
IGH::NSD2 Fusion Gene Transcript as Measurable Residual Disease Marker in Multiple Myeloma

 , , ,
, , ,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
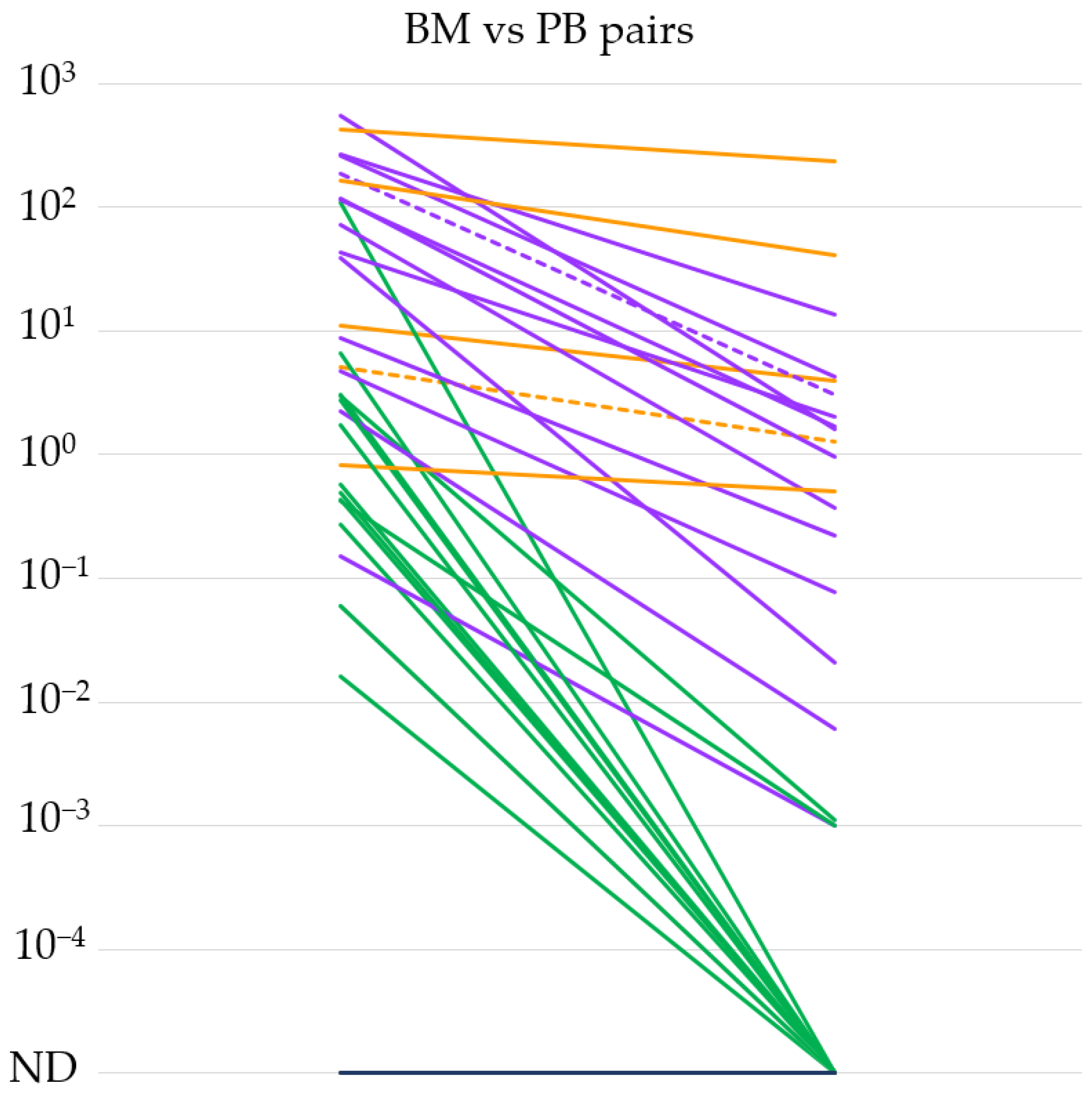
3. Results
3.1. Evaluation of the IGH::NSD2 qPCR System
3.2. Survival Data of Patients According to the Different NSD2 Breakpoint Types
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maura, F.; Bolli, N.; Angelopoulos, N.; Dawson, K.J.; Leongamornlert, D.; Martincorena, I.; Mitchell, T.J.; Fullam, A.; Gonzalez, S.; Szalat, R.; et al. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Nat. Commun. 2019, 10, 3835. [Google Scholar] [CrossRef] [PubMed]
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The genetic architecture of multiple myeloma. Nat. Rev. Cancer 2012, 12, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Zanwar, S.; Kumar, S. Disease heterogeneity, prognostication and the role of targeted therapy in multiple myeloma. Leuk. Lymphoma 2021, 62, 3087–3097. [Google Scholar] [CrossRef] [PubMed]
- Caers, J.; Garderet, L.; Kortüm, K.M.; O’Dwyer, M.E.; van de Donk, N.; Binder, M.; Dold, S.M.; Gay, F.; Corre, J.; Beguin, Y.; et al. European Myeloma Network recommendations on tools for the diagnosis and monitoring of multiple myeloma: What to use and when. Haematologica 2018, 103, 1772–1784. [Google Scholar] [CrossRef] [PubMed]
- Munshi, N.C.; Avet-Loiseau, H.; Rawstron, A.C.; Owen, R.G.; Child, J.A.; Thakurta, A.; Sherrington, P.; Samur, M.K.; Georgieva, A.; Anderson, K.C.; et al. Association of Minimal Residual Disease with Superior Survival Outcomes in Patients with Multiple Myeloma: A Meta-analysis. JAMA Oncol. 2017, 3, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Perrot, A.; Lauwers-Cances, V.; Corre, J.; Robillard, N.; Hulin, C.; Chretien, M.L.; Dejoie, T.; Maheo, S.; Stoppa, A.M.; Pegourie, B.; et al. Minimal residual disease negativity using deep sequencing is a major prognostic factor in multiple myeloma. Blood 2018, 132, 2456–2464. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Orfao, A.; Chim, C.S. Molecular detection of minimal residual disease in multiple myeloma. Br. J. Haematol. 2018, 181, 11–26. [Google Scholar] [CrossRef]
- Romano, A.; Palumbo, G.A.; Parrinello, N.L.; Conticello, C.; Martello, M.; Terragna, C. Minimal Residual Disease Assessment within the Bone Marrow of Multiple Myeloma: A Review of Caveats, Clinical Significance and Future Perspectives. Front. Oncol. 2019, 9, 699. [Google Scholar] [CrossRef]
- Ross, F.M.; Avet-Loiseau, H.; Ameye, G.; Gutiérrez, N.C.; Liebisch, P.; O’Connor, S.; Dalva, K.; Fabris, S.; Testi, A.M.; Jarosova, M.; et al. Report from the European Myeloma Network on interphase FISH in multiple myeloma and related disorders. Haematologica 2012, 97, 1272–1277. [Google Scholar] [CrossRef]
- Bustoros, M.; Mouhieddine, T.H.; Detappe, A.; Ghobrial, I.M. Established and Novel Prognostic Biomarkers in Multiple Myeloma. Am. Soc. Clin. Oncol. Educ. Book 2017, 37, 548–560. [Google Scholar] [CrossRef]
- Lazareth, A.; Song, X.Y.; Coquin, A.; Harel, S.; Karlin, L.; Belhadj, K.; Roos-Weil, D.; Frenzel, L.; Tamburini, J.; Macro, M.; et al. MB4-2 breakpoint in MMSET combined with del(17p) defines a subset of t(4;14) multiple myeloma with very poor prognosis. Haematologica 2015, 100, e471. [Google Scholar] [CrossRef]
- Li, F.; Zhai, Y.P.; Lai, T.; Zhao, Q.; Zhang, H.; Tang, Y.M.; Hou, J. MB4-2/MB4-3 transcripts of IGH-MMSET fusion gene in t(4;14)(pos) multiple myeloma indicate poor prognosis. Oncotarget 2017, 8, 51608–51620. [Google Scholar] [CrossRef]
- Malgeri, U.; Baldini, L.; Perfetti, V.; Fabris, S.; Vignarelli, M.C.; Colombo, G.; Lotti, V.; Compasso, S.; Bogni, S.; Lombardi, L.; et al. Detection of t(4;14)(p16.3;q32) chromosomal translocation in multiple myeloma by reverse transcription-polymerase chain reaction analysis of IGH-MMSET fusion transcripts. Cancer Res. 2000, 60, 4058–4061. [Google Scholar]
- Chandesris, M.O.; Soulier, J.; Labaume, S.; Crinquette, A.; Repellini, L.; Chemin, K.; Malphettes, M.; Fieschi, C.; Asli, B.; Uzunhan, Y.; et al. Detection and follow-up of fibroblast growth factor receptor 3 expression on bone marrow and circulating plasma cells by flow cytometry in patients with t(4;14) multiple myeloma. Br. J. Haematol. 2007, 136, 609–614. [Google Scholar] [CrossRef]
- Gabert, J.; Beillard, E.; van der Velden, V.H.; Bi, W.; Grimwade, D.; Pallisgaard, N.; Barbany, G.; Cazzaniga, G.; Cayuela, J.M.; Cavé, H.; et al. Standardization and quality control studies of ‘real-time’ quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia—A Europe Against Cancer program. Leukemia 2003, 17, 2318–2357. [Google Scholar] [CrossRef]
- Palumbo, A.; Avet-Loiseau, H.; Oliva, S.; Lokhorst, H.M.; Goldschmidt, H.; Rosinol, L.; Richardson, P.; Caltagirone, S.; Lahuerta, J.J.; Facon, T.; et al. Revised International Staging System for Multiple Myeloma: A Report From International Myeloma Working Group. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 2863–2869. [Google Scholar] [CrossRef]
- Rajkumar, S.V. Multiple myeloma: 2020 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2020, 95, 548–567. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, M.; Cairns, D.A.; Lahuerta, J.J.; Wester, R.; Bertsch, U.; Waage, A.; Zamagni, E.; Mateos, M.V.; Dall’Olio, D.; van de Donk, N.; et al. Second Revision of the International Staging System (R2-ISS) for Overall Survival in Multiple Myeloma: A European Myeloma Network (EMN) Report within the HARMONY Project. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2022, 40, 3406–3418. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3--new capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Beillard, E.; Pallisgaard, N.; van der Velden, V.H.; Bi, W.; Dee, R.; van der Schoot, E.; Delabesse, E.; Macintyre, E.; Gottardi, E.; Saglio, G.; et al. Evaluation of candidate control genes for diagnosis and residual disease detection in leukemic patients using ‘real-time’ quantitative reverse-transcriptase polymerase chain reaction (RQ-PCR)—A Europe against cancer program. Leukemia 2003, 17, 2474–2486. [Google Scholar] [CrossRef]
- Topic, E.; Nikolac, N.; Panteghini, M.; Theodorsson, E.; Salvagno, G.L.; Miler, M.; Simundic, A.M.; Infusino, I.; Nordin, G.; Westgard, S. How to assess the quality of your analytical method? Clin. Chem. Lab. Med. 2015, 53, 1707–1718. [Google Scholar] [CrossRef] [PubMed]
- McAvera, R.; Quinn, J.; Murphy, P.; Glavey, S. Genetic Abnormalities in Extramedullary Multiple Myeloma. Int. J. Mol. Sci. 2023, 24, 11259. [Google Scholar] [CrossRef] [PubMed]
- Keats, J.J.; Maxwell, C.A.; Taylor, B.J.; Hendzel, M.J.; Chesi, M.; Bergsagel, P.L.; Larratt, L.M.; Mant, M.J.; Reiman, T.; Belch, A.R.; et al. Overexpression of transcripts originating from the MMSET locus characterizes all t(4;14)(p16;q32)-positive multiple myeloma patients. Blood 2005, 105, 4060–4069. [Google Scholar] [CrossRef]
- Stong, N.; Ortiz-Estévez, M.; Towfic, F.; Samur, M.; Agarwal, A.; Corre, J.; Flynt, E.; Munshi, N.; Avet-Loiseau, H.; Thakurta, A. The location of the t(4;14) translocation breakpoint within the NSD2 gene identifies a subset of patients with high-risk NDMM. Blood 2023, 141, 1574–1583. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Oligonucleotides for IGH (reference sequence: NG_001019.6) | |
| Forward primer for JH region | 5′-GCCCTTGTTAATGGACTTGGA-3′ |
| Hydrolysis probe for JH region | 5′-FAM-TCCATGCCAAAGCTTTGCAAGGC-TAMRA-3′ (for qPCR) |
| 5′-FAM-TCCATGCCAAAGCTTTGCAAGGC-ZEN/lowa Black FQ-3′ (for dPCR) | |
| Oligonucleotides for NSD2 (reference sequence: NG_009269.1) | |
| Reverse primer for exon 2 | 5′-GGATTTCTGGTGCCTGCTTC-3′ |
| Reverse primer for exon 3 | 5′-CCACACCAAATCACCAACGT-3′ |
| Reverse primer for exon 4 | 5′-CTCCTTCAAAAGCTACGAGGC-3′ |
| Overall | MB4-1 | MB4-2 | MB4-3 | p | |
|---|---|---|---|---|---|
| N (%) | 104 | 73 (70.5) | 11 (10.5) | 20 (19) | |
| Sex F/M | 53/51 | 38/35 | 5/6 | 10/10 | 0.916 |
| Age at diagnosis (year) median (interquartile range) | 62 (17) | 62 (17) | 64 (14) | 60 (15) | 0.445 |
| Heavy-chain isotype IgG IgA no heavy chain | 104 50 50 4 | 37 33 3 | 3 8 0 | 10 9 1 | 0.53 |
| Light chain kappa lambda no data | 101 64 37 3 | 43 27 3 | 6 5 0 | 15 5 0 | 0.557 |
| LDH (U/L) median (interquartile range) | 221 (330) | 220 (338) | 266 (371) | 210 (270) | 0.838 |
| Albumin (g/L) median (interquartile range) | 32 (38) | 29 (37) | 34 (44) | 34 (36) | 0.191 |
| Hemoglobin (g/L) median (interquartile range) | 86 (107) | 85 (106) | 91 (118) | 88 (87) | 0.737 |
| beta-2 microglobulin (μg/mL) median (interquartile range) from 85 patients | 4.51 (5.14) | 4.35 (4.98) | 5.25 (13.35) | 3.87 (6.47) | 0.764 |
| ISS | 87 | 60 | 9 | 18 | 0.73 |
| I | 32 (37.6%) | 22 (36.6%) | 3 (33.4%) | 7 (38.9%) | |
| II | 20 (23.5%) | 12 (20%) | 2 (22.2%) | 6 (33.3%) | |
| III | 35 (41.2%) | 26 (43.4%) | 4 (44.4%) | 5 (27.8%) | |
| Additional cytogenetics (1q amplification/17p deletion) | 64 | 42 | 6 | 12 | 0.722 |
| Patients treated with autologous stem cell transplantation | 49/79 | 32/57 | 3/5 | 14/17 | 0.147 |
| Univariate Analyses (n = 104) | Multivariate Analyses (n = 87) | |||
|---|---|---|---|---|
| Hazard ratio (95% CI) | p | Hazard ratio (95% CI) | p | |
| Age at diagnosis (in years) | 1.033 (1.011–1.055) | 0.003 | 1.024 (0.999–1.049) | 0.061 |
| ISS (ISS I as reference) | 1.503 (1.128–2.003) | 0.005 | 1.518 (1.139–2.024) | 0.004 |
| Breakpoint types (MB4-1 as reference) | 0.006 | 0.018 | ||
| MB4-1 vs. MB4-2 | 2.839 (1.442–5.59) | 0.003 | 2.897 (1.345–6.237) | 0.007 |
| MB4-1 vs. MB4-3 | 0.855 (0.484–1.509) | 0.588 | 0.906 (0.488–1.682) | 0.754 |
| Breakpoint Type | Our Cohort n (%) | Keats n (%) [23] | Lazareth n (%) [11] | Li n (%) [12] | Stong n (%) [24] |
|---|---|---|---|---|---|
| MB4-1 | 74 (69.8%) | 32 (71.1) | 159 (62.1) | 25 (47.2) | 66 (60.5) |
| MB4-2 | 11 (10.4%) | 6 (13.3) | 54 (21.1) | 12 (22.6) | 16 (14.7) |
| MB4-3 | 21 (19.8%) | 7 (15.6) | 43 (16.8) | 16 (30.2) | 27 (24.8) |
| Total | 106 | 45 | 256 | 53 | 109 |
| p= | 0.7552 | 0.0531 | 0.016 * | 0.3514 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bors, A.; Kozma, A.; Tomán, Á.; Őrfi, Z.; Kondor, N.; Tasnády, S.; Vályi-Nagy, I.; Reményi, P.; Mikala, G.; Andrikovics, H. IGH::NSD2 Fusion Gene Transcript as Measurable Residual Disease Marker in Multiple Myeloma. Cancers 2024, 16, 283. https://doi.org/10.3390/cancers16020283
Bors A, Kozma A, Tomán Á, Őrfi Z, Kondor N, Tasnády S, Vályi-Nagy I, Reményi P, Mikala G, Andrikovics H. IGH::NSD2 Fusion Gene Transcript as Measurable Residual Disease Marker in Multiple Myeloma. Cancers. 2024; 16(2):283. https://doi.org/10.3390/cancers16020283
Chicago/Turabian StyleBors, András, András Kozma, Ágnes Tomán, Zoltán Őrfi, Nóra Kondor, Szabolcs Tasnády, István Vályi-Nagy, Péter Reményi, Gábor Mikala, and Hajnalka Andrikovics. 2024. "IGH::NSD2 Fusion Gene Transcript as Measurable Residual Disease Marker in Multiple Myeloma" Cancers 16, no. 2: 283. https://doi.org/10.3390/cancers16020283
APA StyleBors, A., Kozma, A., Tomán, Á., Őrfi, Z., Kondor, N., Tasnády, S., Vályi-Nagy, I., Reményi, P., Mikala, G., & Andrikovics, H. (2024). IGH::NSD2 Fusion Gene Transcript as Measurable Residual Disease Marker in Multiple Myeloma. Cancers, 16(2), 283. https://doi.org/10.3390/cancers16020283