

New Insights into Mucosa-Associated Microbiota in Paired Tumor and Non-Tumor Adjacent Mucosal Tissues in Colorectal Cancer Patients

 ,
,  , and
, and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement and Sample Collection
2.2. DNA Extraction and Quantitation
2.3. PCR Amplification
2.4. Library Preparation and Amplicon Sequencing
2.5. Data Acquisition and Sequencing Data Analysis
2.6. Bioinformatics Analysis
2.7. Statistical Analysis
3. Results
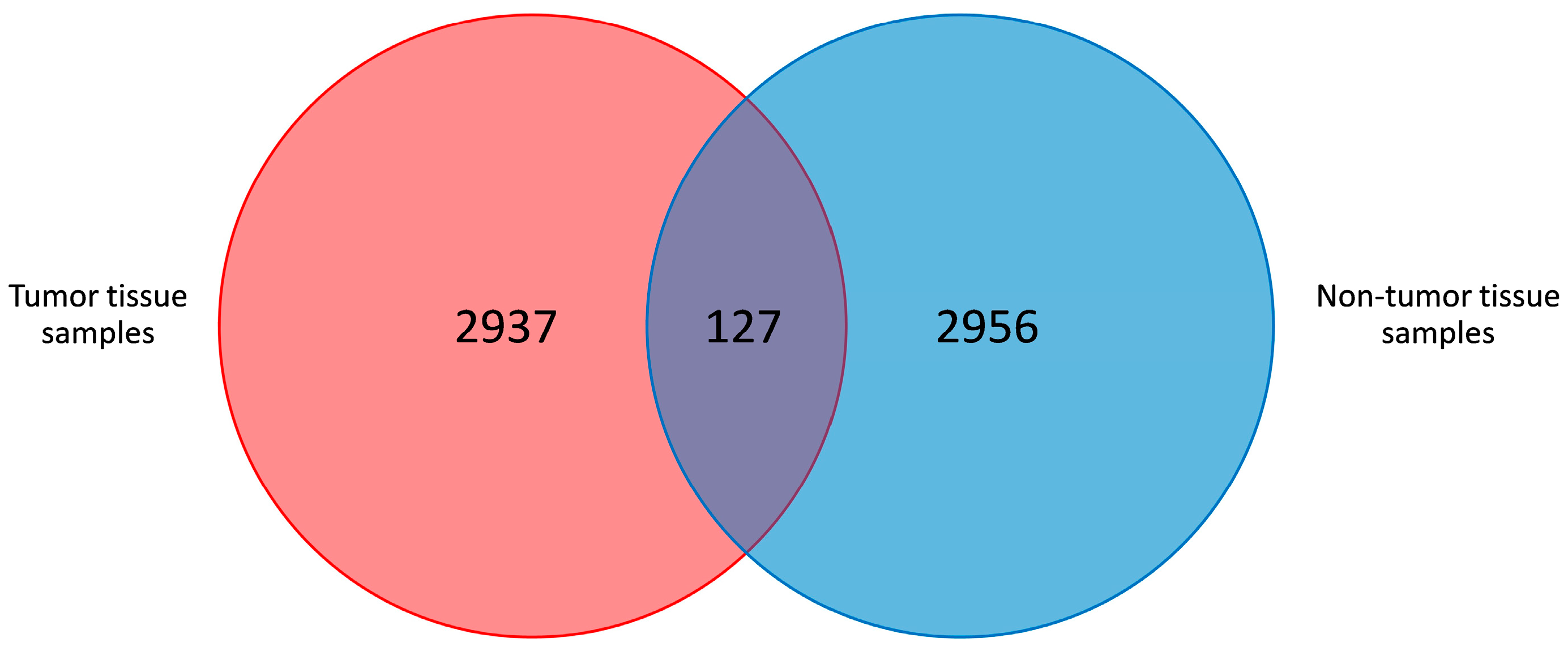
3.1. Sequence Analysis
3.2. α- and β-Diversity
3.2.1. α-Diversity of Microbiota in Tumor Compared to Non-Tumor Adjacent Tissue Samples of CRC Patients
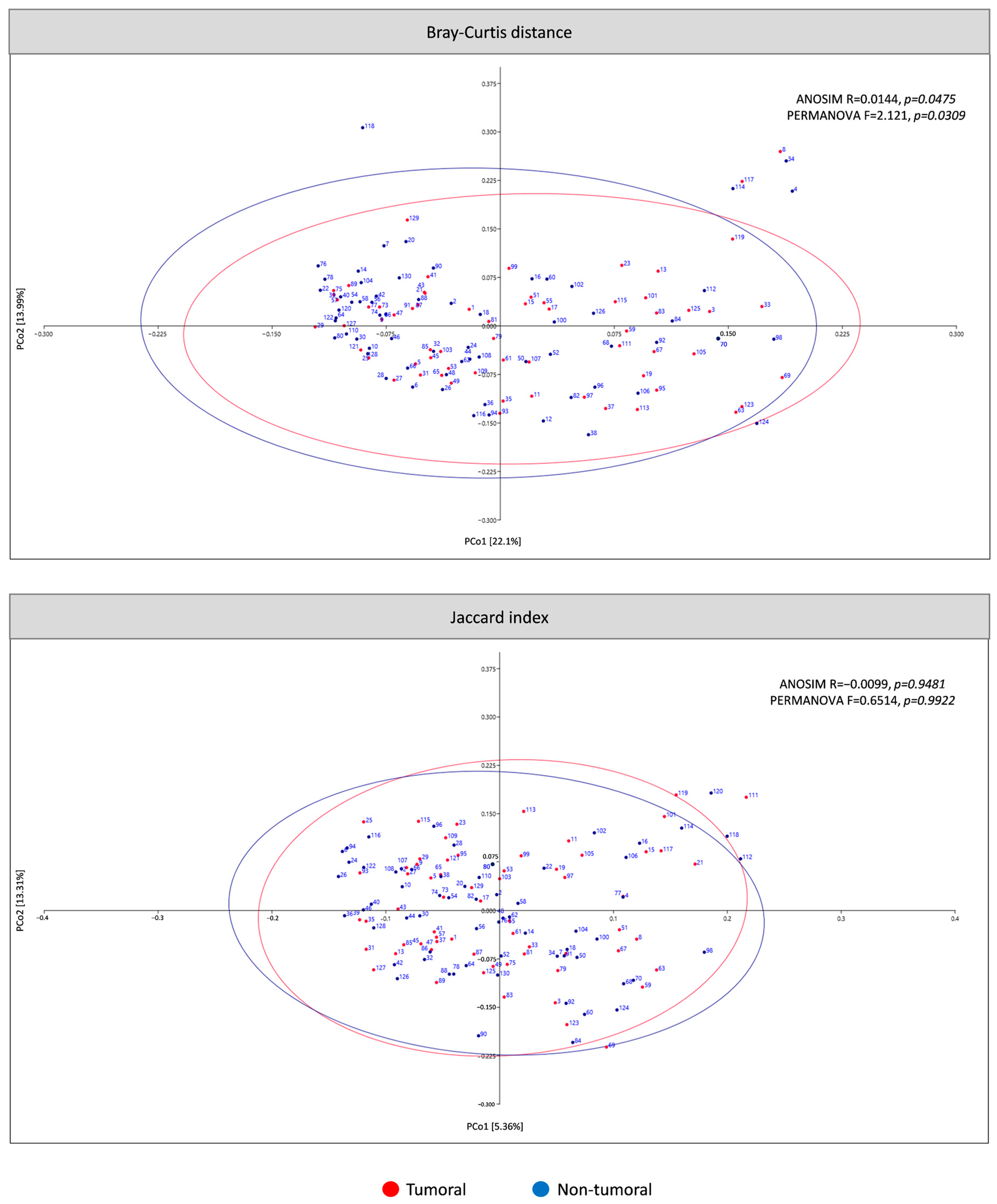
3.2.2. β-Diversity of Microbiota in Tumor Compared to Non-Tumor Adjacent Tissue Samples of CRC Patients
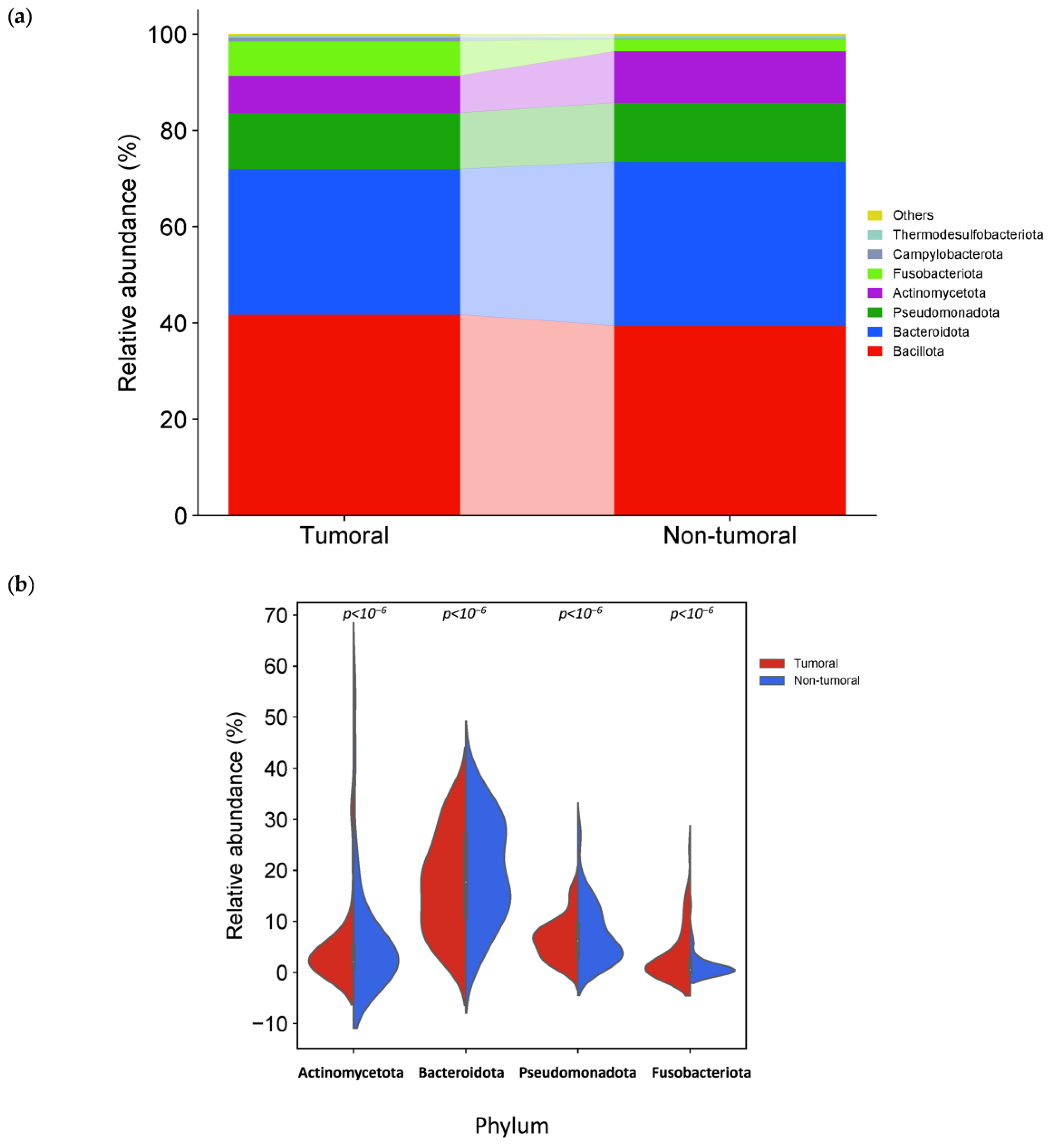
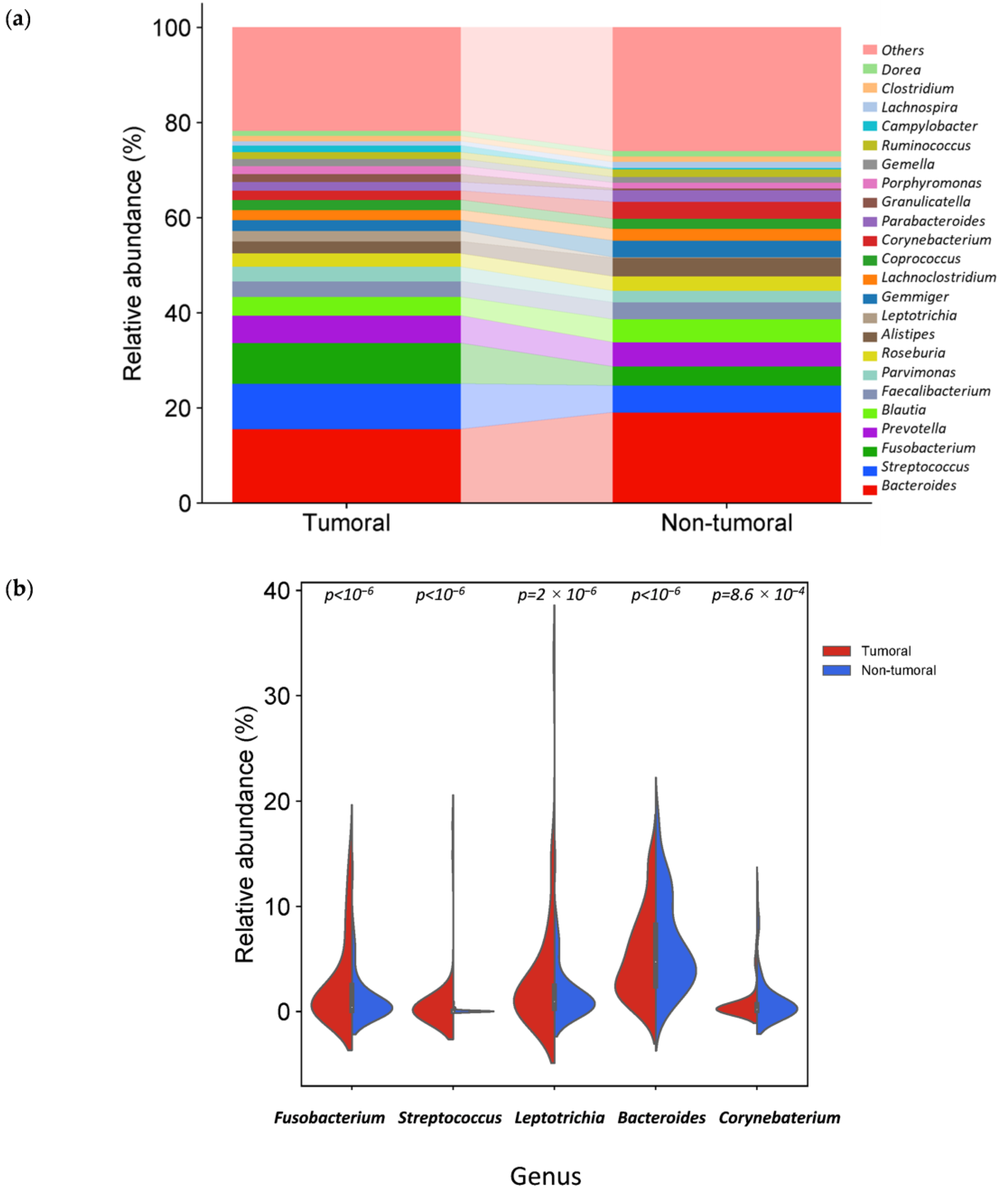
3.3. Specific Microbiota Compositional Differences Between Tumor and Non-Tumor Adjacent Tissue Samples
3.4. Tissue-Associated Microbiota Differences Related to Tumor Location in CRC Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cancer (IARC), T.I.A. for R. on Global Cancer Observatory. Available online: https://gco.iarc.fr/ (accessed on 15 November 2024).
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Jobin, C. Colorectal Cancer: Looking for Answers in the Microbiota. Cancer Discov. 2013, 3, 384–387. [Google Scholar] [CrossRef] [PubMed]
- Rothschild, D.; Weissbrod, O.; Barkan, E.; Kurilshikov, A.; Korem, T.; Zeevi, D.; Costea, P.I.; Godneva, A.; Kalka, I.N.; Bar, N.; et al. Environment Dominates over Host Genetics in Shaping Human Gut Microbiota. Nature 2018, 555, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Janout, V.; Kollárová, H. Epidemiology of Colorectal Cancer. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czechoslov. 2001, 145, 5–10. [Google Scholar] [CrossRef]
- Willett, W.C. Diet and Cancer: An Evolving Picture. JAMA 2005, 293, 233–234. [Google Scholar] [CrossRef]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of Diet in Shaping Gut Microbiota Revealed by a Comparative Study in Children from Europe and Rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef]
- Nakatsu, G.; Andreeva, N.; MacDonald, M.H.; Garrett, W.S. Interactions between Diet and Gut Microbiota in Cancer. Nat. Microbiol. 2024, 9, 1644–1654. [Google Scholar] [CrossRef]
- Casasanta, M.A.; Yoo, C.C.; Udayasuryan, B.; Sanders, B.E.; Umaña, A.; Zhang, Y.; Peng, H.; Duncan, A.J.; Wang, Y.; Li, L.; et al. Fusobacterium nucleatum Host-Cell Binding and Invasion Induces IL-8 and CXCL1 Secretion That Drives Colorectal Cancer Cell Migration. Sci. Signal. 2020, 13, eaba9157. [Google Scholar] [CrossRef]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium nucleatum Promotes Colorectal Carcinogenesis by Modulating E-Cadherin/β-Catenin Signaling via Its FadA Adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef]
- Abdulamir, A.S.; Hafidh, R.R.; Bakar, F.A. Molecular Detection, Quantification, and Isolation of Streptococcus gallolyticus Bacteria Colonizing Colorectal Tumors: Inflammation-Driven Potential of Carcinogenesis via IL-1, COX-2, and IL-8. Mol. Cancer 2010, 9, 249. [Google Scholar] [CrossRef]
- Mazmanian, S.K.; Liu, C.H.; Tzianabos, A.O.; Kasper, D.L. An Immunomodulatory Molecule of Symbiotic Bacteria Directs Maturation of the Host Immune System. Cell 2005, 122, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Long, X.; Wong, C.C.; Tong, L.; Chu, E.S.H.; Ho Szeto, C.; Go, M.Y.Y.; Coker, O.O.; Chan, A.W.H.; Chan, F.K.L.; Sung, J.J.Y.; et al. Peptostreptococcus Anaerobius Promotes Colorectal Carcinogenesis and Modulates Tumour Immunity. Nat. Microbiol. 2019, 4, 2319–2330. [Google Scholar] [CrossRef] [PubMed]
- Drewes, J.L.; Chen, J.; Markham, N.O.; Knippel, R.J.; Domingue, J.C.; Tam, A.J.; Chan, J.L.; Kim, L.; McMann, M.; Stevens, C.; et al. Human Colon Cancer–Derived Clostridioides Difficile Strains Drive Colonic Tumorigenesis in Mice. Cancer Discov. 2022, 12, 1873–1885. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Ling, Z.; Li, L. The Intestinal Microbiota and Colorectal Cancer. Front. Immunol. 2020, 11, 615056. [Google Scholar] [CrossRef]
- Wong, S.H.; Yu, J. Gut Microbiota in Colorectal Cancer: Mechanisms of Action and Clinical Applications. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 690–704. [Google Scholar] [CrossRef]
- González, A.; Fullaondo, A.; Odriozola, A. Microbiota-Associated Mechanisms in Colorectal Cancer. Adv. Genet. 2024, 112, 123–205. [Google Scholar] [CrossRef]
- Sears, C.L.; Pardoll, D.M. Perspective: Alpha-Bugs, Their Microbial Partners, and the Link to Colon Cancer. J. Infect. Dis. 2011, 203, 306–311. [Google Scholar] [CrossRef]
- Tjalsma, H.; Boleij, A.; Marchesi, J.R.; Dutilh, B.E. A Bacterial Driver–Passenger Model for Colorectal Cancer: Beyond the Usual Suspects. Nat. Rev. Microbiol. 2012, 10, 575–582. [Google Scholar] [CrossRef]
- Kiran, N.S.; Yashaswini, C.; Maheshwari, R.; Bhattacharya, S.; Prajapati, B.G. Advances in Precision Medicine Approaches for Colorectal Cancer: From Molecular Profiling to Targeted Therapies. ACS Pharmacol. Transl. Sci. 2024, 7, 967–990. [Google Scholar] [CrossRef]
- da Costa, C.P.; Vieira, P.; Mendes-Rocha, M.; Pereira-Marques, J.; Ferreira, R.M.; Figueiredo, C. The Tissue-Associated Microbiota in Colorectal Cancer: A Systematic Review. Cancers 2022, 14, 3385. [Google Scholar] [CrossRef]
- Taylor, W.S.; Pearson, J.; Miller, A.; Schmeier, S.; Frizelle, F.A.; Purcell, R.V. MinION Sequencing of Colorectal Cancer Tumour Microbiomes—A Comparison with Amplicon-Based and RNA-Sequencing. PLoS ONE 2020, 15, e0233170. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Olsen, H.E.; Paten, B.; Akeson, M. The Oxford Nanopore MinION: Delivery of Nanopore Sequencing to the Genomics Community. Genome Biol. 2016, 17, 239. [Google Scholar] [CrossRef]
- Scholz, M.B.; Lo, C.-C.; Chain, P.S. Next Generation Sequencing and Bioinformatic Bottlenecks: The Current State of Metagenomic Data Analysis. Curr. Opin. Biotechnol. 2012, 23, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Balvočiūtė, M.; Huson, D.H. SILVA, RDP, Greengenes, NCBI and OTT—How Do These Taxonomies Compare? BMC Genom. 2017, 18, 114. [Google Scholar] [CrossRef]
- Stoddard, S.F.; Smith, B.J.; Hein, R.; Roller, B.R.K.; Schmidt, T.M. rrnDB: Improved Tools for Interpreting rRNA Gene Abundance in Bacteria and Archaea and a New Foundation for Future Development. Nucleic Acids Res. 2015, 43, D593–D598. [Google Scholar] [CrossRef]
- Shannon, C.E. A Mathematical Theory of Communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef]
- Simpson, E.H. Measurement of Diversity. Nature 1949, 163, 688. [Google Scholar] [CrossRef]
- Tang, D.; Chen, M.; Huang, X.; Zhang, G.; Zeng, L.; Zhang, G.; Wu, S.; Wang, Y. SRplot: A Free Online Platform for Data Visualization and Graphing. PLoS ONE 2023, 18, e0294236. [Google Scholar] [CrossRef]
- Zhen, J.; Liu, C.; Liao, F.; Zhang, J.; Xie, H.; Tan, C.; Dong, W. The Global Research of Microbiota in Colorectal Cancer Screening: A Bibliometric and Visualization Analysis. Front. Oncol. 2023, 13, 1169369. [Google Scholar] [CrossRef]
- Ternes, D.; Karta, J.; Tsenkova, M.; Wilmes, P.; Haan, S.; Letellier, E. Microbiome in Colorectal Cancer: How to Get from Meta-Omics to Mechanism? Trends Microbiol. 2020, 28, 401–423. [Google Scholar] [CrossRef]
- Allali, I.; Delgado, S.; Marron, P.I.; Astudillo, A.; Yeh, J.J.; Ghazal, H.; Amzazi, S.; Keku, T.; Azcarate-Peril, M.A. Gut Microbiome Compositional and Functional Differences between Tumor and Non-Tumor Adjacent Tissues from Cohorts from the US and Spain. Gut Microbes 2015, 6, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Chung, J.; Cho, M.-L.; Park, D.; Choi, S.S. Analysis of Changes in Microbiome Compositions Related to the Prognosis of Colorectal Cancer Patients Based on Tissue-Derived 16S rRNA Sequences. J. Transl. Med. 2021, 19, 485. [Google Scholar] [CrossRef] [PubMed]
- Cremonesi, E.; Governa, V.; Garzon, J.F.G.; Mele, V.; Amicarella, F.; Muraro, M.G.; Trella, E.; Galati-Fournier, V.; Oertli, D.; Däster, S.R.; et al. Gut Microbiota Modulate T Cell Trafficking into Human Colorectal Cancer. Gut 2018, 67, 1984–1994. [Google Scholar] [CrossRef] [PubMed]
- El Tekle, G.; Andreeva, N.; Garrett, W.S. The Role of the Microbiome in the Etiopathogenesis of Colon Cancer. Annu. Rev. Physiol. 2024, 86, 453–478. [Google Scholar] [CrossRef]
- Wang, N.; Fang, J.-Y. Fusobacterium nucleatum, a Key Pathogenic Factor and Microbial Biomarker for Colorectal Cancer. Trends Microbiol. 2023, 31, 159–172. [Google Scholar] [CrossRef]
- Xu, K.; Jiang, B. Analysis of Mucosa-Associated Microbiota in Colorectal Cancer. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2017, 23, 4422–4430. [Google Scholar] [CrossRef]
- Nakatsu, G.; Li, X.; Zhou, H.; Sheng, J.; Wong, S.H.; Wu, W.K.K.; Ng, S.C.; Tsoi, H.; Dong, Y.; Zhang, N.; et al. Gut Mucosal Microbiome across Stages of Colorectal Carcinogenesis. Nat. Commun. 2015, 6, 8727. [Google Scholar] [CrossRef]
- Fukuoka, H.; Tourlousse, D.M.; Ohashi, A.; Suzuki, S.; Nakagawa, K.; Ozawa, M.; Ishibe, A.; Endo, I.; Sekiguchi, Y. Elucidating Colorectal Cancer-Associated Bacteria through Profiling of Minimally Perturbed Tissue-Associated Microbiota. Front. Cell. Infect. Microbiol. 2023, 13, 1216024. [Google Scholar] [CrossRef]
- Couturier, M.R.; Slechta, E.S.; Goulston, C.; Fisher, M.A.; Hanson, K.E. Leptotrichia Bacteremia in Patients Receiving High-Dose Chemotherapy. J. Clin. Microbiol. 2012, 50, 1228–1232. [Google Scholar] [CrossRef]
- Tee, W.; Midolo, P.; Janssen, P.; Kerr, T.; Dyall-Smith, M. Bacteremia Due to Leptotrichia trevisanii sp. nov. Eur. J. Clin. Microbiol. Infect. Dis. 2001, 20, 765–769. [Google Scholar] [CrossRef]
- Chady, A.; Emmanuel, G.-M.; Antony, S. Leptotrichia trevisanii: Case Report and Review of the Literature on Patients with Leptotrichia trevisanii Bacteremia in Acute Myeloid Leukemia. Infect. Disord. Drug Targets 2023, 23, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Drewes, J.L.; White, J.R.; Dejea, C.M.; Fathi, P.; Iyadorai, T.; Vadivelu, J.; Roslani, A.C.; Wick, E.C.; Mongodin, E.F.; Loke, M.F.; et al. High-Resolution Bacterial 16S rRNA Gene Profile Meta-Analysis and Biofilm Status Reveal Common Colorectal Cancer Consortia. NPJ Biofilms Microbiomes 2017, 3, 34. [Google Scholar] [CrossRef] [PubMed]
- Zeller, G.; Tap, J.; Voigt, A.Y.; Sunagawa, S.; Kultima, J.R.; Costea, P.I.; Amiot, A.; Böhm, J.; Brunetti, F.; Habermann, N.; et al. Potential of Fecal Microbiota for Early-Stage Detection of Colorectal Cancer. Mol. Syst. Biol. 2014, 10, 766. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.A.; Garrett, W.S. Fusobacterium nucleatum—Symbiont, Opportunist and Oncobacterium. Nat. Rev. Microbiol. 2019, 17, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Abed, J.; Maalouf, N.; Manson, A.L.; Earl, A.M.; Parhi, L.; Emgård, J.E.M.; Klutstein, M.; Tayeb, S.; Almogy, G.; Atlan, K.A.; et al. Colon Cancer-Associated Fusobacterium nucleatum May Originate from the Oral Cavity and Reach Colon Tumors via the Circulatory System. Front. Cell. Infect. Microbiol. 2020, 10, 400. [Google Scholar] [CrossRef]
- Mima, K.; Nishihara, R.; Qian, Z.R.; Cao, Y.; Sukawa, Y.; Nowak, J.A.; Yang, J.; Dou, R.; Masugi, Y.; Song, M.; et al. Fusobacterium nucleatum in Colorectal Carcinoma Tissue and Patient Prognosis. Gut 2016, 65, 1973–1980. [Google Scholar] [CrossRef]
- Ganesan, K.; Guo, S.; Fayyaz, S.; Zhang, G.; Xu, B. Targeting Programmed Fusobacterium nucleatum Fap2 for Colorectal Cancer Therapy. Cancers 2019, 11, 1592. [Google Scholar] [CrossRef]
- Zepeda-Rivera, M.; Minot, S.S.; Bouzek, H.; Wu, H.; Blanco-Míguez, A.; Manghi, P.; Jones, D.S.; LaCourse, K.D.; Wu, Y.; McMahon, E.F.; et al. A Distinct Fusobacterium nucleatum Clade Dominates the Colorectal Cancer Niche. Nature 2024, 628, 424–432. [Google Scholar] [CrossRef]
- Tomida, J.; Akiyama-Miyoshi, T.; Tanaka, K.; Hayashi, M.; Kutsuna, R.; Fujiwara, N.; Kawamura, Y. Fusobacterium watanabei sp. nov. As Additional Species within the Genus Fusobacerium, Isolated from Human Clinical Specimens. Anaerobe 2021, 69, 102323. [Google Scholar] [CrossRef]
- Kim, H.-S.; Lee, D.-S.; Chang, Y.-H.; Kim, M.J.; Koh, S.; Kim, J.; Seong, J.-H.; Song, S.K.; Shin, H.S.; Son, J.-B.; et al. Application of rpoB and Zinc Protease Gene for Use in Molecular Discrimination of Fusobacterium nucleatum Subspecies. J. Clin. Microbiol. 2010, 48, 545–553. [Google Scholar] [CrossRef]
- Kook, J.-K.; Park, S.-N.; Lim, Y.K.; Cho, E.; Jo, E.; Roh, H.; Shin, Y.; Paek, J.; Kim, H.-S.; Kim, H.; et al. Genome-Based Reclassification of Fusobacterium nucleatum Subspecies at the Species Level. Curr. Microbiol. 2017, 74, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Komiya, Y.; Shimomura, Y.; Higurashi, T.; Sugi, Y.; Arimoto, J.; Umezawa, S.; Uchiyama, S.; Matsumoto, M.; Nakajima, A. Patients with Colorectal Cancer Have Identical Strains of Fusobacterium nucleatum in Their Colorectal Cancer and Oral Cavity. Gut 2019, 68, 1335–1337. [Google Scholar] [CrossRef] [PubMed]
- Morsi, H.; Golizeh, M.; Brosseau, N.; Janati, A.I.; Emami, E.; Ndao, M.; Tran, S.D. Detection of Fusobacterium nucleatum Subspecies in the Saliva of Pre-Colorectal Cancer Patients, Using Tandem Mass Spectrometry. Arch. Oral Biol. 2022, 134, 105337. [Google Scholar] [CrossRef] [PubMed]
- Borozan, I.; Zaidi, S.H.; Harrison, T.A.; Phipps, A.I.; Zheng, J.; Lee, S.; Trinh, Q.M.; Steinfelder, R.S.; Adams, J.; Banbury, B.L.; et al. Molecular and Pathology Features of Colorectal Tumors and Patient Outcomes Are Associated with Fusobacterium nucleatum and Its Subspecies Animalis. Cancer Epidemiol. Biomark. Prev. 2022, 31, 210–220. [Google Scholar] [CrossRef]
- Ye, X.; Wang, R.; Bhattacharya, R.; Boulbes, D.R.; Fan, F.; Xia, L.; Adoni, H.; Ajami, N.J.; Wong, M.C.; Smith, D.P.; et al. Fusobacterium nucleatum Subspecies Animalis Influences Proinflammatory Cytokine Expression and Monocyte Activation in Human Colorectal Tumors. Cancer Prev. Res. Phila. Pa 2017, 10, 398–409. [Google Scholar] [CrossRef]
- Zhang, H.; Jin, K.; Xiong, K.; Jing, W.; Pang, Z.; Feng, M.; Cheng, X. Disease-Associated Gut Microbiome and Critical Metabolomic Alterations in Patients with Colorectal Cancer. Cancer Med. 2023, 12, 15720–15735. [Google Scholar] [CrossRef]
- Lim, Y.K.; Park, S.-N.; Shin, J.H.; Chang, Y.-H.; Shin, Y.; Paek, J.; Kim, H.; Kook, J.-K. Streptococcus periodonticum sp. nov., Isolated from Human Subgingival Dental Plaque of Periodontitis Lesion. Curr. Microbiol. 2019, 76, 835–841. [Google Scholar] [CrossRef]
- Nagashio, H.; Yoneda, T.; Hoshina, T.; Saito, R.; Kusuhara, K. A Pediatric Case of Previously Unrecognized Streptococcus periodonticum-Associated Meningitis After Cranial Surgery. Pediatr. Infect. Dis. J. 2023, 42, e99. [Google Scholar] [CrossRef]
- Edwards, A.M.; Grossman, T.J.; Rudney, J.D. Fusobacterium nucleatum Transports Noninvasive Streptococcus cristatus into Human Epithelial Cells. Infect. Immun. 2006, 74, 654–662. [Google Scholar] [CrossRef]
- Warren, R.L.; Freeman, D.J.; Pleasance, S.; Watson, P.; Moore, R.A.; Cochrane, K.; Allen-Vercoe, E.; Holt, R.A. Co-Occurrence of Anaerobic Bacteria in Colorectal Carcinomas. Microbiome 2013, 1, 16. [Google Scholar] [CrossRef]
- Zhang, J.; Tao, J.; Gao, R.-N.; Wei, Z.-Y.; He, Y.-S.; Ren, C.-Y.; Li, Q.-C.; Liu, Y.-S.; Wang, K.-W.; Yang, G.; et al. Cytotoxic T-Cell Trafficking Chemokine Profiles Correlate with Defined Mucosal Microbial Communities in Colorectal Cancer. Front. Immunol. 2021, 12, 715559. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Guo, B.; Gao, R.; Zhu, Q.; Qin, H. Microbiota Disbiosis Is Associated with Colorectal Cancer. Front. Microbiol. 2015, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.A.; Zhu, C.; Li, J.; LaComb, J.F.; Denoya, P.I.; Kravets, I.; Miller, J.D.; Yang, J.; Kramer, M.; McCombie, W.R.; et al. Impact of Preoperative Antibiotics and Other Variables on Integrated Microbiome-Host Transcriptomic Data Generated from Colorectal Cancer Resections. World J. Gastroenterol. 2021, 27, 1465–1482. [Google Scholar] [CrossRef] [PubMed]
- Niccolai, E.; Russo, E.; Baldi, S.; Ricci, F.; Nannini, G.; Pedone, M.; Stingo, F.C.; Taddei, A.; Ringressi, M.N.; Bechi, P.; et al. Significant and Conflicting Correlation of IL-9 with Prevotella and Bacteroides in Human Colorectal Cancer. Front. Immunol. 2020, 11, 573158. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ye, J.; Fang, D.; Lv, L.; Wu, W.; Shi, D.; Li, Y.; Yang, L.; Bian, X.; Wu, J.; et al. Multi-Omic Profiling Reveals Associations between the Gut Mucosal Microbiome, the Metabolome, and Host DNA Methylation Associated Gene Expression in Patients with Colorectal Cancer. BMC Microbiol. 2020, 20, 83. [Google Scholar] [CrossRef]
- Shin, N.-R.; Whon, T.W.; Bae, J.-W. Proteobacteria: Microbial Signature of Dysbiosis in Gut Microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef]
- de Carvalho, A.C.; de Mattos Pereira, L.; Datorre, J.G.; Dos Santos, W.; Berardinelli, G.N.; Matsushita, M.d.M.; Oliveira, M.A.; Durães, R.O.; Guimarães, D.P.; Reis, R.M. Microbiota Profile and Impact of Fusobacterium nucleatum in Colorectal Cancer Patients of Barretos Cancer Hospital. Front. Oncol. 2019, 9, 813. [Google Scholar] [CrossRef]
- Loke, M.F.; Chua, E.G.; Gan, H.M.; Thulasi, K.; Wanyiri, J.W.; Thevambiga, I.; Goh, K.L.; Wong, W.F.; Vadivelu, J. Metabolomics and 16S rRNA Sequencing of Human Colorectal Cancers and Adjacent Mucosa. PLoS ONE 2018, 13, e0208584. [Google Scholar] [CrossRef]
- Okuda, S.; Shimada, Y.; Tajima, Y.; Yuza, K.; Hirose, Y.; Ichikawa, H.; Nagahashi, M.; Sakata, J.; Ling, Y.; Miura, N.; et al. Profiling of Host Genetic Alterations and Intra-Tumor Microbiomes in Colorectal Cancer. Comput. Struct. Biotechnol. J. 2021, 19, 3330–3338. [Google Scholar] [CrossRef]
- Zhu, Q.; Jin, Z.; Wu, W.; Gao, R.; Guo, B.; Gao, Z.; Yang, Y.; Qin, H. Analysis of the Intestinal Lumen Microbiota in an Animal Model of Colorectal Cancer. PLoS ONE 2014, 9, e90849. [Google Scholar] [CrossRef]
- Kim, S.; Lee, M.; Kim, N.-Y.; Kwon, Y.-S.; Nam, G.S.; Lee, K.; Kwon, K.M.; Kim, D.K.; Hwang, I.H. Oxidative Tryptamine Dimers from Corynebacterium durum Directly Target Survivin to Induce AIF-Mediated Apoptosis in Cancer Cells. Biomed. Pharmacother. 2024, 173, 116335. [Google Scholar] [CrossRef] [PubMed]
- Cruse, J.P.; Lewin, M.R.; Clark, C.G. Corynebacterium Parvum Enhances Colonic Cancer in Dimethylhydrazine-Treated Rats. Br. J. Cancer 1978, 37, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Wirth, U.; Garzetti, D.; Jochum, L.M.; Spriewald, S.; Kühn, F.; Ilmer, M.; Lee, S.M.L.; Niess, H.; Bazhin, A.V.; Andrassy, J.; et al. Microbiome Analysis from Paired Mucosal and Fecal Samples of a Colorectal Cancer Biobank. Cancers 2020, 12, 3702. [Google Scholar] [CrossRef] [PubMed]
- Debesa-Tur, G.; Pérez-Brocal, V.; Ruiz-Ruiz, S.; Castillejo, A.; Latorre, A.; Soto, J.L.; Moya, A. Metagenomic Analysis of Formalin-Fixed Paraffin-Embedded Tumor and Normal Mucosa Reveals Differences in the Microbiome of Colorectal Cancer Patients. Sci. Rep. 2021, 11, 391. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhang, X.; Xu, H.; Li, S.; Lau, H.C.-H.; Chen, Q.; Zhang, B.; Zhao, L.; Chen, H.; Sung, J.J.-Y.; et al. Microbial Community Heterogeneity Within Colorectal Neoplasia and Its Correlation with Colorectal Carcinogenesis. Gastroenterology 2021, 160, 2395–2408. [Google Scholar] [CrossRef]
- Sheng, Q.-S.; He, K.-X.; Li, J.-J.; Zhong, Z.-F.; Wang, F.-X.; Pan, L.-L.; Lin, J.-J. Comparison of Gut Microbiome in Human Colorectal Cancer in Paired Tumor and Adjacent Normal Tissues. OncoTargets Ther. 2020, 13, 635–646. [Google Scholar] [CrossRef]
- Burns, M.B.; Lynch, J.; Starr, T.K.; Knights, D.; Blekhman, R. Virulence Genes Are a Signature of the Microbiome in the Colorectal Tumor Microenvironment. Genome Med. 2015, 7, 55. [Google Scholar] [CrossRef]
- Leung, P.H.M.; Subramanya, R.; Mou, Q.; Lee, K.T.-W.; Islam, F.; Gopalan, V.; Lu, C.-T.; Lam, A.K.-Y. Characterization of Mucosa-Associated Microbiota in Matched Cancer and Non-Neoplastic Mucosa from Patients with Colorectal Cancer. Front. Microbiol. 2019, 10, 1317. [Google Scholar] [CrossRef]
- Richard, M.L.; Liguori, G.; Lamas, B.; Brandi, G.; da Costa, G.; Hoffmann, T.W.; Pierluigi Di Simone, M.; Calabrese, C.; Poggioli, G.; Langella, P.; et al. Mucosa-Associated Microbiota Dysbiosis in Colitis Associated Cancer. Gut Microbes 2018, 9, 131–142. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The Consensus Molecular Subtypes of Colorectal Cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Tesolato, S.; Ortega-Hernández, A.; Gómez-Garre, D.; Claver, P.; Juan, C.D.; la Serna, S.D.; Paz, M.; Domínguez-Serrano, I.; Dziakova, J.; Rivera, D.; et al. Gut Microbiota Profiles in Feces and Paired Tumor and Non-Tumor Tissues from Colorectal Cancer Patients. Relationship to the Body Mass Index. PLoS ONE 2023, 18, e0292551. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Coker, O.O.; Nakatsu, G.; Wu, W.K.K.; Zhao, L.; Chen, Z.; Chan, F.K.L.; Kristiansen, K.; Sung, J.J.Y.; Wong, S.H.; et al. Multi-Cohort Analysis of Colorectal Cancer Metagenome Identified Altered Bacteria across Populations and Universal Bacterial Markers. Microbiome 2018, 6, 70. [Google Scholar] [CrossRef] [PubMed]
- Valciukiene, J.; Strupas, K.; Poskus, T. Tissue vs. Fecal-Derived Bacterial Dysbiosis in Precancerous Colorectal Lesions: A Systematic Review. Cancers 2023, 15, 1602. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, G.; Chen, J.; Chen, X.; Chia, N.; O’Connor, H.M.; Wolf, P.G.; Gaskins, H.R.; Bharucha, A.E. Relationship Between Microbiota of the Colonic Mucosa vs. Feces and Symptoms, Colonic Transit, and Methane Production in Female Patients with Chronic Constipation. Gastroenterology 2016, 150, 367–379.e1. [Google Scholar] [CrossRef]
- Zoetendal, E.G.; von Wright, A.; Vilpponen-Salmela, T.; Ben-Amor, K.; Akkermans, A.D.L.; de Vos, W.M. Mucosa-Associated Bacteria in the Human Gastrointestinal Tract Are Uniformly Distributed along the Colon and Differ from the Community Recovered from Feces. Appl. Environ. Microbiol. 2002, 68, 3401–3407. [Google Scholar] [CrossRef]
- Chen, W.; Liu, F.; Ling, Z.; Tong, X.; Xiang, C. Human Intestinal Lumen and Mucosa-Associated Microbiota in Patients with Colorectal Cancer. PLoS ONE 2012, 7, e39743. [Google Scholar] [CrossRef]
- Álvarez-Herms, J.; Burtscher, M.; González-Benito, A.; Corbi, F.; Odriozola-Martínez, A. The Gut Microbiota Characterization of a World-Class Mountain Trail Runner during a Complete Competition Season: A Case Report. J. Athl. Train. 2024; ahead of print. [Google Scholar] [CrossRef]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun Metagenomics, from Sampling to Analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef]
- Osman, M.-A.; Neoh, H.; Ab Mutalib, N.-S.; Chin, S.-F.; Jamal, R. 16S rRNA Gene Sequencing for Deciphering the Colorectal Cancer Gut Microbiome: Current Protocols and Workflows. Front. Microbiol. 2018, 9, 767. [Google Scholar] [CrossRef]
- Federhen, S. The NCBI Taxonomy Database. Nucleic Acids Res. 2012, 40, D136–D143. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxa Name | Rank | Mean of Tumor Relative Abundance (%) | Mean of Non-Tumor Relative Abundance (%) | p-Value | Log 2 Fold Change | Occurrence in Tumor (%) | Occurrence in Non-Tumor (%) |
|---|---|---|---|---|---|---|---|
| Fusobacteriota | Phylum | 3.49 | 1.38 | <10−6 | 1.33 | 100 | 100 |
| Bacilli | Class | 4.09 | 2.47 | <10−6 | 0.72 | 100 | 100 |
| Fusobacteriia | Class | 3.48 | 1.39 | <10−6 | 1.33 | 100 | 100 |
| Fusobacteriales | Order | 3.49 | 1.39 | <10−6 | 1.33 | 100 | 100 |
| Lactobacillales | Order | 4.25 | 2.44 | <10−6 | 0.80 | 100 | 100 |
| Fusobacteriaceae | Family | 2.18 | 1.01 | <10−6 | 1.11 | 100 | 100 |
| Leptotrichiaceae | Family | 0.91 | 0.08 | <10−6 | 3.58 | 66.1 | 57.6 |
| Streptococcaceae | Family | 3.17 | 1.89 | <10−6 | 0.75 | 100 | 100 |
| Fusobacterium | Genus | 2.89 | 1.34 | <10−6 | 1.10 | 100 | 100 |
| Leptotrichia | Genus | 0.75 | 0.06 | 2 × 10−6 | 3.59 | 66.1 | 57.6 |
| Streptococcus | Genus | 3.23 | 1.90 | <10−6 | 0.77 | 100 | 100 |
| Fusobacterium nucleatum | Species | 2.34 | 1.03 | <10−6 | 1.19 | 100 | 100 |
| Fusobacterium polymorphum | Species | 0.62 | 0.13 | 5.3 × 10−3 | 2.29 | 76.3 | 72.9 |
| Streptococcus periodonticum | Species | 2.25 | 0.99 | <10−6 | 1.19 | 100 | 100 |
| Taxa Name | Rank | Mean of Tumor Relative Abundance (%) | Mean of Non-Tumor Relative Abundance (%) | p-Value | Log 2 Fold Change | Occurrence in Tumor (%) | Occurrence in Non-Tumor (%) |
|---|---|---|---|---|---|---|---|
| Actinomycetota | Phylum | 4.76 | 7.39 | <10−6 | −0.64 | 100 | 100 |
| Bacteroidota | Phylum | 17.12 | 20.08 | <10−6 | −0.23 | 100 | 100 |
| Pseudomonadota | Phylum | 6.60 | 7.71 | <10−6 | −0.22 | 100 | 100 |
| Actinomycetes | Class | 2.72 | 4.39 | <10−6 | −0.69 | 100 | 98.3 |
| Alphaproteobacteria | Class | 0.50 | 1.31 | <10−6 | −1.40 | 81.4 | 88.1 |
| Bacteroidia | Class | 11.42 | 13.35 | <10−6 | −0.23 | 100 | 100 |
| Betaproteobacteria | Class | 1.72 | 2.66 | <10−6 | −0.62 | 98.3 | 98.3 |
| Clostridia | Class | 18.19 | 20.25 | <10−6 | −0.15 | 100 | 100 |
| Bacteroidales | Order | 11.75 | 13.74 | <10−6 | −0.23 | 100 | 100 |
| Eubacteriales | Order | 14.16 | 15.77 | <10−6 | −0.15 | 100 | 100 |
| Mycobacteriales | Order | 1.08 | 2.03 | <10−6 | −0.92 | 96.6 | 98.3 |
| Bacteroidaceae | Family | 4.36 | 5.32 | <10−6 | −0.29 | 100 | 100 |
| Corynebacteriaceae | Family | 0.64 | 1.19 | 8.6 × 10−4 | −0.89 | 96.6 | 96.6 |
| Lachnospiraceae | Family | 7.48 | 8.46 | <10−6 | −0.18 | 100 | 100 |
| Propionibacteriaceae | Family | 1.20 | 1.85 | 1 × 10−5 | −0.62 | 88.1 | 93.2 |
| Bacteroides | Genus | 5.25 | 6.41 | <10−6 | −0.29 | 100 | 100 |
| Corynebacterium | Genus | 0.64 | 1.19 | 8.6 × 10−4 | −0.89 | 96.6 | 96.6 |
| β-Diversity | ||||||
|---|---|---|---|---|---|---|
| Colon vs. Rectum | Colon vs. Sigmoid Colon | Rectum vs. Sigmoid Colon | ||||
| Metric | ANOSIM | PERMANOVA | ANOSIM | PERMANOVA | ANOSIM | PERMANOVA |
| Bray–Curtis | R = 0.02825 p = 0.0978 | F = 1.821 p = 0.0624 | R = 0.1895 p = 0.054 | F = 1.087 p = 0.335 | R = 0.099 p = 0.1587 | F = 0.7444 p = 0.6527 |
| Jaccard | R = 0.05181 p = 0.0285 | F = 1.34 p = 0.0649 | R = 0.3916 p = 0.0016 | F = 1.422 p = 0.0534 | R = 0.2703 p = 0.0095 | F = 1.131 p = 0.2193 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González, A.; Fullaondo, A.; Navarro, D.; Rodríguez, J.; Tirnauca, C.; Odriozola, A. New Insights into Mucosa-Associated Microbiota in Paired Tumor and Non-Tumor Adjacent Mucosal Tissues in Colorectal Cancer Patients. Cancers 2024, 16, 4008. https://doi.org/10.3390/cancers16234008
González A, Fullaondo A, Navarro D, Rodríguez J, Tirnauca C, Odriozola A. New Insights into Mucosa-Associated Microbiota in Paired Tumor and Non-Tumor Adjacent Mucosal Tissues in Colorectal Cancer Patients. Cancers. 2024; 16(23):4008. https://doi.org/10.3390/cancers16234008
Chicago/Turabian StyleGonzález, Adriana, Asier Fullaondo, David Navarro, Javier Rodríguez, Cristina Tirnauca, and Adrian Odriozola. 2024. "New Insights into Mucosa-Associated Microbiota in Paired Tumor and Non-Tumor Adjacent Mucosal Tissues in Colorectal Cancer Patients" Cancers 16, no. 23: 4008. https://doi.org/10.3390/cancers16234008
APA StyleGonzález, A., Fullaondo, A., Navarro, D., Rodríguez, J., Tirnauca, C., & Odriozola, A. (2024). New Insights into Mucosa-Associated Microbiota in Paired Tumor and Non-Tumor Adjacent Mucosal Tissues in Colorectal Cancer Patients. Cancers, 16(23), 4008. https://doi.org/10.3390/cancers16234008