Simple Summary
Conjunctival melanoma (CM) is an aggressive cancer with an unmet need for prognostic biomarkers and more effective treatments. In recent years, a rapid increase in available epigenetic technologies and epigenetic modulation-based treatment options has led to the development/implementation of various epi-drugs in the cancer field. Like other cancers, CM occurrence and prognosis are also believed to be influenced by multiple genetic and epigenetic factors. While the genetic understanding of CM has been significantly improved in recent decades, the epigenetic understanding of CM remains limited. A relatively small number of CM epigenetics studies published to date have revealed some potential biomarkers and/or therapeutic targets; however, their results warrant replication in independent and larger studies/samples. Furthermore, not all epigenetic aspects of CM have been investigated in published studies; hence, an in-depth understanding of CM epigenetics remains largely incomplete. Additional studies are therefore urgently needed to advance our epigenetic understanding of CM and improve clinical outcomes by taking advantage of new therapy options driven by epigenetic knowledge.
Abstract
The purpose of this article is to provide a literature review of the epigenetic understanding of conjunctival melanoma (CM), with a primary focus on current gaps in knowledge and future directions in research. CM is a rare aggressive cancer that predominantly affects older adults. Local recurrences and distant metastases commonly occur in CM patients; however, their prediction and management remain challenging. Hence, there is currently an unmet need for useful biomarkers and more effective treatments to improve the clinical outcomes of these patients. Like other cancers, CM occurrence and prognosis are believed to be influenced by multiple genetic and epigenetic factors that contribute to tumor development/progression/recurrence/spread, immune evasion, and primary/acquired resistance to therapies. Epigenetic alterations may involve changes in chromatin conformation/accessibility, post-translational histone modifications or the use of histone variants, changes in DNA methylation, alterations in levels/functions of short (small) or long non-coding RNAs (ncRNAs), or RNA modifications. While recent years have witnessed a rapid increase in available epigenetic technologies and epigenetic modulation-based treatment options, which has enabled the development/implementation of various epi-drugs in the cancer field, the epigenetic understanding of CM remains limited due to a relatively small number of epigenetic studies published to date. These studies primarily investigated DNA methylation, ncRNA (e.g., miRNA or circRNA) expression, or RNA methylation. While these initial epigenetic investigations have revealed some potential biomarkers and/or therapeutic targets, they had various limitations, and their findings warrant replication in independent and larger studies/samples. In summary, an in-depth understanding of CM epigenetics remains largely incomplete but essential for advancing our molecular knowledge and improving clinical management/outcomes of this aggressive disease.
1. Background Information on Conjunctival Melanoma
Conjunctival melanoma (CM) is a rare cancer occurring on the surface of the eye. CM arises from melanocytes located in the basal layer of the conjunctival epithelium [1] and most commonly occurs in the bulbar conjunctiva [2], which is the part of the conjunctiva that is directly exposed to sunlight [3]. The patients affected by this cancer are predominantly of European descent and >50 years of age [4,5]. While some studies reported more common occurrence in men, others did not find significant sex-related differences [5,6,7,8,9,10]. CM is rare with incidence rate estimates ranging from 0.23 to 1.11 cases per million persons per year in Western countries [5,8]. While earlier studies (using data from before 2008) suggested a rising incidence of CM, more recent studies do not suggest the continuation of that trend, although geographical differences may exist [5,6,7,8,10,11]. Overall, the CM cases account for about 5% of all ocular melanoma cases and about 0.25% of all melanoma cases [12,13,14]. CM represents a biologically distinct type of melanoma that bears certain similarities to both sun-exposed cutaneous melanomas and other (non-sun-exposed) mucosal melanomas, as supported by the genetic changes that overlap with those seen in either (or both) of these two major melanoma types [15,16].
There are two known precursor conditions, conjunctival nevus and primary acquired melanosis (PAM) with atypia, which can lead to the development of CM. While PAM with atypia is responsible for up to about 75% of CM cases, the remaining arise from conjunctival nevi or de novo [2,14,17,18]. Conjunctival nevi are benign lesions that uncommonly increase the risk of developing CM [2,14,17,18]. De novo cases account for about 20% of all CM cases and are usually associated with less favorable outcomes [2,14,17,18].
CM is initially diagnosed by clinical examination and is subsequently confirmed by excisional biopsy and histopathological examination [19]. The current standard of care for CM involves local excision followed by adjuvant therapy such as focal cryotherapy, topical chemotherapy, or brachytherapy [1,19]. Advanced CMs may require orbital exenteration, which is an extensive and disfiguring surgical procedure [1,19]. CM is very prone to local spread/recurrence and distant metastasis through mainly the lymphatic but also vascular system [14]. Currently, 10 years after the initial CM diagnosis, the local recurrence rate is up to about 50% and the mortality rate is up to around 30% [20,21]. As suggested by these rates, a better understanding of CM development and progression (i.e., underlying molecular mechanisms) is warranted to inform future prevention and management strategies and help to improve clinical outcomes.
CM occurs sporadically, and like other sporadic cancers, its initiation and progression are believed to be influenced by multiple somatic genetic and epigenetic alterations. Similar to cutaneous and other mucosal melanomas, CM is characterized by overactive mitogen-activated protein kinase (MAPK) signaling (also known as RAS-RAF-MEK-ERK signaling) and/or phosphatidylinositol 3-kinase (P13K)-AKT-mTOR signaling pathway(s) [14,16,22,23]. These two biological pathways are responsible for transmitting the growth signals to the nucleus and altering the expression of multiple genes involved in cellular differentiation, proliferation, and survival [14,23]. The well-known CM-related genes involved in the MAPK and/or P13K-AKT-mTOR pathway(s) include the proto-oncogenes BRAF, NRAS, and KIT, and the tumor-suppressor genes NF1 and PTEN (reviewed recently by Chang et al. [16]). In addition, the genes involved in telomere maintenance, including TERT and ATRX, are believed to play important roles in CM pathogenesis [16]. Maintenance of telomere length through TERT activation or ATRX loss helps cancer cells achieve immortality by preventing progressive telomere shortening in successive cell divisions. Furthermore, ATRX is known to contribute to epigenetic regulation, and several other newly implicated genes (pending confirmation in additional CM studies) also appear to play important roles in epigenetic mechanisms [16].
Genetic alterations observed in CMs include somatic mutations affecting the abovementioned genes/pathways as well as structural variations occurring throughout the genome. The UV mutational signature (often associated with high tumor mutational burden) is also frequently observed in CMs, especially if/when subjected to DNA damage caused by chronic sun exposure [16]. All these improvements in our understanding of CM genetics have opened the door to new systemic therapies for better CM management, such as targeted therapies for tumors with specific oncogenic mutations (e.g., BRAF/MEK inhibitors for tumors with BRAF mutations) and immunotherapies (e.g., for tumors with high mutational burden) [9,14,24,25,26]. Recent years have witnessed a rapid increase in available epigenetic technologies and epigenetic modulation-based treatment options, which has enabled the development/implementation of various epi-drugs with promising results in cancer therapies. In order to also take advantage of these novel therapeutic approaches for better CM management, an in-depth understanding of CM epigenetics is urgently needed but currently limited, as summarized in this review.
2. Background Information on Epigenetic Regulation Mechanisms
Epigenetics refers to the molecular mechanisms that regulate gene expression without altering the DNA sequence. Epigenetic regulation can occur either at the transcriptional or post-transcriptional level (Figure 1 and Figure 2), through alterations in chromatin conformation/accessibility, post-translational histone modifications or the use of histone variants, changes in DNA methylation, alterations in levels/functions of short (small) or long non-coding RNAs, or RNA modifications [27,28,29,30,31]. Various interactions, either synergistic or antagonistic, can also occur among these different epigenetic mechanisms [32,33,34,35,36]. Physiologically, these mechanisms are necessary for healthy development, maintenance of cell type/tissue-specific gene expression, and adaptive responses to internal and external changes/stimuli. In addition to gene expression regulation, epigenetic mechanisms are also important for DNA repair and replication [28,29,31,37,38,39]. Accordingly, accumulating evidence supports a major role of epigenetic alterations in tumor development/progression/recurrence/spread, tumor–immune system interactions, and response/resistance to cancer treatments [27,28,29,30,31,40,41,42,43,44,45]. Indeed, epigenetic reprogramming constitutes one of the hallmarks of sporadic cancer and is believed to be driven/influenced by accumulating genetic and epigenetic alterations and environmental exposures [46,47]. Due to their reversible and usually dynamic nature, epigenetic alterations represent attractive targets for new therapeutic options [27,30,45,48].
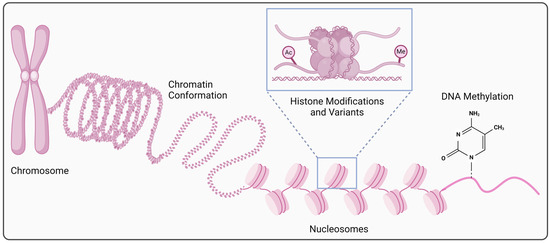
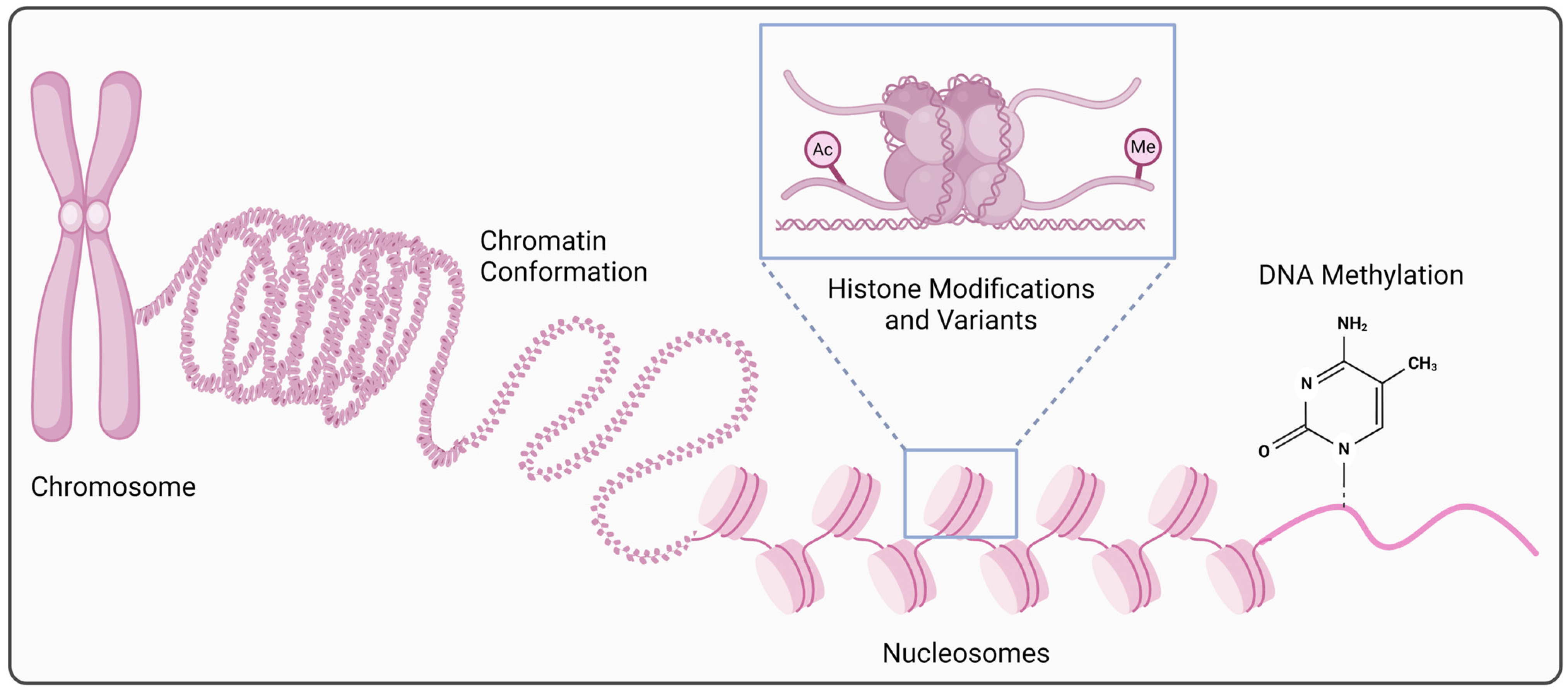
Figure 1.
Epigenetic regulation at the transcriptional level. Alterations in chromatin structure and organization (conformation/accessibility), post-translational histone modifications (acetylation, methylation, etc.) or the use of histone variants, and changes in DNA methylation are involved in the cell type-, time-, or context-specific gene expression regulation at the transcriptional level (created in BioRender. https://BioRender.com/x40t223, accessed on 22 October 2024).
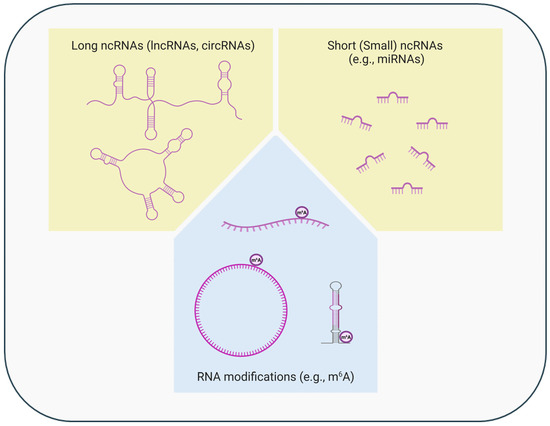
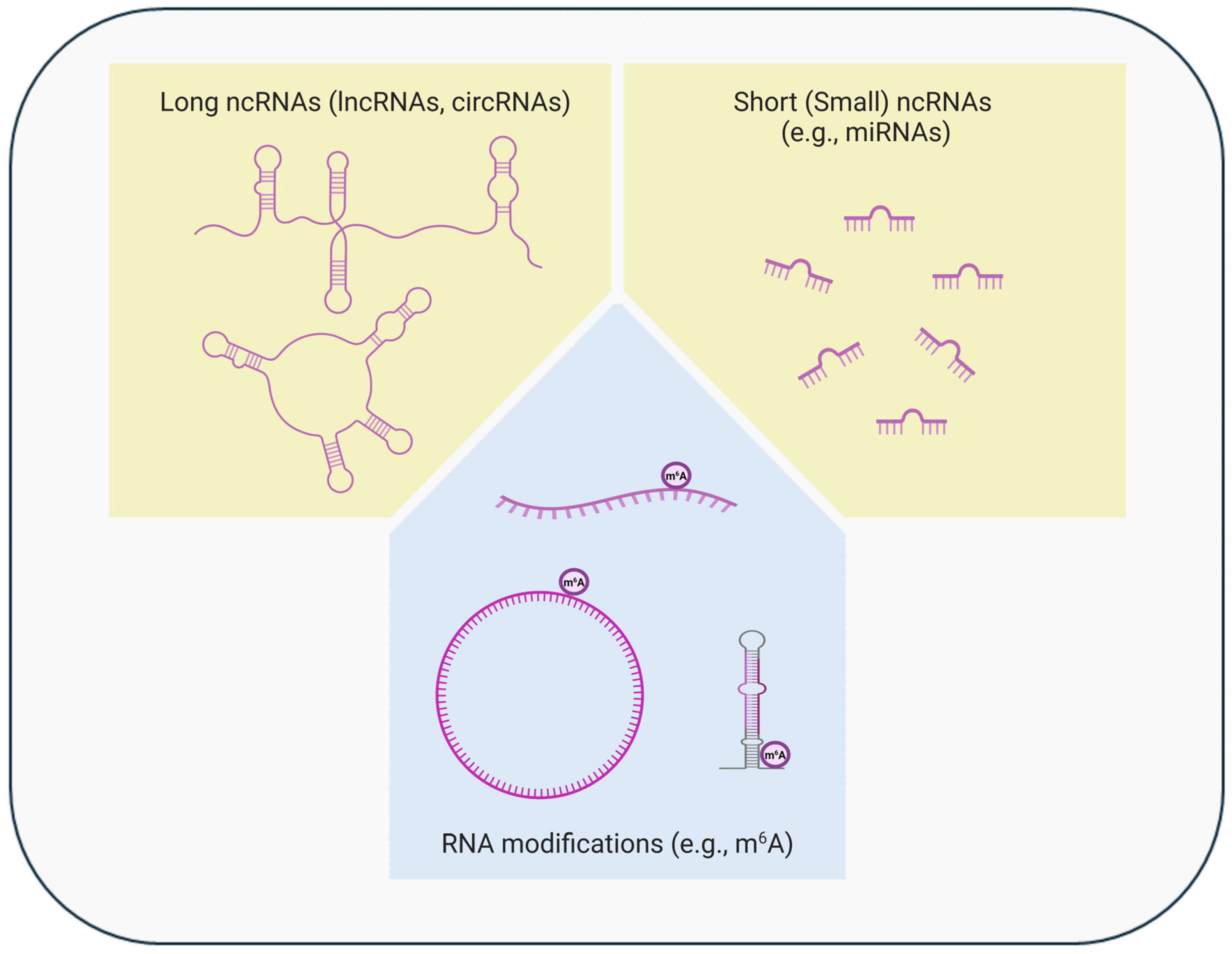
Figure 2.
Epigenetic regulation at the post-transcriptional level. Alterations in levels/functions of long or short (small) non-coding RNAs (ncRNAs) and chemical RNA modifications (e.g., methylation) are involved in the cell type-, time-, or context-specific gene expression regulation at the post-transcriptional level (created in BioRender. https://BioRender.com/x92h200, accessed on 22 October 2024). The ncRNAs, especially the long ncRNAs and their modifications, can regulate gene expression also at the transcriptional level.
2.1. Chromatin Conformation and Accessibility
Genome activity is mediated by changes in the structure and conformation (spatial organization) of the chromatin, which is composed of nucleic acids and associated proteins [28]. Chromatin accessibility (closed/repressive, permissive/bivalent, or open/active chromatin) is regulated by the enzymes that are involved in DNA methylation or histone modifications, the proteins that can recognize these chemical modifications, and ATP-dependent remodeling complexes that can mediate the swapping of histones/variant histones into/out of nucleosomes or the (re)arrangement (sliding/repositioning or eviction) of nucleosomes [30,37,49,50,51]. All these chromatin modifiers (writers, erasers, and readers) play important roles in epigenetic regulation at the transcriptional level (Figure 1) and are often found mutated/altered in cancers [52,53].
2.2. Histone Modifications and Variants
Nucleosomes constitute the basic and repeating units of chromatin structure and organization (Figure 1). Each nucleosome is comprised of eight histone proteins (two copies of four core histones) and ~150 base pairs of the DNA double-wrapped around them [30,50,51]. Post-translational histone modifications can influence chromatin accessibility by affecting chromatin’s structure and compaction, leading to changes in its transcriptional state. These modifications (acetylation, methylation, as well as other alterations at specific amino acidic residues) predominantly occur at the N-terminal tail of the histones and can lead to either activation or repression of transcription by enabling or preventing the access of regulatory proteins to DNA elements [27,28,30,50,51]. Major enzyme groups responsible for the dynamic regulation of these modifications include “histone acetyltransferases (HATs), histone deacetylases (HDACs), histone methyltransferases (HMTs), and histone demethylases (HDMs)” [27,28,30,50,51]. Alterations in expression/function of these enzymes are often observed in cancers [52,54,55]. Histone variants, which differ from their canonical counterparts in their protein sequences, can replace the core histones in nucleosomes in specific genomic regions and contribute to genome integrity/stability [56,57,58].
2.3. DNA Methylation
DNA methylation typically involves the modification of cytosine (C) of Cytosine-phosphate-Guanine (CpG) dinucleotides by the addition of a methyl group to the 5-carbon position in the pyrimidine ring [5-methylcytosine (5-mC)] (Figure 1) [29,32,59]. DNA methylation is a repressive epigenetic mark as it leads to gene expression inhibition by attracting specific methyl-CpG-binding proteins for chromatin reconfiguration and preventing the transcription factors and other regulatory molecules from accessing the DNA [30,32]. Thus, “DNA methyltransferases (DNMTs) and methyl-binding domain (MBD) proteins” play major roles in transcriptional repression. While the majority of CpG sites are generally methylated in the human genome, the CpG islands (CpG-rich and mostly hypomethylated DNA sequences) are found in about 60–70% of gene promoters, of which the methylation state determines the gene activity status [29,59,60]. DNA methylation is usually a stable epigenetic mark as it leads to long-term gene silencing as related to its critical role in embryonic development, cell-type/tissue-specific gene regulation, genomic imprinting, X chromosome inactivation, and transposable elements silencing [30,32]. DNA demethylation can occur either passively (due to the failure of DNMT1 to maintain methylation through DNA replication) or actively (by removal of the methyl group from 5-mC through a chain of reactions catalyzed by TET proteins) [31,50,61,62,63]. An intermediary molecule generated during this chain of reactions is 5-hydroxymethylcytosine (5-hmC), the loss of which is implicated in cancer biology along with the mutations/dysregulation of TET genes/proteins [31,50,61,62,63]. Alterations in DNA methylation states are frequently observed in cancers including the focal promoter hypermethylation leading to tumor suppressor gene inactivation and the global hypomethylation leading to chromosomal instability, proto-oncogene activation, and drug resistance [29,59,64].
2.4. Non-Coding RNAs
Non-coding RNAs (ncRNAs) have mainly regulatory functions and are divided into two major groups: “(i) short (small) ncRNAs (sncRNAs) [microRNAs (miRNAs), short interfering RNAs (siRNAs), and P-element-induced wimpy testis (PIWI)-interacting RNAs (piRNAs)] and (ii) long ncRNAs (lncRNAs) [linear lncRNAs and circular RNAs (circRNAs)]” (Figure 2) [50,59,65,66,67,68].
Among the sncRNAs, the most studied are the miRNAs (~18 to 25 nucleotides in length), which are typically known to downregulate gene expression post-transcriptionally via degradation of their target mRNAs or repression of their translation [65,67]. A miRNA can target different mRNAs by recognizing and binding to their 3′ untranslated region (UTR), and the same mRNA can be targeted by different miRNAs [67]. Aberrantly expressed miRNAs can function as either onco-suppressors or oncomiRs when involved in oncogenesis [27,67]. MiRNA-mediated repression of tumor suppressor transcripts or lack of miRNA-mediated degradation of oncogenic transcripts can play active roles in cancers [27,50,65,67].
LncRNAs (>200 nucleotides in length) represent a large group of transcripts that can interact with various types of molecules (DNA, other RNAs, and proteins) and can cis/trans-regulate gene expression at the transcriptional and/or post-transcriptional level [27,29,65,69]. LncRNAs can also contribute to miRNA biogenesis or function as miRNA sponges [28,29,65]. As part of their involvement in cancer, dysregulated lncRNAs can act as tumor-suppressive or oncogenic molecules depending on their functions and interactions [65,67,70,71]. Non-coding circRNAs are covalently closed lncRNAs (their 5′ and 3′ ends are covalently linked by back-splicing), and they are involved in multiple biological processes similar to their linear counterparts; however, they are usually more stable due to their circular structure [28,65,67].
2.5. RNA Modifications
Both coding and non-coding RNAs can undergo diverse post-transcriptional chemical modifications; one of the most abundant being the methylation of adenosine residue at the N-6 position [N6-methyladenosine (m6A)], which can occur in 5′ and 3′ UTRs and internal long exons [30,51,72,73,74,75]. This modification can influence RNA processing/structure/function including its splicing, polyadenylation, stability, interactions, nuclear export, and translation [28,30,72,73]. The m6A modification is generated by N6-methyltransferases (the METTL3-METTL14 complex), removed by demethylases (ALKBH5 or FTO enzymes), and recognized by m6A-binding proteins [30,51,73]. Being studied increasingly, misregulated RNA modifiers (writers, erasers, and readers) and aberrant RNA modification profiles are emerging as important players in cancer biology (initiation, progression, spread, immune evasion, and resistance to therapy) [28,30,51,72,74,76,77,78].
3. Brief Overview of Melanoma Epigenetics
Epigenetic alterations are common in melanomas and can be influenced by both genetic and non-genetic factors affecting the tumor cells or their microenvironment. These alterations can contribute to melanoma initiation, progression, recurrence, and metastatic potential as well as the tumor’s interactions with the immune system and response to therapy [27,29,59,79]. Most of the current knowledge on melanoma-associated epigenetic alterations comes from the studies of cutaneous melanoma, as it represents the most common melanoma type. While various epigenetic mechanisms have increasingly been studied in melanomas, DNA methylation and miRNA expression are among the most frequently investigated.
Chromatin structure/conformation can be influenced by changes in levels/functions of the components of chromatin remodeling complexes, and altered chromatin remodeling can play an important role in melanomagenesis [29,59], as exemplified by the frequently dysregulated activity of the Switch/sucrose-non-fermentable (SWI/SNF) complexes. The components of these complexes are involved in DNA transcription, replication, repair, and genomic stability [80,81]. Of those components, the ones commonly altered in melanomas include the members of the ARID and SMARC protein families [80,81].
Aberrant patterns of histone modifications (e.g., hypoacetylation and hypermethylation) play active roles in melanomagenesis, mainly by silencing the tumor suppressor genes and leading to the dysregulation of signaling pathways, cell cycle progression, and apoptosis [29,59]. Consistent with these findings, altered levels/functions of histone modifier enzymes (e.g., various HDACs, HMTs, and HDMs) are often observed in melanomas [27,29,41,79,82]. Altered histone modifications can also influence the expression of genes that contribute to tumor immunogenicity and/or anti-tumor immunity within the tumor microenvironment [79]. Aberrant expression of histone variants, such as loss of macroH2A or overexpression of H2A.Z.2, have also been reported in correlation with melanoma progression as a result of cell cycle dysregulation and increased metastatic potential [29,59,83].
DNA methylation aberrations have frequently been studied in melanomas for their pathogenetic or prognostic influences. Focal hypermethylation of specific tumor suppressor gene promoters (e.g., PTEN, CDKN2A, and APC promoters) is commonly seen in melanomas, resulting in dysregulated intracellular signaling, cell cycle progression, apoptosis, and DNA repair [27,29,41,59]. In addition to known tumor suppressor genes, multiple other genes involved in cell differentiation/survival/growth or tumor immunogenicity and anti-tumor immunity have also been found aberrantly methylated in melanomas [29,59,79]. Some melanomas can exhibit a “CpG island methylator phenotype”, which is characterized by widespread CpG island hypermethylation associated with worse clinical outcomes [29,59,84,85]. Melanomas can also often show partial/complete loss of 5-hmC, which usually results from the deficiency of enzymes/cofactors (e.g., TETs) involved in active DNA demethylation [31,86,87]. Furthermore, melanomas can commonly exhibit global DNA hypomethylation, which can lead to genomic instability and oncogene activation [27,31].
Aberrant expression of miRNAs and lncRNAs (linear lncRNAs or circRNAs) has been increasingly studied in recent decades as related to melanoma pathogenesis and prognosis [27,41,59,88,89]. Multiple miRNAs have been found dysregulated in melanomas, leading to oncogenic, anti-apoptotic, and/or pro-metastatic properties, or influencing immune evasion and/or resistance to therapy [27,41,88,89]. Likewise, several lncRNAs have been found dysregulated in melanomas (e.g., HOTAIR and MALAT1), leading to oncogenic, anti-apoptotic, and/or pro-metastatic properties, or influencing immune evasion and/or drug resistance [27,41,88,89,90]. Melanoma-associated ncRNAs have therefore increasingly been investigated as potential biomarkers or new therapeutic targets [27,41].
Aberrant m6A RNA methylation has also been increasingly recognized for its involvement in melanoma, by influencing disease development/progression/metastasis, tumor immunogenicity or functions of tumor-infiltrating immune cells, and/or response to therapy [41,79,91]. Consistent with these observations, m6A-related modulators (writers, erasers, and readers) are frequently found altered in melanomas [91,92].
4. Current Knowledge of Conjunctival Melanoma Epigenetics
Only a handful of studies have been published to date concerning the epigenetic analyses of primary CMs, and they have mainly focused on DNA methylation [93,94], ncRNA expression (miRNAs or circRNAs) [95,96,97,98], or RNA methylation [99,100,101]. While chromatin conformation and histone modification analyses of CMs are currently lacking, alterations of the genes involved in the regulation of these processes have increasingly been implicated in CM, including altered chromatin remodelers (ATRX and ARID2) and altered histone modifiers (some HDAC, SETD, and KMT genes) [16].
As stated earlier and reviewed recently [16], CMs show genetic similarities to cutaneous and other mucosal melanomas, and the initial results from published CM epigenetics studies (reviewed below) similarly suggest an overlap between epigenetic alterations observed in CM and those reported in cutaneous and other mucosal melanomas.
4.1. DNA Methylation Studies of CM
A recent study by Jurmeister et al. [93] investigated the DNA methylation landscape of 107 melanomas originating from different primary sites (25 cutaneous and 82 non-cutaneous melanomas including 9 CMs) along with their genome-wide copy number analyses. Their results have revealed that conjunctival and other mucosal melanomas share a common global DNA methylation profile with cutaneous melanomas; however, the primary tumor sites differ in promotor methylation status and copy number changes of cancer-related genes. While the promoter methylation of tumor suppressor APC was the most common in CMs (6/9; 67%) compared to cutaneous (7/25; 28%) and other mucosal (8/63; 13%) melanomas, the promoter methylation of tumor suppressor CDKN2A was commonly present in both CMs (4/9; 44%) and cutaneous melanomas (9/25; 36%) but less commonly in other mucosal melanomas (6/63; 10%). The promoter methylation rate was comparable across these melanoma types for tumor suppressors WIF1 (33–57%) and RASSF1 (44–51%); however, it was relatively lower in CMs (11%) compared to cutaneous and other mucosal melanomas (24–33%) for tumor suppressor PTEN. The CDKN2A and PTEN tumor-suppressor genes were found to be inactivated in melanomas by either promoter methylation or focal deletion, the two events occurring in a mutually exclusive manner [93].
Given the molecular similarities between cutaneous and conjunctival melanomas, Stahl et al. [94] investigated whether CMs show a global 5-hydroxymethylcytosine (5-hmC) loss compared to the nevi, similar to that previously reported in cutaneous melanomas [86,87]. For this purpose, the authors performed immunohistochemistry and RNA in situ hybridization (RNA ISH) in 37 CMs and 40 conjunctival nevi to assess the expression of 5-mC, 5-hmC, and TET2 (a methylcytosine dioxygenase that catalyzes the conversion of 5-mC to 5-hmC). The loss of or reduction in 5-hmC has previously been linked to TET inactivation or downregulation [87]. Stahl et al. [94] were able to detect 5-hmC and TET2 in 54% and 35.1% of CMs and in 100% and 95% of conjunctival nevi, respectively, suggesting a significant loss of 5-hmC and TET2 occurring in CMs similar to cutaneous melanomas. While the levels/functions of TET proteins can be influenced by various factors, TET2 mutations were identified in a subset of CMs analyzed by comprehensive genetic studies [15,102]. In addition, mutations in IDH genes, which can influence the functions of TET proteins [87,103], were also found in a subset of comprehensively analyzed CMs [15,104,105].
4.2. MicroRNA Expression Studies of CM
MicroRNAs are among the most studied epigenetic regulators in CM, investigated in three studies published to date [95,96,97]. In an initial 2016 study, Larsen et al. [97] performed a “microarray-based miRNA expression profiling” (for 2578 mature miRNAs) to compare archived [formalin-fixed, paraffin-embedded (FFPE)] CM samples (n = 37) with normal conjunctiva samples (n = 7). In addition, the authors tested the association of miRNA expression patterns with TNM stage, local recurrence, and distant metastases in CM. Their study identified 25 dysregulated (24 upregulated and 1 downregulated) miRNAs in CM compared to normal conjunctiva, including many miRNAs with known roles in cutaneous melanoma (most noteworthy being miR-20b-5p, miR-146a-5p, miR-146b-5p, miR-506-3p, and miR-509-3p). Furthermore, the authors were able to link seven upregulated miRNAs to the T stage and increased tumor thickness, and two upregulated miRNAs (miR-3687 and miR-3916) to local recurrence risk, but found no link to metastasis risk or mortality in CM. Moreover, when they analyzed/compared fresh frozen samples of CM (n = 6) with those of head and neck mucosal melanomas (n = 4), they found a resemblance in miRNA expression patterns. The authors thus concluded that CMs show a similar pattern of differentially expressed miRNAs compared to other melanomas, and some of the dysregulated miRNAs could potentially function as prognostic biomarkers or future therapeutic targets. In three patients, the authors were also able to compare fresh frozen and archived samples obtained from the same CMs and observe a good correlation in miRNA results, suggesting that archived (FFPE) samples could provide reliable results for miRNA expression analysis.
In a later study published by the same research group, Mikkelsen et al. [96] performed a “microarray-based miRNA expression profiling” of primary CMs with or without subsequent metastases to identify prognostic miRNAs associated with metastatic spread. The study analyzed archived (FFPE) samples from 25 patients with nonmetastatic CM and 13 patients with metastatic CM. When the authors compared primary CMs (with or without subsequent metastases) with normal conjunctiva, they identified several hundred differentially expressed miRNAs, including two (miR-509-3p and miR-181b-5p) similarly found upregulated in their previous study [97]. Pathway analysis of these two miRNAs linked them to Hippo and p53 signaling pathways as relevant to tumorigenesis. When the authors compared primary CMs with or without subsequent metastases, they discovered 15 differentially expressed miRNAs, including 9 upregulated and 6 downregulated in primary CMs with subsequent metastases. Of these 15 miRNAs associated with metastasis risk in CM, 4 (mir-575, mir-622, miR-1270, and miR-1290) were previously linked to other cancers. Furthermore, when the authors compared primary CMs with their pair-matched metastases in 13 patients, they detected 6 differentially expressed miRNAs, including 2 upregulated (miR-1246 and miR-302d-5p) and 4 downregulated (mir-6084, miR-184, mir-658, and mir-4427) in distant metastases, 3 of which (miR-1246, miR-184, and mir-658) were previously linked to other cancers. Using quantitative PCR (qPCR), the authors were able to confirm the downregulation of miR-184 in metastatic vs. primary tumors from the same patients and nominate this extracellular matrix (ECM)-interaction pathway-related miRNA as a potential candidate for therapy. Pathway analysis of miRNAs dysregulated either in metastasis-prone primary CMs or in metastatic lesions linked these miRNAs to “cancers, cell growth and death, signal transduction, metabolism, and immune system”. While acknowledging their small sample size due to the rarity of CM, the authors concluded that the primary CMs with or without subsequent metastases show distinct global miRNA expression profiles, but larger studies are necessary to characterize the specific set of miRNAs predictive of metastatic potential in CM. Because of the poor correlation between the microarray and qPCR results, however, the authors could not recommend one method or another for future miRNA expression studies of CM.
In a more recent study from another research group, van Ipenburg et al. [95] used “TaqMan Low-Density Array Card A for miRNA expression profiling” to compare benign vs. malignant conjunctival melanocytic lesions, as well as the primary CMs with vs. without metastases, in order to identify the discriminating miRNAs between these study groups. Their study included a discovery cohort of 6 conjunctival nevi and 20 CMs and a replication cohort of 13 conjunctival nevi and 19 CMs. Both study cohorts were comprised of archived (FFPE) samples, and the study analysis primarily focused on miRNAs upregulated in CMs. While the authors found no miRNA expression differences between the primary CMs with vs. without metastases, they were able to identify and replicate a set of five miRNAs upregulated in primary CMs compared to the nevi, including “miR-9-5p, miR-196b-5p, miR-450a-5p, miR-501-5p, and miR-615-3p”. Three of these miRNAs (miR-196b-5p, miR-615-3p, and miR-9-5p) represented the best-performing miRNAs possibly involved in a shared pathway of homeobox gene clusters. The authors concluded that while further studies are needed to identify the miRNAs that could predict the metastatic potential of CM, their results suggest a potential benefit of miRNA profiling to discriminate benign vs. malignant lesions, especially in situations where the limited tissue availability can make such distinction difficult using routine methods.
4.3. CircRNA Expression Studies of CM
In a single study published to date on the role of circRNAs in CM, Shang et al. [98] performed RNA-seq to evaluate the expression of circRNAs in three CM samples compared to the paired adjacent normal tissues, and they identified a large number of potentially CM-relevant circRNA candidates. Upon further investigation with functional analyses, the authors characterized CircMTUS1 (a circRNA derived from exons 2-3 of MTUS1) as an oncogenic circRNA upregulated in CM. CircMTUS1 appeared to function as a miRNA sponge for miR-622 and miR-1208 to promote tumorigenesis by modulating several tumor-associated biological pathways (such as ErbB, MAPK, and Wnt signaling pathways). Both the host gene of this circRNA (MTUS1) and miR-622 targeted by this circRNA are known to function as tumor suppressors in several cancers [106,107].
4.4. RNA Modification Studies of CM
Two recent studies [100,101] investigated the role of RNA methylation (m6A) in ocular melanomas by analyzing intraocular (uveal) and ocular surface (conjunctival) melanomas together. After evaluating 88 ocular melanoma and 28 normal melanocyte tissues using various analyses (including m6A assay in a subset; n = 14), Jia et al. [101] detected decreased levels of global m6A (associated with tumor progression) and reduced methylation of tumor-suppressor HINT2 mRNA (hampering its translation efficiency) in ocular melanomas. Decreased global m6A levels appeared to be mediated by downregulation of m6A ‘writer’ METTL3 and upregulation of m6A ‘eraser’ ALKBH5, both associated with poor prognosis. The authors demonstrated that, in normal conditions, the methylation of HINT2 mRNA facilitated its recognition and promotion for translation by YTHDF1, an m6A ‘reader’ protein with both m6A-containing RNA and ribosome binding activities. This observation provided a mechanistic insight into the role of m6A modification in tumorigenesis as a modulator of translation. In a later study that examined 47 ocular melanoma and 27 normal melanocyte tissues, He et al. [100] demonstrated the upregulation of BACE2 mRNA, which appeared to be mediated by increased m6A RNA methylation. Upon further functional analysis, the authors found that the BACE2 mRNA upregulation was associated with intracellular calcium release leading to ocular melanoma progression.
In a more recent study by Liao et al. [99], the authors studied 41 CM and 11 melanocytic nevi samples, and upon conducting multi-omic and functional analyses, they showed that the m6A ‘eraser’ FTO was upregulated in cancer-associated fibroblasts (CAFs) of the CM microenvironment that displayed pronounced proangiogenic potential (through the activation of VEGFA and EGR1). The authors demonstrated that the increased elimination of m6A modifications from VEGFA and EGR1 by upregulated FTO prevented the YTHDF2-mediated mRNA decay, which in turn led to increased stability and upregulation of VEGFA and EGR1. Given the pivotal role of FTO in CAF-mediated angiogenesis and the critical role of angiogenesis in tumor progression and spread, the authors proposed FTO as a promising antiangiogenic therapeutic target to improve CM treatment. Interestingly, in addition to somatic FTO changes similarly shown to play a role in cutaneous melanomagenesis [108], recent genome-wide association studies have implicated germline FTO polymorphisms in genetic susceptibility to develop cutaneous melanoma [109,110,111].
5. Gaps in Knowledge of Conjunctival Melanoma Epigenetics and Future Directions
The emerging epi-drugs for cancer therapy (as a single agent or combined with other drugs/therapies) currently include those targeting chromatin remodelers (e.g., SMARC inhibitors), histone modifiers or readers (e.g., HDAC, HMT, HDM, or BET inhibitors), DNA methylation-related enzymes (e.g., DNMT inhibitors), ncRNAs (e.g., lncRNA or miRNA mimics or antagonists), or RNA-modifying enzymes (e.g., ALKBH5 or FTO inhibitors) [30,32,43,44,53,112,113,114,115]. The promising results from the latest clinical studies/trials on combination therapies (epi-drugs with other treatments) are especially encouraging given the commonly observed primary/acquired resistance to existing cancer therapies (immunotherapies, targeted therapies, or conventional therapies), as epigenetic reprogramming appears to play a crucial role in the development of such resistance [30,42,43,44,53,112,113]. Furthermore, some chemotherapy drugs (e.g., curaxins) that have been found to show their anti-cancer activity through chromatin damage rather than DNA damage (e.g., CBL0137 causing nucleosome destabilization and histone eviction) are currently tested in melanoma clinical trials as safer drug options [52]. Lastly, the recently emerging CRISPR-based targeted epigenetic editing tools provide exciting opportunities for future clinical interventions [116]. To be able to take advantage of these emerging drugs and therapeutic opportunities for better CM management, we first need to improve our understanding of epigenetic alterations contributing to CM development/progression and its microenvironment by conducting additional studies aiming to fill the gaps in our knowledge of the epigenetic makeup of CM.
Currently, epigenetic studies on chromatin conformation/structure and histone modifications are completely lacking in CM. A recent study [93] on DNA methylation described shared global but different promoter methylation patterns between CM and other mucosal and cutaneous melanomas; however, it included only nine CM samples and thus warrants replication in larger studies. Another study [94] reported a significant loss of 5-hmC and TET2 in 37 CMs vs. 40 conjunctival nevi, a finding that awaits confirmation in independent studies. A few studies that explored RNA methylation [99,100,101] or circRNAs [98] in primary CMs or their microenvironment have revealed some interesting findings, which again warrant replication in independent and larger studies. The studies on linear lncRNAs, on the other hand, are currently completely lacking in CM.
Three studies published on CM-related miRNA expression profiles (investigated by either microarray-based or TaqMan methods) [95,96,97] have revealed some dysregulated miRNAs, which were associated with CM development (compared to normal conjunctiva), CM recurrence or metastasis risk (prognostic potential), or discrimination of CMs from nevi (diagnostic potential); however, the findings were not very reproducible across studies or screening platforms. Methodological differences, the use of FFPE rather than fresh frozen tissues, and small sample sizes were proposed as potential factors that might have contributed to observed inconsistencies. Therefore, additional studies are needed using larger cohorts and employing the latest technologies, such as miRNA-seq, to further explore the miRNA dysregulation in CM for clinical biomarker and therapeutic target discovery. Moreover, investigating ‘circulating’ miRNAs in biofluids (e.g., in tear or serum/plasma samples) from CM patients could be an alternative and attractive strategy for biomarker discovery/development as compared to tissue-based analyses [95]. While the initial studies have looked at the miRNA expression differences between CM and nevi, studies on comparisons between CM and PAM (a precursor with a greater risk for malignant transformation) are currently lacking [95]. Since PAM with atypia is an important risk factor for CM, molecular characterization of predictive factors of PAM to CM transformation could have important implications for better follow-up and early treatment of CM patients [95]. There is also a need for more advanced-stage tumor-related research to identify differentially expressed miRNAs associated with advanced CM [97]. Identification of advanced CM-associated molecular factors could aid the prognostication efforts for better follow-up and timely treatment of CM patients [97].
6. Conclusions
CM is a rare but highly aggressive cancer. While local recurrences and distant metastases commonly occur in CM patients, their prediction and management remain difficult. Predictive biomarkers and more effective treatments are therefore urgently needed to help reduce the recurrence and metastasis rates and improve clinical outcomes in CM patients. CM is an age-related condition, and as the population ages and medical advances prolong the average lifespan, more CM cases may occur in the future and benefit from improved clinical management.
Like other cancers, CM initiation and progression are believed to be influenced by multiple genetic and epigenetic factors that contribute to tumorigenesis, recurrence/spread, immune evasion, and primary/acquired therapy resistance. While our genetic understanding of CM has been significantly improved in recent decades, our epigenetic understanding of CM remains largely incomplete due to a limited number of studies published to date. Recent years have witnessed a rapid increase in available epigenetic technologies and epigenetic modulation-based treatment options in the cancer field. In light of these advancements and emerging new therapies, an in-depth understanding of CM epigenetics has become essential to help improve the clinical outcomes in CM patients. Identifying epigenetic factors promoting CM development, progression, and spread can allow patients to receive more effective treatments through the use of epi-drugs, alone or in combination with other therapies, and lead to more favorable outcomes. Apart from the potential therapeutic benefits, advanced knowledge of CM epigenetics can also guide the diagnostic/prognostic biomarker development efforts for this aggressive disease and help implement more effective follow-up and timely treatment strategies.
Author Contributions
Conceptualization, H.D. and F.Y.D.; Writing—original draft, K.M.F. and F.Y.D.; and Writing—review and editing, K.M.F., H.D. and F.Y.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Vora, G.K.; Demirci, H.; Marr, B.; Mruthyunjaya, P. Advances in the management of conjunctival melanoma. Surv. Ophthalmol. 2017, 62, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Shields, C.L.; Markowitz, J.S.; Belinsky, I.; Schwartzstein, H.; George, N.S.; Lally, S.E.; Mashayekhi, A.; Shields, J.A. Conjunctival melanoma: Outcomes based on tumor origin in 382 consecutive cases. Ophthalmology 2011, 118, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Shumway, C.L.; Motlagh, M.; Wade, M. Anatomy, Head and Neck, Eye Conjunctiva; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Shields, C.L.; Demirci, H.; Karatza, E.; Shields, J.A. Clinical survey of 1643 melanocytic and nonmelanocytic conjunctival tumors. Ophthalmology 2004, 111, 1747–1754. [Google Scholar] [CrossRef] [PubMed]
- Weppelmann, T.A.; Zimmerman, K.T.; Rashidi, V. Trends in Incidence of Conjunctival Melanoma in the US. JAMA Netw. Open 2022, 5, e2237229. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.P.; Hu, D.N.; McCormick, S.; Finger, P.T. Conjunctival melanoma: Is it increasing in the United States? Am. J. Ophthalmol. 2003, 135, 800–806. [Google Scholar] [CrossRef]
- Triay, E.; Bergman, L.; Nilsson, B.; All-Ericsson, C.; Seregard, S. Time trends in the incidence of conjunctival melanoma in Sweden. Br. J. Ophthalmol. 2009, 93, 1524–1528. [Google Scholar] [CrossRef]
- Virgili, G.; Parravano, M.; Gatta, G.; Capocaccia, R.; Mazzini, C.; Mallone, S.; Botta, L.; Group, R.A.W. Incidence and Survival of Patients With Conjunctival Melanoma in Europe. JAMA Ophthalmol. 2020, 138, 601–608. [Google Scholar] [CrossRef]
- Brouwer, N.J.; Verdijk, R.M.; Heegaard, S.; Marinkovic, M.; Esmaeli, B.; Jager, M.J. Conjunctival melanoma: New insights in tumour genetics and immunology, leading to new therapeutic options. Prog. Retin. Eye Res. 2022, 86, 100971. [Google Scholar] [CrossRef]
- Nowak, M.S.; Romanowska-Dixon, B.; Grabska-Liberek, I.; Zurek, M. Incidence and survival of ocular melanoma in National Cancer Registry of Poland in 2010–2017. Adv. Clin. Exp. Med. 2022, 31, 615–621. [Google Scholar] [CrossRef]
- Lim, J.Z.; Misra, S.L.; Gokul, A.; Hadden, P.W.; Cavadino, A.; McGhee, C.N.J. Conjunctival Melanoma in Aotearoa-New Zealand: A 21-Year Analysis of Incidence and Survival. Asia Pac. J. Ophthalmol. 2023, 12, 273–278. [Google Scholar] [CrossRef]
- Chang, A.E.; Karnell, L.H.; Menck, H.R. The National Cancer Data Base report on cutaneous and noncutaneous melanoma: A summary of 84,836 cases from the past decade. The American College of Surgeons Commission on Cancer and the American Cancer Society. Cancer 1998, 83, 1664–1678. [Google Scholar] [CrossRef]
- McLaughlin, C.C.; Wu, X.C.; Jemal, A.; Martin, H.J.; Roche, L.M.; Chen, V.W. Incidence of noncutaneous melanomas in the U.S. Cancer 2005, 103, 1000–1007. [Google Scholar] [CrossRef]
- Gkiala, A.; Palioura, S. Conjunctival Melanoma: Update on Genetics, Epigenetics and Targeted Molecular and Immune-Based Therapies. Clin. Ophthalmol. 2020, 14, 3137–3152. [Google Scholar] [CrossRef] [PubMed]
- Gardrat, S.; Houy, A.; Brooks, K.; Cassoux, N.; Barnhill, R.; Dayot, S.; Bieche, I.; Raynal, V.; Baulande, S.; Marais, R.; et al. Definition of Biologically Distinct Groups of Conjunctival Melanomas According to Etiological Factors and Implications for Precision Medicine. Cancers 2021, 13, 3836. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.; Demirci, H.; Demirci, F.Y. Genetic Aspects of Conjunctival Melanoma: A Review. Genes 2023, 14, 1668. [Google Scholar] [CrossRef]
- Shields, C.L.; Fasiuddin, A.F.; Mashayekhi, A.; Shields, J.A. Conjunctival nevi: Clinical features and natural course in 410 consecutive patients. Arch. Ophthalmol. 2004, 122, 167–175. [Google Scholar] [CrossRef]
- Pacheco, R.R.; Yaghy, A.; Dalvin, L.A.; Vaidya, S.; Perez, A.L.; Lally, S.E.; Shields, J.A.; Shields, C.L. Conjunctival melanoma: Outcomes based on tumour origin in 629 patients at a single ocular oncology centre. Eye 2022, 36, 603–611. [Google Scholar] [CrossRef]
- Wong, J.R.; Nanji, A.A.; Galor, A.; Karp, C.L. Management of conjunctival malignant melanoma: A review and update. Expert. Rev. Ophthalmol. 2014, 9, 185–204. [Google Scholar] [CrossRef]
- Shields, C.L.; Shields, J.A.; Gunduz, K.; Cater, J.; Mercado, G.V.; Gross, N.; Lally, B. Conjunctival melanoma: Risk factors for recurrence, exenteration, metastasis, and death in 150 consecutive patients. Arch. Ophthalmol. 2000, 118, 1497–1507. [Google Scholar] [CrossRef]
- Jain, P.; Finger, P.T.; Fili, M.; Damato, B.; Coupland, S.E.; Heimann, H.; Kenawy, N.; Brouwer, N.J.; Marinkovic, M.; Van Duinen, S.G.; et al. Conjunctival melanoma treatment outcomes in 288 patients: A multicentre international data-sharing study. Br. J. Ophthalmol. 2021, 105, 1358–1364. [Google Scholar] [CrossRef]
- Bol, K.F.; Donia, M.; Heegaard, S.; Kiilgaard, J.F.; Svane, I.M. Genetic Biomarkers in Melanoma of the Ocular Region: What the Medical Oncologist Should Know. Int. J. Mol. Sci. 2020, 21, 5231. [Google Scholar] [CrossRef]
- Rossi, E.; Schinzari, G.; Maiorano, B.A.; Pagliara, M.M.; Di Stefani, A.; Bria, E.; Peris, K.; Blasi, M.A.; Tortora, G. Conjunctival Melanoma: Genetic and Epigenetic Insights of a Distinct Type of Melanoma. Int. J. Mol. Sci. 2019, 20, 5447. [Google Scholar] [CrossRef] [PubMed]
- Demirci, H.; Demirci, F.Y.; Ciftci, S.; Elner, V.M.; Wu, Y.M.; Ning, Y.; Chinnaiyan, A.; Robinson, D.R. Integrative Exome and Transcriptome Analysis of Conjunctival Melanoma and Its Potential Application for Personalized Therapy. JAMA Ophthalmol. 2019, 137, 1444–1448. [Google Scholar] [CrossRef] [PubMed]
- Sa, H.S.; Daniel, C.; Esmaeli, B. Update on Immune Checkpoint Inhibitors for Conjunctival Melanoma. J. Ophthalmic Vis. Res. 2022, 17, 405–412. [Google Scholar] [CrossRef]
- Serbest Ceylanoglu, K.; Guneri Beser, B.; Singalavanija, T.; Juntipwong, S.; Worden, F.P.; Demirci, H. Targeted Therapy and Immunotherapy for Advanced Malignant Conjunctival Tumors: Systematic Review. Ophthalmic Plast. Reconstr. Surg. 2024, 40, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Anestopoulos, I.; Kyriakou, S.; Tragkola, V.; Paraskevaidis, I.; Tzika, E.; Mitsiogianni, M.; Deligiorgi, M.V.; Petrakis, G.; Trafalis, D.T.; Botaitis, S.; et al. Targeting the epigenome in malignant melanoma: Facts, challenges and therapeutic promises. Pharmacol. Ther. 2022, 240, 108301. [Google Scholar] [CrossRef] [PubMed]
- Chai, P.; Jia, R.; Li, Y.; Zhou, C.; Gu, X.; Yang, L.; Shi, H.; Tian, H.; Lin, H.; Yu, J.; et al. Regulation of epigenetic homeostasis in uveal melanoma and retinoblastoma. Prog. Retin. Eye Res. 2022, 89, 101030. [Google Scholar] [CrossRef]
- Giunta, E.F.; Arrichiello, G.; Curvietto, M.; Pappalardo, A.; Bosso, D.; Rosanova, M.; Diana, A.; Giordano, P.; Petrillo, A.; Federico, P.; et al. Epigenetic Regulation in Melanoma: Facts and Hopes. Cells 2021, 10, 2048. [Google Scholar] [CrossRef]
- Lu, Y.; Chan, Y.T.; Tan, H.Y.; Li, S.; Wang, N.; Feng, Y. Epigenetic regulation in human cancer: The potential role of epi-drug in cancer therapy. Mol. Cancer 2020, 19, 79. [Google Scholar] [CrossRef]
- Moran, B.; Silva, R.; Perry, A.S.; Gallagher, W.M. Epigenetics of malignant melanoma. Semin. Cancer Biol. 2018, 51, 80–88. [Google Scholar] [CrossRef]
- Lee, A.V.; Nestler, K.A.; Chiappinelli, K.B. Therapeutic targeting of DNA methylation alterations in cancer. Pharmacol. Ther. 2024, 258, 108640. [Google Scholar] [CrossRef] [PubMed]
- Janssen, S.M.; Lorincz, M.C. Interplay between chromatin marks in development and disease. Nat. Rev. Genet. 2022, 23, 137–153. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Peng, Y.; Panchenko, A.R. DNA methylation: Precise modulation of chromatin structure and dynamics. Curr. Opin. Struct. Biol. 2022, 75, 102430. [Google Scholar] [CrossRef] [PubMed]
- Saviana, M.; Le, P.; Micalo, L.; Del Valle-Morales, D.; Romano, G.; Acunzo, M.; Li, H.; Nana-Sinkam, P. Crosstalk between miRNAs and DNA Methylation in Cancer. Genes 2023, 14, 1075. [Google Scholar] [CrossRef]
- Yang, Z.; Xu, F.; Teschendorff, A.E.; Zhao, Y.; Yao, L.; Li, J.; He, Y. Insights into the role of long non-coding RNAs in DNA methylation mediated transcriptional regulation. Front. Mol. Biosci. 2022, 9, 1067406. [Google Scholar] [CrossRef]
- Eustermann, S.; Patel, A.B.; Hopfner, K.P.; He, Y.; Korber, P. Energy-driven genome regulation by ATP-dependent chromatin remodellers. Nat. Rev. Mol. Cell Biol. 2024, 25, 309–332. [Google Scholar] [CrossRef]
- Corcoran, E.T.; Jacob, Y. Direct assessment of histone function using histone replacement. Trends Biochem. Sci. 2023, 48, 53–70. [Google Scholar] [CrossRef]
- Millán-Zambrano, G.; Burton, A.; Bannister, A.J.; Schneider, R. Histone post-translational modifications—Cause and consequence of genome function. Nat. Rev. Genet. 2022, 23, 563–580. [Google Scholar] [CrossRef]
- Chaudhri, A.; Lizee, G.; Hwu, P.; Rai, K. Chromatin Remodelers Are Regulators of the Tumor Immune Microenvironment. Cancer Res. 2024, 84, 965–976. [Google Scholar] [CrossRef]
- Gibson, F.; Hanly, A.; Fisher, R.; Wyant, W.A.; Wu, M.; Collard, M.; Alani, R.M. Epigenetics and cutaneous neoplasms: From mechanism to therapy. Epigenomics 2023, 15, 167–187. [Google Scholar] [CrossRef]
- Rubanov, A.; Berico, P.; Hernando, E. Epigenetic Mechanisms Underlying Melanoma Resistance to Immune and Targeted Therapies. Cancers 2022, 14, 5858. [Google Scholar] [CrossRef] [PubMed]
- Hanly, A.; Gibson, F.; Nocco, S.; Rogers, S.; Wu, M.; Alani, R.M. Drugging the Epigenome: Overcoming Resistance to Targeted and Immunotherapies in Melanoma. JID Innov. 2022, 2, 100090. [Google Scholar] [CrossRef] [PubMed]
- Gracia-Hernandez, M.; Munoz, Z.; Villagra, A. Enhancing Therapeutic Approaches for Melanoma Patients Targeting Epigenetic Modifiers. Cancers 2021, 13, 6180. [Google Scholar] [CrossRef]
- Xiao, Y.; Xia, Y.; Wang, Y.; Xue, C. Pathogenic roles of long noncoding RNAs in melanoma: Implications in diagnosis and therapies. Genes. Dis. 2023, 10, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Takeshima, H.; Ushijima, T. Accumulation of genetic and epigenetic alterations in normal cells and cancer risk. NPJ Precis. Oncol. 2019, 3, 7. [Google Scholar] [CrossRef]
- Griazeva, E.D.; Fedoseeva, D.M.; Radion, E.I.; Ershov, P.V.; Meshkov, I.O.; Semyanihina, A.V.; Makarova, A.S.; Makarov, V.V.; Yudin, V.S.; Keskinov, A.A.; et al. Current Approaches to Epigenetic Therapy. Epigenomes 2023, 7, 23. [Google Scholar] [CrossRef]
- Gourisankar, S.; Krokhotin, A.; Wenderski, W.; Crabtree, G.R. Context-specific functions of chromatin remodellers in development and disease. Nat. Rev. Genet. 2024, 25, 340–361. [Google Scholar] [CrossRef]
- Martinez-Useros, J.; Martin-Galan, M.; Florez-Cespedes, M.; Garcia-Foncillas, J. Epigenetics of Most Aggressive Solid Tumors: Pathways, Targets and Treatments. Cancers 2021, 13, 3209. [Google Scholar] [CrossRef]
- Sang, Y.; Deng, Y. Current insights into the epigenetic mechanisms of skin cancer. Dermatol. Ther. 2019, 32, e12964. [Google Scholar] [CrossRef]
- Neefjes, J.; Gurova, K.; Sarthy, J.; Szabó, G.; Henikoff, S. Chromatin as an old and new anticancer target. Trends Cancer 2024, 10, 696–707. [Google Scholar] [CrossRef] [PubMed]
- Malone, H.A.; Roberts, C.W.M. Chromatin remodellers as therapeutic targets. Nat. Rev. Drug Discov. 2024, 23, 661–681. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, M.; Wang, Y. The roles of histone modifications in tumorigenesis and associated inhibitors in cancer therapy. J. Natl. Cancer Cent. 2022, 2, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Shilatifard, A. Epigenetic modifications of histones in cancer. Genome Biol. 2019, 20, 245. [Google Scholar] [CrossRef] [PubMed]
- Phillips, E.O.N.; Gunjan, A. Histone variants: The unsung guardians of the genome. DNA Repair 2022, 112, 103301. [Google Scholar] [CrossRef]
- Kurumizaka, H.; Kujirai, T.; Takizawa, Y. Contributions of Histone Variants in Nucleosome Structure and Function. J. Mol. Biol. 2021, 433, 166678. [Google Scholar] [CrossRef]
- Martire, S.; Banaszynski, L.A. The roles of histone variants in fine-tuning chromatin organization and function. Nat. Rev. Mol. Cell Biol. 2020, 21, 522–541. [Google Scholar] [CrossRef]
- Mannavola, F.; D’Oronzo, S.; Cives, M.; Stucci, L.S.; Ranieri, G.; Silvestris, F.; Tucci, M. Extracellular Vesicles and Epigenetic Modifications Are Hallmarks of Melanoma Progression. Int. J. Mol. Sci. 2019, 21, 52. [Google Scholar] [CrossRef]
- Angeloni, A.; Bogdanovic, O. Sequence determinants, function, and evolution of CpG islands. Biochem. Soc. Trans. 2021, 49, 1109–1119. [Google Scholar] [CrossRef]
- Joshi, K.; Liu, S.; Breslin, S.J.P.; Zhang, J. Mechanisms that regulate the activities of TET proteins. Cell Mol. Life Sci. 2022, 79, 363. [Google Scholar] [CrossRef]
- Turpin, M.; Salbert, G. 5-methylcytosine turnover: Mechanisms and therapeutic implications in cancer. Front. Mol. Biosci. 2022, 9, 976862. [Google Scholar] [CrossRef] [PubMed]
- Bray, J.K.; Dawlaty, M.M.; Verma, A.; Maitra, A. Roles and Regulations of TET Enzymes in Solid Tumors. Trends Cancer 2021, 7, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, S.E.; Gladyshev, V.N.; Aryee, M.J.; Bernstein, B.E. Epigenetic clocks, aging, and cancer. Science 2022, 378, 1276–1277. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, K.; Bayraktar, R.; Ferracin, M.; Calin, G.A. Non-coding RNAs in disease: From mechanisms to therapeutics. Nat. Rev. Genet. 2024, 25, 211–232. [Google Scholar] [CrossRef] [PubMed]
- Loganathan, T.; Doss, C.G. Non-coding RNAs in human health and disease: Potential function as biomarkers and therapeutic targets. Funct. Integr. Genomics 2023, 23, 33. [Google Scholar] [CrossRef]
- Goodall, G.J.; Wickramasinghe, V.O. RNA in cancer. Nat. Rev. Cancer 2021, 21, 22–36. [Google Scholar] [CrossRef]
- Qin, T.; Li, J.; Zhang, K.Q. Structure, Regulation, and Function of Linear and Circular Long Non-Coding RNAs. Front. Genet. 2020, 11, 150. [Google Scholar] [CrossRef]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Segal, D.; Dostie, J. The Talented LncRNAs: Meshing into Transcriptional Regulatory Networks in Cancer. Cancers 2023, 15, 3433. [Google Scholar] [CrossRef]
- Liu, S.J.; Dang, H.X.; Lim, D.A.; Feng, F.Y.; Maher, C.A. Long noncoding RNAs in cancer metastasis. Nat. Rev. Cancer 2021, 21, 446–460. [Google Scholar] [CrossRef]
- Delaunay, S.; Helm, M.; Frye, M. RNA modifications in physiology and disease: Towards clinical applications. Nat. Rev. Genet. 2024, 25, 104–122. [Google Scholar] [CrossRef] [PubMed]
- Boulias, K.; Greer, E.L. Biological roles of adenine methylation in RNA. Nat. Rev. Genet. 2023, 24, 143–160. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhao, W.; Zhang, L.; Wang, X. The emerging roles of N6-methyladenosine (m6A)-modified long non-coding RNAs in human cancers. Cell Death Discov. 2022, 8, 255. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Peng, J.; Yi, C. The epitranscriptome of small non-coding RNAs. Noncoding RNA Res. 2021, 6, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Tang, L.; Min, Q.; Tian, H.; Li, L.; Zhao, Y.; Wu, X.; Li, M.; Du, F.; Chen, Y.; et al. Emerging role of RNA modification and long noncoding RNA interaction in cancer. Cancer Gene Ther. 2024, 31, 816–830. [Google Scholar] [CrossRef]
- Deng, X.; Qing, Y.; Horne, D.; Huang, H.; Chen, J. The roles and implications of RNA m(6)A modification in cancer. Nat. Rev. Clin. Oncol. 2023, 20, 507–526. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, J.; Yang, H.; Yang, X.; Zhang, Y.; Yu, X.; Li, Y.; Chen, G.; Yang, Z. The potential role of m6A reader YTHDF1 as diagnostic biomarker and the signaling pathways in tumorigenesis and metastasis in pan-cancer. Cell Death Discov. 2023, 9, 34. [Google Scholar] [CrossRef]
- Chen, Y.; Yi, X.; Sun, N.; Guo, W.; Li, C. Epigenetics Regulates Antitumor Immunity in Melanoma. Front. Immunol. 2022, 13, 868786. [Google Scholar] [CrossRef]
- Mollapour Sisakht, M.; Amirkhani, M.A.; Nilforoushzadeh, M.A. SWI/SNF complex, promising target in melanoma therapy: Snapshot view. Front. Med. 2023, 10, 1096615. [Google Scholar] [CrossRef]
- Dreier, M.R.; de la Serna, I.L. SWI/SNF Chromatin Remodeling Enzymes in Melanoma. Epigenomes 2022, 6, 10. [Google Scholar] [CrossRef]
- Sutopo, N.C.; Kim, J.H.; Cho, J.Y. Role of histone methylation in skin cancers: Histone methylation-modifying enzymes as a new class of targets for skin cancer treatment. Biochim. Biophys. Acta Rev. Cancer 2023, 1878, 188865. [Google Scholar] [CrossRef]
- Konstantinov, N.K.; Ulff-Møller, C.J.; Dimitrov, S. Histone variants and melanoma: Facts and hypotheses. Pigment. Cell Melanoma Res. 2016, 29, 426–433. [Google Scholar] [CrossRef]
- Bustos, M.A.; Hoon, D.S.B. Prognostic Utility of CpG Island Hypermethylated Phenotype in Early-Stage Invasive Primary Melanomas. J. Investig. Dermatol. 2022, 142, 1770–1772. [Google Scholar] [CrossRef]
- Conway, K.; Tsai, Y.S.; Edmiston, S.N.; Parker, J.S.; Parrish, E.A.; Hao, H.; Kuan, P.F.; Scott, G.A.; Frank, J.S.; Googe, P.; et al. Characterization of the CpG Island Hypermethylated Phenotype Subclass in Primary Melanomas. J. Investig. Dermatol. 2022, 142, 1869–1881.e10. [Google Scholar] [CrossRef]
- Li, F.J.; Li, L.M.; Zhang, R.H.; Xu, C.; Zhou, P.; Long, J.; Hu, G.; Jiang, M.J. The role of 5-hydroxymethylcytosine in melanoma. Melanoma Res. 2017, 27, 175–179. [Google Scholar] [CrossRef]
- Lian, C.G.; Xu, Y.; Ceol, C.; Wu, F.; Larson, A.; Dresser, K.; Xu, W.; Tan, L.; Hu, Y.; Zhan, Q.; et al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell 2012, 150, 1135–1146. [Google Scholar] [CrossRef]
- Natarelli, N.; Boby, A.; Aflatooni, S.; Tran, J.T.; Diaz, M.J.; Taneja, K.; Forouzandeh, M. Regulatory miRNAs and lncRNAs in Skin Cancer: A Narrative Review. Life 2023, 13, 1696. [Google Scholar] [CrossRef] [PubMed]
- Grafanaki, K.; Grammatikakis, I.; Ghosh, A.; Gopalan, V.; Olgun, G.; Liu, H.; Kyriakopoulos, G.C.; Skeparnias, I.; Georgiou, S.; Stathopoulos, C.; et al. Noncoding RNA circuitry in melanoma onset, plasticity, and therapeutic response. Pharmacol. Ther. 2023, 248, 108466. [Google Scholar] [CrossRef] [PubMed]
- Melixetian, M.; Pelicci, P.G.; Lanfrancone, L. Regulation of LncRNAs in Melanoma and Their Functional Roles in the Metastatic Process. Cells 2022, 11, 577. [Google Scholar] [CrossRef] [PubMed]
- Ran, Y.; Yan, Z.; Jiang, B.; Liang, P. N6-methyladenosine functions and its role in skin cancer. Exp. Dermatol. 2023, 32, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Zhang, K.; Guo, Y.; Shen, C.; Liu, X.; Huang, H.; Dou, X.; Yu, B. The crucial roles of m(6)A RNA modifications in cutaneous cancers: Implications in pathogenesis, metastasis, drug resistance, and targeted therapies. Genes. Dis. 2023, 10, 2320–2330. [Google Scholar] [CrossRef] [PubMed]
- Jurmeister, P.; Wrede, N.; Hoffmann, I.; Vollbrecht, C.; Heim, D.; Hummel, M.; Wolkenstein, P.; Koch, I.; Heynol, V.; Schmitt, W.D.; et al. Mucosal melanomas of different anatomic sites share a common global DNA methylation profile with cutaneous melanoma but show location-dependent patterns of genetic and epigenetic alterations. J. Pathol. 2022, 256, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Stahl, A.; Riggi, N.; Nardou, K.; Nicolas, M.; Kaya, G.; Moulin, A. 5-Hydroxymethylcytosine Loss in Conjunctival Melanoma. Dermatopathology 2021, 8, 176–184. [Google Scholar] [CrossRef] [PubMed]
- van Ipenburg, J.A.; Gillis Ing, A.J.M.; Dorssers, L.C.J.; van den Bosch, Q.C.C.; van Ginderdeuren, R.; Missotten, G.S.; Naus, N.; Paridaens, D.; Looijenga, L.H.J.; Verdijk, R.M. MicroRNA Profiling in Benign and Malignant Conjunctival Melanocytic Lesions. Ophthalmology 2020, 127, 432–434. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, L.H.; Andersen, M.K.; Andreasen, S.; Larsen, A.C.; Tan, Q.; Toft, P.B.; Wadt, K.; Heegaard, S. Global microRNA profiling of metastatic conjunctival melanoma. Melanoma Res. 2019, 29, 465–473. [Google Scholar] [CrossRef]
- Larsen, A.C.; Mikkelsen, L.H.; Borup, R.; Kiss, K.; Toft, P.B.; von Buchwald, C.; Coupland, S.E.; Prause, J.U.; Heegaard, S. MicroRNA Expression Profile in Conjunctival Melanoma. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4205–4212. [Google Scholar] [CrossRef]
- Shang, Q.; Li, Y.; Wang, H.; Ge, S.; Jia, R. Altered expression profile of circular RNAs in conjunctival melanoma. Epigenomics 2019, 11, 787–804. [Google Scholar] [CrossRef]
- Liao, Q.; Shi, H.; Yang, J.; Ge, S.; Jia, R.; Song, X.; Chai, P.; Jia, R. FTO elicits tumor neovascularization in cancer-associated fibroblasts through eliminating m(6)A modifications of multiple pro-angiogenic factors. Cancer Lett. 2024, 592, 216911. [Google Scholar] [CrossRef]
- He, F.; Yu, J.; Yang, J.; Wang, S.; Zhuang, A.; Shi, H.; Gu, X.; Xu, X.; Chai, P.; Jia, R. m(6)A RNA hypermethylation-induced BACE2 boosts intracellular calcium release and accelerates tumorigenesis of ocular melanoma. Mol. Ther. 2021, 29, 2121–2133. [Google Scholar] [CrossRef]
- Jia, R.; Chai, P.; Wang, S.; Sun, B.; Xu, Y.; Yang, Y.; Ge, S.; Jia, R.; Yang, Y.G.; Fan, X. m(6)A modification suppresses ocular melanoma through modulating HINT2 mRNA translation. Mol. Cancer. 2019, 18, 161. [Google Scholar] [CrossRef]
- Swaminathan, S.S.; Field, M.G.; Sant, D.; Wang, G.; Galor, A.; Dubovy, S.R.; Harbour, J.W.; Karp, C.L. Molecular Characteristics of Conjunctival Melanoma Using Whole-Exome Sequencing. JAMA Ophthalmol. 2017, 135, 1434–1437. [Google Scholar] [CrossRef] [PubMed]
- Gagné, L.M.; Boulay, K.; Topisirovic, I.; Huot, M.; Mallette, F.A. Oncogenic Activities of IDH1/2 Mutations: From Epigenetics to Cellular Signaling. Trends Cell Biol. 2017, 27, 738–752. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, L.H.; Maag, E.; Andersen, M.K.; Kruhoffer, M.; Larsen, A.C.; Melchior, L.C.; Toft, P.B.; von Buchwald, C.; Wadt, K.; Heegaard, S. The molecular profile of mucosal melanoma. Melanoma Res. 2020, 30, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Cisarova, K.; Folcher, M.; El Zaoui, I.; Pescini-Gobert, R.; Peter, V.G.; Royer-Bertrand, B.; Zografos, L.; Schalenbourg, A.; Nicolas, M.; Rimoldi, D.; et al. Genomic and transcriptomic landscape of conjunctival melanoma. PLoS Genet. 2020, 16, e1009201. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Xie, Z.; Xiao, Z.; Zhu, D. The expression and function of miR-622 in a variety of tumors. Biomed. Pharmacother. 2022, 146, 112544. [Google Scholar] [CrossRef]
- Zhang, C.; Hu, J.; Yu, Y. CircRNA Is a Rising Star in Researches of Ocular Diseases. Front. Cell Dev. Biol. 2020, 8, 850. [Google Scholar] [CrossRef]
- Yang, S.; Wei, J.; Cui, Y.H.; Park, G.; Shah, P.; Deng, Y.; Aplin, A.E.; Lu, Z.; Hwang, S.; He, C.; et al. m(6)A mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat. Commun. 2019, 10, 2782. [Google Scholar] [CrossRef]
- Iles, M.M.; Law, M.H.; Stacey, S.N.; Han, J.; Fang, S.; Pfeiffer, R.; Harland, M.; Macgregor, S.; Taylor, J.C.; Aben, K.K.; et al. A variant in FTO shows association with melanoma risk not due to BMI. Nat. Genet. 2013, 45, 428–432.432e1. [Google Scholar]
- Landi, M.T.; Bishop, D.T.; MacGregor, S.; Machiela, M.J.; Stratigos, A.J.; Ghiorzo, P.; Brossard, M.; Calista, D.; Choi, J.; Fargnoli, M.C.; et al. Genome-wide association meta-analyses combining multiple risk phenotypes provide insights into the genetic architecture of cutaneous melanoma susceptibility. Nat. Genet. 2020, 52, 494–504. [Google Scholar] [CrossRef]
- Zheng, Q.K.; Ma, C.; Ullah, I.; Hu, K.; Ma, R.J.; Zhang, N.; Sun, Z.G. Roles of N6-Methyladenosine Demethylase FTO in Malignant Tumors Progression. Onco Targets Ther. 2021, 14, 4837–4846. [Google Scholar] [CrossRef]
- Mabe, N.W.; Perry, J.A.; Malone, C.F.; Stegmaier, K. Pharmacological targeting of the cancer epigenome. Nat. Cancer 2024, 5, 844–865. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zhao, H.; Wang, R.; Chen, Y.; Ouyang, X.; Li, W.; Sun, Y.; Peng, A. Cancer epigenetics: From laboratory studies and clinical trials to precision medicine. Cell Death Discov. 2024, 10, 28. [Google Scholar] [CrossRef] [PubMed]
- Coan, M.; Haefliger, S.; Ounzain, S.; Johnson, R. Targeting and engineering long non-coding RNAs for cancer therapy. Nat. Rev. Genet. 2024, 25, 578–595. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Jing, Q.; Li, Y.; Han, J. RNA modification: Mechanisms and therapeutic targets. Mol. Biomed. 2023, 4, 25. [Google Scholar] [CrossRef]
- Roth, G.V.; Gengaro, I.R.; Qi, L.S. Precision epigenetic editing: Technological advances, enduring challenges, and therapeutic applications. Cell Chem. Biol. 2024. Online ahead of print. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).