.; Berdel, W.E.; Lenz, G.; et al. Blinatumomab or Inotuzumab Ozogamicin as Bridge to Allogeneic Stem Cell Transplantation for Relapsed or Refractory B-Lineage Acute Lymphoblastic Leukemia: A Retrospective Single-Center Analysis. Clin. Lymphoma. Myeloma Leuk. 2020, 20, e724–e733. [Google Scholar] [CrossRef] [PubMed]
- Curran, E.; O’Brien, M. Role of Blinatumomab, Inotuzumab, and CAR T-Cells: Which to Choose and How to Sequence for Patients with Relapsed Disease. Semin. Hematol. 2020, 57, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Wudhikarn, K.; King, A.C.; Geyer, M.B.; Roshal, M.; Bernal, Y.; Gyurkocza, B.; Perales, M.A.; Park, J.H. Outcomes of Relapsed B-Cell Acute Lymphoblastic Leukemia after Sequential Treatment with Blinatumomab and Inotuzumab. Blood Adv. 2022, 6, 1432–1443. [Google Scholar] [CrossRef]
- Kebriaei, P.; Cutler, C.; De Lima, M.; Giralt, S.; Lee, S.J.; Marks, D.; Merchant, A.; Stock, W.; Van Besien, K.; Stelljes, M. Management of Important Adverse Events Associated with Inotuzumab Ozogamicin: Expert Panel Review. Bone Marrow Transplant. 2018, 53, 449. [Google Scholar] [CrossRef] [PubMed]
- Kayser, S.; Sartor, C.; Giglio, F.; Bruno, A.; Webster, J.; Chiusolo, P.; Saraceni, F.; Guerzoni, S.; Pochintesta, L.; Borlenghi, E.; et al. Impact of Inotuzumab Ozogamicin on Outcome in Relapsed or Refractory Acute B-Cell Lymphoblastic Leukemia Patients Prior to Allogeneic Hematopoietic Stem Cell Transplantation and Risk of Sinusoidal Obstruction Syndrome/Venous Occlusive Disease. Haematologica 2024, 109, 1385–1392. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; Boissel, N.; Papayannidis, C.; Luskin, M.R.; Stelljes, M.; Advani, A.S.; Jabbour, E.J.; Ribera, J.M.; Marks, D.I. Inotuzumab Ozogamicin in Adult Acute Lymphoblastic Leukemia: Development, Current Status, and Future Directions. Cancer 2024, 130, 3631–3646. [Google Scholar] [CrossRef]
- Wenge, D.V.; Wethmar, K.; Klar, C.A.; Kolve, H.; Sauer, T.; Angenendt, L.; Evers, G.; Call, S.; Kerkhoff, A.; Khandanpour, C.; et al. Characteristics and Outcome of Elderly Patients (>55 Years) with Acute Lymphoblastic Leukemia. Cancers 2022, 14, 565. [Google Scholar] [CrossRef] [PubMed]
- Stelljes, M.; Raffel, S.; Alakel, N.; Wäsch, R.; Kondakci, M.; Scholl, S.; Rank, A.; Hänel, M.; Spriewald, B.; Hanoun, M.; et al. Inotuzumab Ozogamicin as Induction Therapy for Patients Older than 55 Years with Philadelphia Chromosome–Negative B-Precursor ALL. J. Clin. Oncol. 2024, 42, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D. Polatuzumab Vedotin: First Global Approval. Drugs 2019, 79, 1467–1475. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, M.; Zheng, B.; Erdmann, T.; Koeppen, H.; Mccord, R.; Grau, M.; Staiger, A.; Chai, A.; Sandmann, T.; Madle, H.; et al. Anti-CD22 and Anti-CD79B Antibody Drug Conjugates Are Active in Different Molecular Diffuse Large B-Cell Lymphoma Subtypes. Leukemia 2015, 29, 1578–1586. [Google Scholar] [CrossRef]
- Sehn, L.H.; Herrera, A.F.; Flowers, C.R.; Kamdar, M.K.; McMillan, A.; Hertzberg, M.; Assouline, S.; Kim, T.M.; Kim, W.S.; Ozcan, M.; et al. Polatuzumab Vedotin in Relapsed or Refractory Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2020, 38, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Flowers, C.R.; Matasar, M.J.; Herrera, A.F.; Hertzberg, M.; Assouline, S.; Demeter, J.; McMillan, A.; Mehta, A.; Opat, S.; Trněný, M.; et al. Polatuzumab Vedotin plus Bendamustine and Rituximab or Obinutuzumab in Relapsed/Refractory Follicular Lymphoma: A Phase Ib/II Study. Haematologica 2024, 109, 1194–1205. [Google Scholar] [CrossRef] [PubMed]
- Tilly, H.; Morschhauser, F.; Sehn, L.H.; Friedberg, J.W.; Trněný, M.; Sharman, J.P.; Herbaux, C.; Burke, J.M.; Matasar, M.; Rai, S.; et al. Polatuzumab Vedotin for the Front-Line Treatment of Diffuse Large B-Cell Lymphoma: A New Standard of Care? J. Adv. Pract. Oncol. 2023, 14, 67–72. [Google Scholar] [CrossRef]
- Tilly, H.; Morschhauser, F.; Sehn, L.H.; Friedberg, J.W.; Trněný, M.; Sharman, J.P.; Herbaux, C.; Burke, J.M.; Matasar, M.; Rai, S.; et al. Polatuzumab Vedotin in Previously Untreated Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2022, 386, 351–363. [Google Scholar] [CrossRef]
- Sarraf Yazdy, M.; Kasamon, Y.L.; Gu, W.; Rodriguez, L.R.; Jin, S.; Bhatnagar, V.; Richardson, N.C.; Theoret, M.R.; Pazdur, R.; Gormley, N.J. FDA Approval Summary: Polatuzumab Vedotin in the First-Line Treatment of Select Large B-Cell Lymphomas. Clin. Cancer Res. 2024. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Belantamab Mafodotin: First Approval. Drugs 2020, 80, 1607–1613. [Google Scholar] [CrossRef] [PubMed]
- Nooka, A.K.; Cohen, A.; Lee, H.C.; Badros, A.Z.; Suvannasankha, A.; Callander, N.; Abdallah, A.-O.; Trudel, S.; Chari, A.; Libby, E.; et al. Single-Agent Belantamab Mafodotin in Patients with Relapsed or Refractory Multiple Myeloma: Final Analysis of the DREAMM-2 Trial. Blood 2022, 140, 7301–7303. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Hungria, V.T.M.; Radinoff, A.; Delimpasi, S.; Mikala, G.; Masszi, T.; Li, J.; Capra, M.; Maiolino, A.; Pappa, V.; et al. Efficacy and Safety of Single-Agent Belantamab Mafodotin versus Pomalidomide plus Low-Dose Dexamethasone in Patients with Relapsed or Refractory Multiple Myeloma (DREAMM-3): A Phase 3, Open-Label, Randomised Study. Lancet Haematol. 2023, 10, e801–e812. [Google Scholar] [CrossRef]
- Mateos, M.-V.; Robak, P.; Hus, M.; Xia, Z.; Zherebtsova, V.; Ward, C.; Ho, P.J.; Hajek, R.; Kim, K.; Dimopoulos, M.A.; et al. Results from the Randomized Phase III DREAMM-7 Study of Belantamab Mafodotin (Belamaf) + Bortezomib, and Dexamethasone (BVd) vs Daratumumab, Bortezomib, and Dexamethasone (DVd) in Relapsed/Refractory Multiple Myeloma (RRMM). J. Clin. Oncol. 2024, 42, 439572. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Beksac, M.; Pour, L.; Delimpasi, S.; Vorobyev, V.; Quach, H.; Spicka, I.; Radocha, J.; Robak, P.; Kim, K.; et al. Belantamab Mafodotin, Pomalidomide, and Dexamethasone in Multiple Myeloma. N. Engl. J. Med. 2024, 391, 408–421. [Google Scholar] [CrossRef] [PubMed]
- Morè, S.; Offidani, M.; Corvatta, L.; Petrucci, M.T.; Fazio, F. Belantamab Mafodotin: From Clinical Trials Data to Real-Life Experiences. Cancers 2023, 15, 2948. [Google Scholar] [CrossRef]
- Lee, A. Loncastuximab Tesirine: First Approval. Drugs 2021, 81, 1229–1233. [Google Scholar] [CrossRef]
- Hamadani, M.; Radford, J.; Carlo-Stella, C.; Caimi, P.F.; Reid, E.; O’Connor, O.A.; Feingold, J.M.; Ardeshna, K.M.; Townsend, W.; Solh, M.; et al. Final Results of a Phase 1 Study of Loncastuximab Tesirine in Relapsed/Refractory B-Cell Non-Hodgkin Lymphoma. Blood 2021, 137, 2634–2645. [Google Scholar] [CrossRef]
- Caimi, P.F.; Ai, W.; Alderuccio, J.P.; Ardeshna, K.M.; Hamadani, M.; Hess, B.; Kahl, B.S.; Radford, J.; Solh, M.; Stathis, A.; et al. Loncastuximab Tesirine in Relapsed or Refractory Diffuse Large B-Cell Lymphoma (LOTIS-2): A Multicentre, Open-Label, Single-Arm, Phase 2 Trial. Lancet Oncol. 2021, 22, 790–800. [Google Scholar] [CrossRef]
- Markham, A.; Al-Salama, Z.T. Loncastuximab Tesirine in Relapsed or Refractory Diffuse Large B-Cell Lymphoma: A Profile of Its Use in the USA. Drugs Ther. Perspect. 2022, 38, 261–267. [Google Scholar] [CrossRef]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Flies, D.B. Molecular Mechanisms of T Cell Co-Stimulation and Co-Inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Xu, C. Immune Checkpoint Signaling and Cancer Immunotherapy. Cell Res. 2020, 30, 660–669. [Google Scholar] [CrossRef]
- Iranzo, P.; Callejo, A.; Assaf, J.D.; Molina, G.; Lopez, D.E.; Garcia-Illescas, D.; Pardo, N.; Navarro, A.; Martinez-Marti, A.; Cedres, S.; et al. Overview of Checkpoint Inhibitors Mechanism of Action: Role of Immune-Related Adverse Events and Their Treatment on Progression of Underlying Cancer. Front. Med. 2022, 9, 875974. [Google Scholar] [CrossRef] [PubMed]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune Checkpoint Blockade Therapy for Cancer: An Overview of FDA-Approved Immune Checkpoint Inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Petitprez, F.; Meylan, M.; de Reyniès, A.; Sautès-Fridman, C.; Fridman, W.H. The Tumor Microenvironment in the Response to Immune Checkpoint Blockade Therapies. Front. Immunol. 2020, 11, 784. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Monti, S.; Rodig, S.J.; Juszczynski, P.; Currie, T.; O’Donnell, E.; Chapuy, B.; Takeyama, K.; Neuberg, D.; Golub, T.R.; et al. Integrative Analysis Reveals Selective 9p24.1 Amplification, Increased PD-1 Ligand Expression, and Further Induction via JAK2 in Nodular Sclerosing Hodgkin Lymphoma and Primary Mediastinal Large B-Cell Lymphoma. Blood 2010, 116, 3268. [Google Scholar] [CrossRef]
- Pianko, M.J.; Moskowitz, A.J.; Lesokhin, A.M. Immunotherapy of Lymphoma and Myeloma: Facts and Hopes. Clin. Cancer Res. 2018, 24, 1002–1010. [Google Scholar] [CrossRef] [PubMed]
- Armand, P.; Engert, A.; Younes, A.; Fanale, M.; Santoro, A.; Zinzani, P.L.; Timmerman, J.M.; Collins, G.P.; Ramchandren, R.; Cohen, J.B.; et al. Nivolumab for Relapsed/Refractory Classic Hodgkin Lymphoma After Failure of Autologous Hematopoietic Cell Transplantation: Extended Follow-Up of the Multicohort Single-Arm Phase II CheckMate 205 Trial. J. Clin. Oncol. 2018, 36, 1428–1439. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Bröckelmann, P.J.; von Keudell, G.; Lee, H.J.; Santoro, A.; Zinzani, P.L.; Collins, G.P.; Cohen, J.B.; de Boer, J.P.; Kuruvilla, J.; et al. Nivolumab for Relapsed/Refractory Classical Hodgkin Lymphoma: 5-Year Survival from the Pivotal Phase 2 CheckMate 205 Study. Blood Adv. 2023, 7, 6266–6274. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Santoro, A.; Shipp, M.; Zinzani, P.L.; Timmerman, J.M.; Ansell, S.; Armand, P.; Fanale, M.; Ratanatharathorn, V.; Kuruvilla, J.; et al. Nivolumab for Classical Hodgkin’s Lymphoma after Failure of Both Autologous Stem-Cell Transplantation and Brentuximab Vedotin: A Multicentre, Multicohort, Single-Arm Phase 2 Trial. Lancet. Oncol. 2016, 17, 1283–1294. [Google Scholar] [CrossRef]
- Kasamon, Y.L.; Claro, R.A.d.; Wang, Y.; Shen, Y.L.; Farrell, A.T.; Pazdur, R. FDA Approval Summary: Nivolumab for the Treatment of Relapsed or Progressive Classical Hodgkin Lymphoma. Oncologist 2017, 22, 585. [Google Scholar] [CrossRef] [PubMed]
- Herrera, A.F.; LeBlanc, M.; Castellino, S.M.; Li, H.; Rutherford, S.C.; Evens, A.M.; Davison, K.; Punnett, A.; Parsons, S.K.; Ahmed, S.; et al. Nivolumab+AVD in Advanced-Stage Classic Hodgkin’s Lymphoma. N. Engl. J. Med. 2024, 391, 1379–1389. [Google Scholar] [CrossRef]
- Armand, P.; Shipp, M.A.; Ribrag, V.; Michot, J.M.; Zinzani, P.L.; Kuruvilla, J.; Snyder, E.S.; Ricart, A.D.; Balakumaran, A.; Rose, S.; et al. Programmed Death-1 Blockade with Pembrolizumab in Patients with Classical Hodgkin Lymphoma after Brentuximab Vedotin Failure. J. Clin. Oncol. 2016, 34, 3733–3739. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zinzani, P.L.; Fanale, M.A.; Armand, P.; Johnson, N.A.; Brice, P.; Radford, J.; Ribrag, V.; Molin, D.; Vassilakopoulos, T.P.; et al. Phase II Study of the Efficacy and Safety of Pembrolizumab for Relapsed/Refractory Classic Hodgkin Lymphoma. J. Clin. Oncol. 2017, 35, 2125–2132. [Google Scholar] [CrossRef]
- Kuruvilla, J.; Ramchandren, R.; Santoro, A.; Paszkiewicz-Kozik, E.; Gasiorowski, R.; Johnson, N.A.; Fogliatto, L.M.; Goncalves, I.; de Oliveira, J.S.R.; Buccheri, V.; et al. Pembrolizumab versus Brentuximab Vedotin in Relapsed or Refractory Classical Hodgkin Lymphoma (KEYNOTE-204): An Interim Analysis of a Multicentre, Randomised, Open-Label, Phase 3 Study. Lancet. Oncol. 2021, 22, 512–524. [Google Scholar] [CrossRef] [PubMed]
- Voorhees, T.J.; Beaven, A.W. Therapeutic Updates for Relapsed and Refractory Classical Hodgkin Lymphoma. Cancers 2020, 12, 2887. [Google Scholar] [CrossRef] [PubMed]
- Armand, P.; Chen, Y.B.; Redd, R.A.; Joyce, R.M.; Bsat, J.; Jeter, E.; Merryman, R.W.; Coleman, K.C.; Dahi, P.B.; Nieto, Y.; et al. PD-1 Blockade with Pembrolizumab for Classical Hodgkin Lymphoma after Autologous Stem Cell Transplantation. Blood 2019, 134, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Mei, M.; Palmer, J.; Tsai, N.-C.; Lee, H.J.; Isufi, I.; Popplewell, L.L.; Smith, L.; Peters, L.; Rodriguez, L.; Godfrey, J.; et al. Nivolumab Plus ICE As First Salvage Therapy in High-Risk Relapsed/Refractory Hodgkin Lymphoma. Blood 2022, 140, 774–776. [Google Scholar] [CrossRef]
- Mei, M.G.; Lee, H.J.; Palmer, J.M.; Chen, R.; Tsai, N.C.; Chen, L.; McBride, K.; Smith, D.L.; Melgar, I.; Song, J.Y.; et al. Response-Adapted Anti-PD-1–Based Salvage Therapy for Hodgkin Lymphoma with Nivolumab Alone or in Combination with ICE. Blood 2022, 139, 3605–3616. [Google Scholar] [CrossRef] [PubMed]
- Zinzani, P.L.; Ribrag, V.; Moskowitz, C.H.; Michot, J.M.; Kuruvilla, J.; Balakumaran, A.; Zhang, Y.; Chlosta, S.; Shipp, M.A.; Armand, P. Safety and Tolerability of Pembrolizumab in Patients with Relapsed/Refractory Primary Mediastinal Large B-Cell Lymphoma. Blood 2017, 130, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; LaPlant, B.R.; Call, T.G.; Parikh, S.A.; Leis, J.F.; He, R.; Shanafelt, T.D.; Sinha, S.; Le-Rademacher, J.; Feldman, A.L.; et al. Pembrolizumab in Patients with CLL and Richter Transformation or with Relapsed CLL. Blood 2017, 129, 3419–3427. [Google Scholar] [CrossRef]
- Melani, C.; Major, A.; Schowinsky, J.; Roschewski, M.; Pittaluga, S.; Jaffe, E.S.; Pack, S.D.; Abdullaev, Z.; Ahlman, M.A.; Kwak, J.J.; et al. PD-1 Blockade in Mediastinal Gray-Zone Lymphoma. N. Engl. J. Med. 2017, 377, 89–91. [Google Scholar] [CrossRef]
- Tomassetti, S.; Chen, R.; Dandapani, S. The Role of Pembrolizumab in Relapsed/Refractory Primary Mediastinal Large B-Cell Lymphoma. Ther. Adv. Hematol. 2019, 10, 204062071984159. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Senapati, J.; Thakral, B.; Ferrajoli, A.; Thompson, P.; Burger, J.; Basu, S.; Kadia, T.; Daver, N.; Borthakur, G.; et al. A Phase 2 Study of Nivolumab Combined with Ibrutinib in Patients with Diffuse Large B-Cell Richter Transformation of CLL. Blood Adv. 2023, 7, 1958–1966. [Google Scholar] [CrossRef]
- Joshi, M.; Ansell, S.M. Activating the Antitumor Immune Response in Non-Hodgkin Lymphoma Using Immune Checkpoint Inhibitors. J. Immunol. Res. 2020, 2020. [Google Scholar] [CrossRef] [PubMed]
- Apostolidis, J.; Sayyed, A.; Darweesh, M.; Kaloyannidis, P.; Al Hashmi, H. Current Clinical Applications and Future Perspectives of Immune Checkpoint Inhibitors in Non-Hodgkin Lymphoma. J. Immunol. Res. 2020, 2020, 8820377. [Google Scholar] [CrossRef]
- Chari, A.; Suvannasankha, A.; Fay, J.W.; Arnulf, B.; Kaufman, J.L.; Ifthikharuddin, J.J.; Weiss, B.M.; Krishnan, A.; Lentzsch, S.; Comenzo, R.; et al. Daratumumab plus Pomalidomide and Dexamethasone in Relapsed and/or Refractory Multiple Myeloma. Blood 2017, 130, 974–981. [Google Scholar] [CrossRef] [PubMed]
- Mateos, M.V.; Blacklock, H.; Schjesvold, F.; Oriol, A.; Simpson, D.; George, A.; Goldschmidt, H.; Larocca, A.; Chanan-Khan, A.; Sherbenou, D.; et al. Pembrolizumab plus Pomalidomide and Dexamethasone for Patients with Relapsed or Refractory Multiple Myeloma (KEYNOTE-183): A Randomised, Open-Label, Phase 3 Trial. Lancet. Haematol. 2019, 6, e459–e469. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Xu, X.; Liu, H.; Zhao, X.; Yang, C.; Fu, R. Immune Checkpoint Inhibitors for Multiple Myeloma Immunotherapy. Exp. Hematol. Oncol. 2023, 12, 99. [Google Scholar] [CrossRef]
- Mun, J.Y.; Leem, S.H.; Lee, J.H.; Kim, H.S. Dual Relationship Between Stromal Cells and Immune Cells in the Tumor Microenvironment. Front. Immunol. 2022, 13, 864739. [Google Scholar] [CrossRef]
- Li, Z.W.; Dalton, W.S. Tumor Microenvironment and Drug Resistance in Hematologic Malignancies. Blood Rev. 2006, 20, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Arandi, N.; Dehghani, M. Immune Microenvironment in Hematologic Malignancies. Iran. J. Med. Sci. 2023, 48, 1. [Google Scholar] [CrossRef] [PubMed]
- Kotlov, N.; Bagaev, A.; Revuelta, M.V.; Phillip, J.M.; Cacciapuoti, M.T.; Antysheva, Z.; Svekolkin, V.; Tikhonova, E.; Miheecheva, N.; Kuzkina, N.; et al. Clinical and Biological Subtypes of B-Cell Lymphoma Revealed by Microenvironmental Signatures. Cancer Discov. 2021, 11, 1468–1489. [Google Scholar] [CrossRef]
- Höpken, U.E.; Rehm, A. Targeting the Tumor Microenvironment of Leukemia and Lymphoma. Trends Cancer 2019, 5, 351–364. [Google Scholar] [CrossRef] [PubMed]
- Bilotta, M.T.; Antignani, A.; Fitzgerald, D.J. Managing the TME to Improve the Efficacy of Cancer Therapy. Front. Immunol. 2022, 13, 954992. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. Cancer Clin. Trials 2016, 39, 98–106. [Google Scholar] [CrossRef]
- Sinkarevs, S.; Strumfs, B.; Volkova, S.; Strumfa, I. Tumour Microenvironment: The General Principles of Pathogenesis and Implications in Diffuse Large B Cell Lymphoma. Cells 2024, 13, 1057. [Google Scholar] [CrossRef]
- Mulder, T.A.; Wahlin, B.E.; Österborg, A.; Palma, M. Targeting the Immune Microenvironment in Lymphomas of B-Cell Origin: From Biology to Clinical Application. Cancers 2019, 11, 915. [Google Scholar] [CrossRef]
- Li, Y.; Wu, Y.; Hu, Y. Metabolites in the Tumor Microenvironment Reprogram Functions of Immune Effector Cells Through Epigenetic Modifications. Front. Immunol. 2021, 12, 641883. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A Guide to Cancer Immunotherapy: From T Cell Basic Science to Clinical Practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Gulla, A.; Anderson, K.C. Multiple Myeloma: The (r)Evolution of Current Therapy and a Glance into the Future. Haematologica 2020, 105, 2358–2367. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Pagel, J.M. Current and Future Treatment Strategies in Chronic Lymphocytic Leukemia. J. Hematol. Oncol. 2021, 14, 69. [Google Scholar] [CrossRef] [PubMed]
- Hallek, M.; Al-Sawaf, O. Chronic Lymphocytic Leukemia: 2022 Update on Diagnostic and Therapeutic Procedures. Am. J. Hematol. 2021, 96, 1679–1705. [Google Scholar] [CrossRef] [PubMed]
- Tavarozzi, R.; Zacchi, G.; Pietrasanta, D.; Catania, G.; Castellino, A.; Monaco, F.; Gandolfo, C.; Rivela, P.; Sofia, A.; Schiena, N.; et al. Changing Trends in B-Cell Non-Hodgkin Lymphoma Treatment: The Role of Novel Monoclonal Antibodies in Clinical Practice. Cancers 2023, 15, 5397. [Google Scholar] [CrossRef]
- Shepard, H.M.; Phillips, G.L.; Thanos, C.D.; Feldmann, M. Developments in Therapy with Monoclonal Antibodies and Related Proteins. Clin. Med. 2017, 17, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Chiu, M.L.; Goulet, D.R.; Teplyakov, A.; Gilliland, G.L. Antibody Structure and Function: The Basis for Engineering Therapeutics. Antibodies 2019, 8, 55. [Google Scholar] [CrossRef]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody Therapy of Cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef]
- Isabwe, G.A.C.; Garcia Neuer, M.; de las Vecillas Sanchez, L.; Lynch, D.M.; Marquis, K.; Castells, M. Hypersensitivity Reactions to Therapeutic Monoclonal Antibodies: Phenotypes and Endotypes. J. Allergy Clin. Immunol. 2018, 142, 159–170.e2. [Google Scholar] [CrossRef] [PubMed]
- Gelis, S.; Verdesoto, J.T.; Pascal, M.; Muñoz-Cano, R.M. Hypersensitivity Reactions to Monoclonal Antibodies: New Approaches. Curr. Treat. Options Allergy 2022, 9, 394–408. [Google Scholar] [CrossRef]
- Picard, M.; Galvão, V.R. Current Knowledge and Management of Hypersensitivity Reactions to Monoclonal Antibodies. J. Allergy Clin. Immunol. Pract. 2017, 5, 600–609. [Google Scholar] [CrossRef]
- Isabwe, G.A.C.; Sanchez, L.D.L.V.; Castells, M. Management of Adverse Reactions to Biologic Agents. Allergy Asthma Proc. 2017, 38, 409–418. [Google Scholar] [CrossRef]
- Pavanello, F.; Zucca, E.; Ghielmini, M. Rituximab: 13 Open Questions after 20years of Clinical Use. Cancer Treat. Rev. 2017, 53, 38–46. [Google Scholar] [CrossRef]
- Leipold, D.; Prabhu, S. Pharmacokinetic and Pharmacodynamic Considerations in the Design of Therapeutic Antibodies. Clin. Transl. Sci. 2019, 12, 130. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chen, Y.; Park, J.; Liu, X.; Hu, Y.; Wang, T.; McFarland, K.; Betenbaugh, M.J. Design and Production of Bispecific Antibodies. Antibodies 2019, 8, 43. [Google Scholar] [CrossRef]
- Jureczek, J.; Bergmann, R.; Berndt, N.; Koristka, S.; Kegler, A.; Puentes-Cala, E.; Soto, J.A.; Arndt, C.; Bachmann, M.; Feldmann, A. An Oligo-His-Tag of a Targeting Module Does Not Influence Its Biodistribution and the Retargeting Capabilities of UniCAR T Cells. Sci. Rep. 2019, 9, 10547. [Google Scholar] [CrossRef] [PubMed]
- Strohl, W.R. Fusion Proteins for Half-Life Extension of Biologics as a Strategy to Make Biobetters. BioDrugs 2015, 29, 215–239. [Google Scholar] [CrossRef] [PubMed]
- Jureczek, J.; Feldmann, A.; Bergmann, R.; Arndt, C.; Berndt, N.; Koristka, S.; Loureiro, L.R.; Mitwasi, N.; Hoffmann, A.; Kegler, A.; et al. Highly Efficient Targeting of EGFR-Expressing Tumor Cells with UNiCAR T Cells via Target Modules Based on Cetuximab®. Onco. Targets. Ther. 2020, 13, 5515–5527. [Google Scholar] [CrossRef]
- Elshiaty, M.; Schindler, H.; Christopoulos, P. Principles and Current Clinical Landscape of Multispecific Antibodies against Cancer. Int. J. Mol. Sci. 2021, 22, 5632. [Google Scholar] [CrossRef]
- Surowka, M.; Klein, C. A Pivotal Decade for Bispecific Antibodies? MAbs 2024, 16, 2321635. [Google Scholar] [CrossRef]
- Cassanello, G.; Luna de Abia, A.; Falchi, L. Trial Watch: Bispecific Antibodies for the Treatment of Relapsed or Refractory Large B-Cell Lymphoma. Oncoimmunology 2024, 13, 2321648. [Google Scholar] [CrossRef] [PubMed]
- Caraccio, C.; Krishna, S.; Phillips, D.J.; Schürch, C.M. Bispecific Antibodies for Multiple Myeloma: A Review of Targets, Drugs, Clinical Trials, and Future Directions. Front. Immunol. 2020, 11, 501. [Google Scholar] [CrossRef] [PubMed]
- Dickopf, S.; Georges, G.J.; Brinkmann, U. Format and Geometries Matter: Structure-Based Design Defines the Functionality of Bispecific Antibodies. Comput. Struct. Biotechnol. J. 2020, 18, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Mo, Y.; Tang, M.; Shen, J.; Qi, Y.; Zhao, W.; Huang, Y.; Xu, Y.; Qian, C. Bispecific Antibodies: From Research to Clinical Application. Front. Immunol. 2021, 12, 626616. [Google Scholar] [CrossRef] [PubMed]
- Gerber, H.P.; Gangwar, S.; Betts, A. Therapeutic Index Improvement of Antibody-Drug Conjugates. MAbs 2023, 15, 2230618. [Google Scholar] [CrossRef]
- Dean, A.Q.; Luo, S.; Twomey, J.D.; Zhang, B. Targeting Cancer with Antibody-Drug Conjugates: Promises and Challenges. MAbs 2021, 13, 1951427. [Google Scholar] [CrossRef]
- Weggen, J.T.; Bean, R.; Hui, K.; Wendeler, M.; Hubbuch, J. Kinetic Models towards an Enhanced Understanding of Diverse ADC Conjugation Reactions. Front. Bioeng. Biotechnol. 2024, 12, 1403644. [Google Scholar] [CrossRef] [PubMed]
- Fuentes-Antrás, J.; Genta, S.; Vijenthira, A.; Siu, L.L. Antibody-Drug Conjugates: In Search of Partners of Choice. Trends Cancer 2023, 9, 339–354. [Google Scholar] [CrossRef]
- Cuesta-Mateos, C.; Alcaraz-Serna, A.; Somovilla-Crespo, B.; Muñoz-Calleja, C. Monoclonal Antibody Therapies for Hematological Malignancies: Not Just Lineage-Specific Targets. Front. Immunol. 2018, 8, 313558. [Google Scholar] [CrossRef] [PubMed]
- Molica, M.; Perrone, S.; Andriola, C.; Rossi, M. Immunotherapy with Monoclonal Antibodies for Acute Myeloid Leukemia: A Work in Progress. Cancers 2023, 15, 5060. [Google Scholar] [CrossRef]
- D’Alò, F.; Bellesi, S.; Maiolo, E.; Alma, E.; Bellisario, F.; Malafronte, R.; Viscovo, M.; Campana, F.; Hohaus, S. Novel Targets and Advanced Therapies in Diffuse Large B Cell Lymphomas. Cancers 2024, 16, 2243. [Google Scholar] [CrossRef]
- Crombie, J.L.; Graff, T.; Falchi, L.; Karimi, Y.H.; Bannerji, R.; Nastoupil, L.; Thieblemont, C.; Ursu, R.; Bartlett, N.; Nachar, V.; et al. Consensus Recommendations on the Management of Toxicity Associated with CD3×CD20 Bispecific Antibody Therapy. Blood 2024, 143, 1565–1575. [Google Scholar] [CrossRef] [PubMed]
- Madsen, A.V.; Pedersen, L.E.; Kristensen, P.; Goletz, S. Design and Engineering of Bispecific Antibodies: Insights and Practical Considerations. Front. Bioeng. Biotechnol. 2024, 12, 1352014. [Google Scholar] [CrossRef]

{kind=link}
| Name | Brand Name | Targeted Antigen | Indication | Year Approved FDA | Year Approved EMA |
|---|---|---|---|---|---|
| rituximab | Rituxan | CD20 | CLL, DLBCL, BL, BLL, B-ALL | 1997 | 1998 |
| ofatumumab | Arzerra | CD20 | CLL | 2009 | 2010 * |
| obinutuzumab | Gazyvaro/Gazyva | CD20 | CLL, FL | 2013 | 2014 |
| elotuzumab | Empliciti | SLAMF7 | R/R MM | 2015 | 2016 |
| daratumumab | Darzalex | CD38 | MM | 2016 | 2016 |
| isatuximab | Sarclisa | CD38 | MM | 2020 | 2020 |
| tafasitamab | Monjuvi | CD19 | DLBCL | 2020 | 2021 |
| Name | Brand Name | Targeted Antigen | Indication | Year Approved FDA | Year Approved EMA |
|---|---|---|---|---|---|
| blinatumomab | Blincyto | CD3xCD19 | B-ALL Ph− | 2014 | 2015 |
| mosunetuzumab | Lunsumio | CD3xCD20 | R/R FL | 2022 | 2022 |
| teclistamab | Tecvayli | CD3xBCMA | R/R MM | 2022 | 2022 |
| epcoritamab | Epkinly | CD3xCD20 | R/R DLBCL | 2023 | 2023 |
| glofitamab | Columvi | CD3xCD20 | R/R DLBCL, LBL | 2023 | 2023 |
| elranatamab | Elrexfio | CD3xBCMA | R/R MM | 2023 | 2023 |
| talquetamab | Talvey | CD3xGPRC5D | R/R MM | 2023 | 2023 |
| Name | Brand Name | Targeted Antigen | Payload | Indication | Year Approved FDA | Year Approved EMA |
|---|---|---|---|---|---|---|
| gemtuzumab ozogamicin | Mylotarg | CD33 | calicheamicin | AML | 2000, 2017 | 2018 |
| brentuximab vedotin | Adcetris | CD30 | monomethyl auristatin E | HL | 2011 | 2012 |
| inotuzumab ozogamicin | Besponsa | CD22 | calicheamicin | R/R CD22+ B-ALL | 2017 | 2017 |
| polatuzumab vedotin | Polivy | CD79b | monomethyl auristatin E | DLBCL | 2019 | 2020 |
| belantamab mafodotin | Blenrep | BCMA | monomethyl auristatin F | R/R MM | 2020 * | 2020 * |
| loncastuximab tesirine | Zynlonta | CD19 | pyrrolobenzodiazepine dimer SG3199 | R/R DLBCL, R/R HGBL | 2021 | 2022 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jureczek, J.; Kałwak, K.; Dzięgiel, P. Antibody-Based Immunotherapies for the Treatment of Hematologic Malignancies. Cancers 2024, 16, 4181. https://doi.org/10.3390/cancers16244181
Jureczek J, Kałwak K, Dzięgiel P. Antibody-Based Immunotherapies for the Treatment of Hematologic Malignancies. Cancers. 2024; 16(24):4181. https://doi.org/10.3390/cancers16244181
Chicago/Turabian StyleJureczek, Justyna, Krzysztof Kałwak, and Piotr Dzięgiel. 2024. "Antibody-Based Immunotherapies for the Treatment of Hematologic Malignancies" Cancers 16, no. 24: 4181. https://doi.org/10.3390/cancers16244181
APA StyleJureczek, J., Kałwak, K., & Dzięgiel, P. (2024). Antibody-Based Immunotherapies for the Treatment of Hematologic Malignancies. Cancers, 16(24), 4181. https://doi.org/10.3390/cancers16244181