Inorganic Polyphosphate Promotes Colorectal Cancer Growth via TRPM8 Receptor Signaling Pathway

 , , , ,
, , , ,  ,
,  ,
,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients’ Samples
2.2. Tissue Preparation
2.3. Inorganic Polyphosphate Detection Assay
2.4. Cell Culture and Reagents
2.5. Cellular Treatments
2.6. Immunoblotting
2.7. Knockdown of TRPM8 with siRNA
2.8. Crystal Violet Assay
2.9. Immunofluorescence Microscopy
2.10. CRC Tumor Organoids
2.11. CRC Tumor Organoid Growth Test
2.12. Cell Lines-Derived Spheroids
2.13. Gene Expression Analysis by Real-Time Quantitative Reverse Transcription PCR
2.14. Cell-Cycle Assay
2.15. Statistical Analysis
3. Results
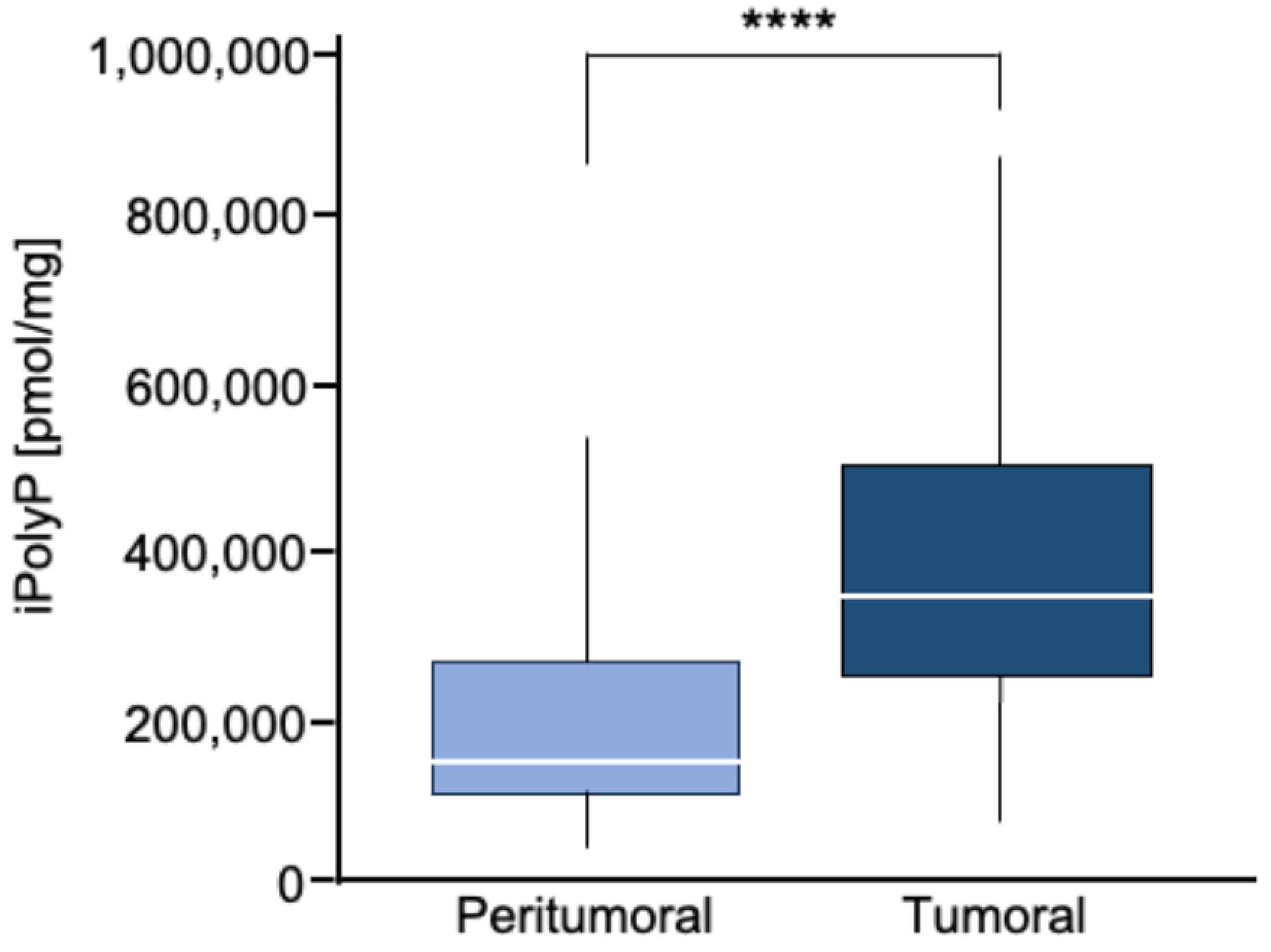
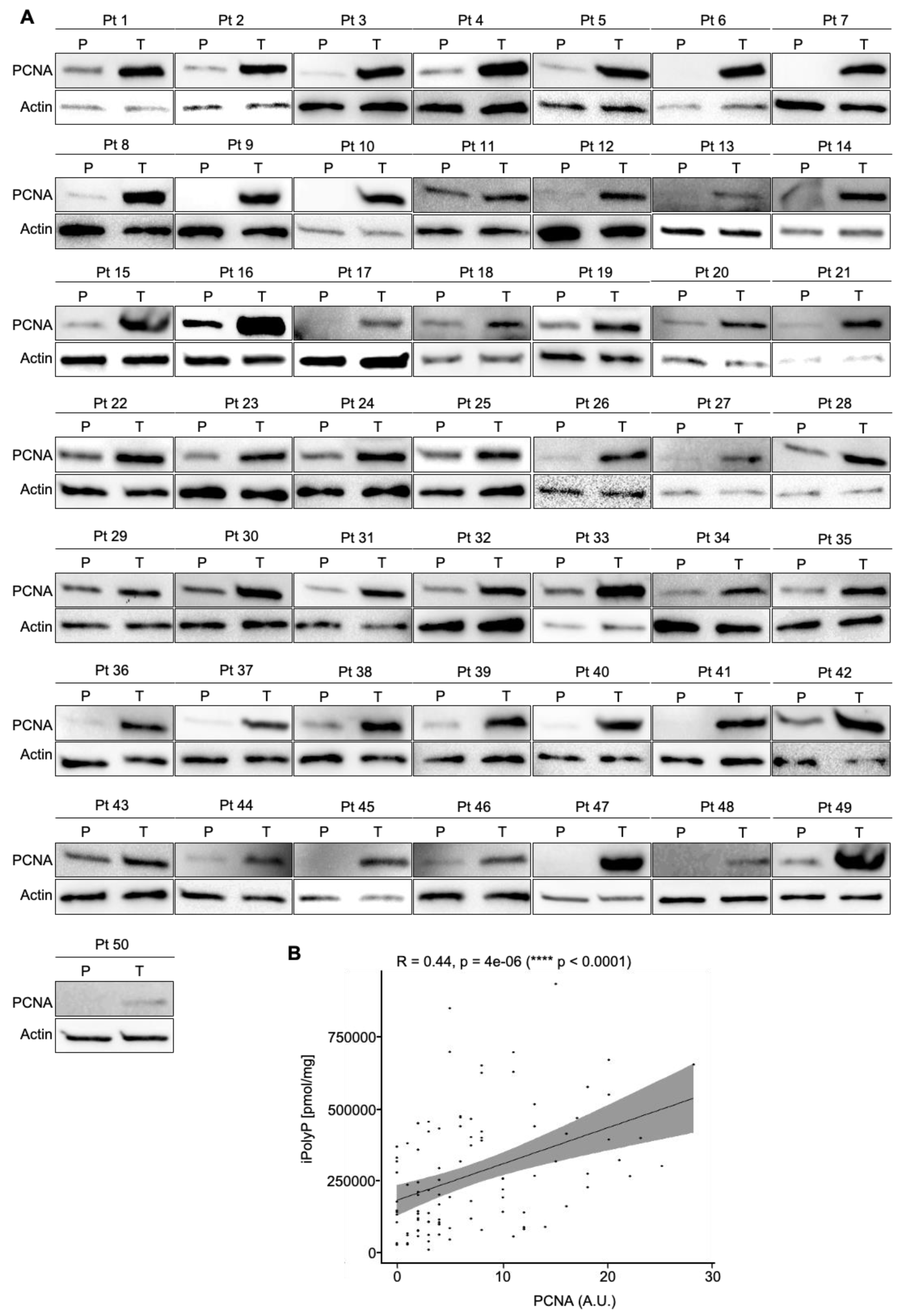
3.1. Human Colorectal Cancer Tissue Displays Enhanced Levels of iPolyP That Are Correlated with the Proliferation Marker PCNA
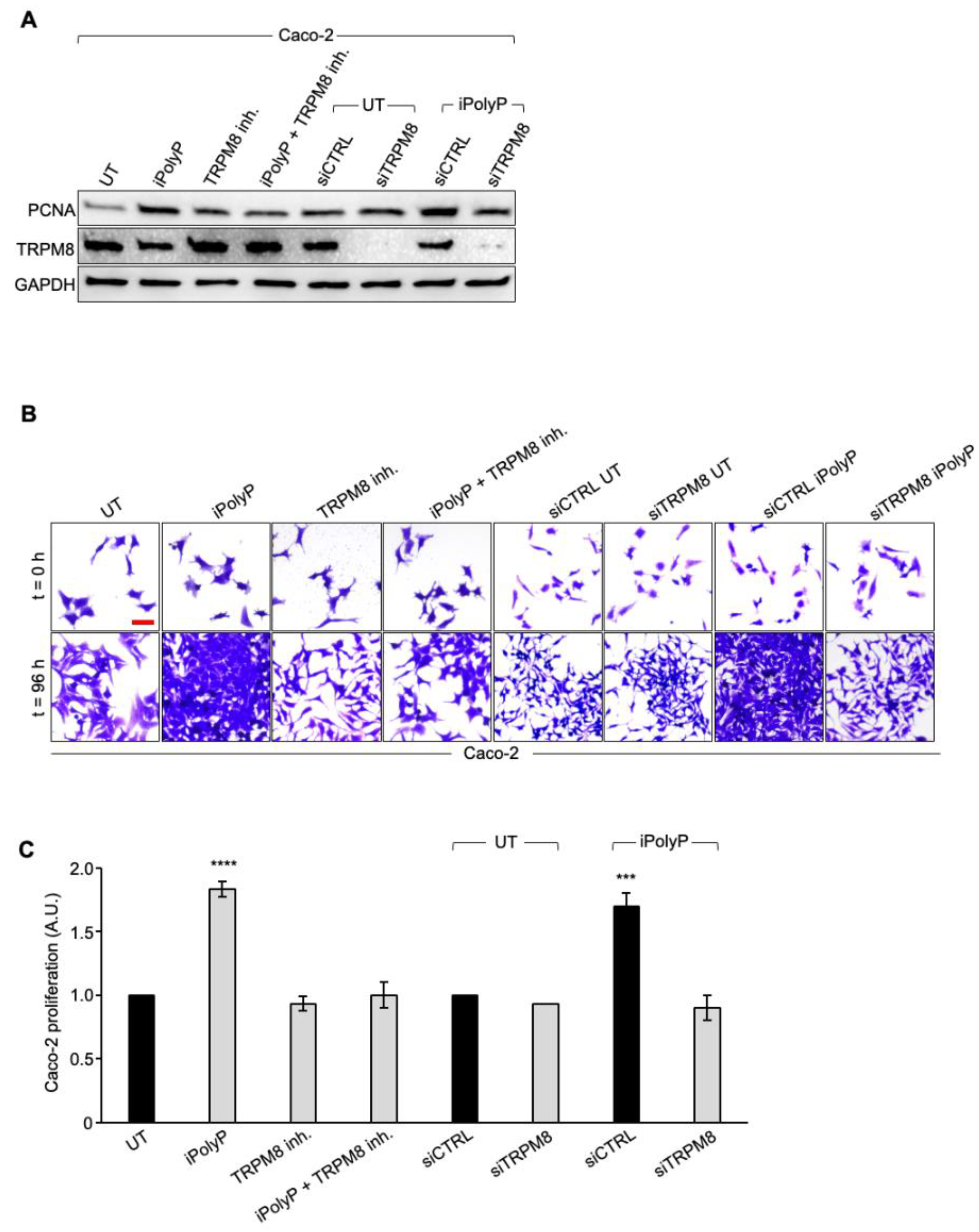
3.2. The iPolyP–TRPM8 Signaling Axis Sustains Colorectal Cancer Cell Proliferation
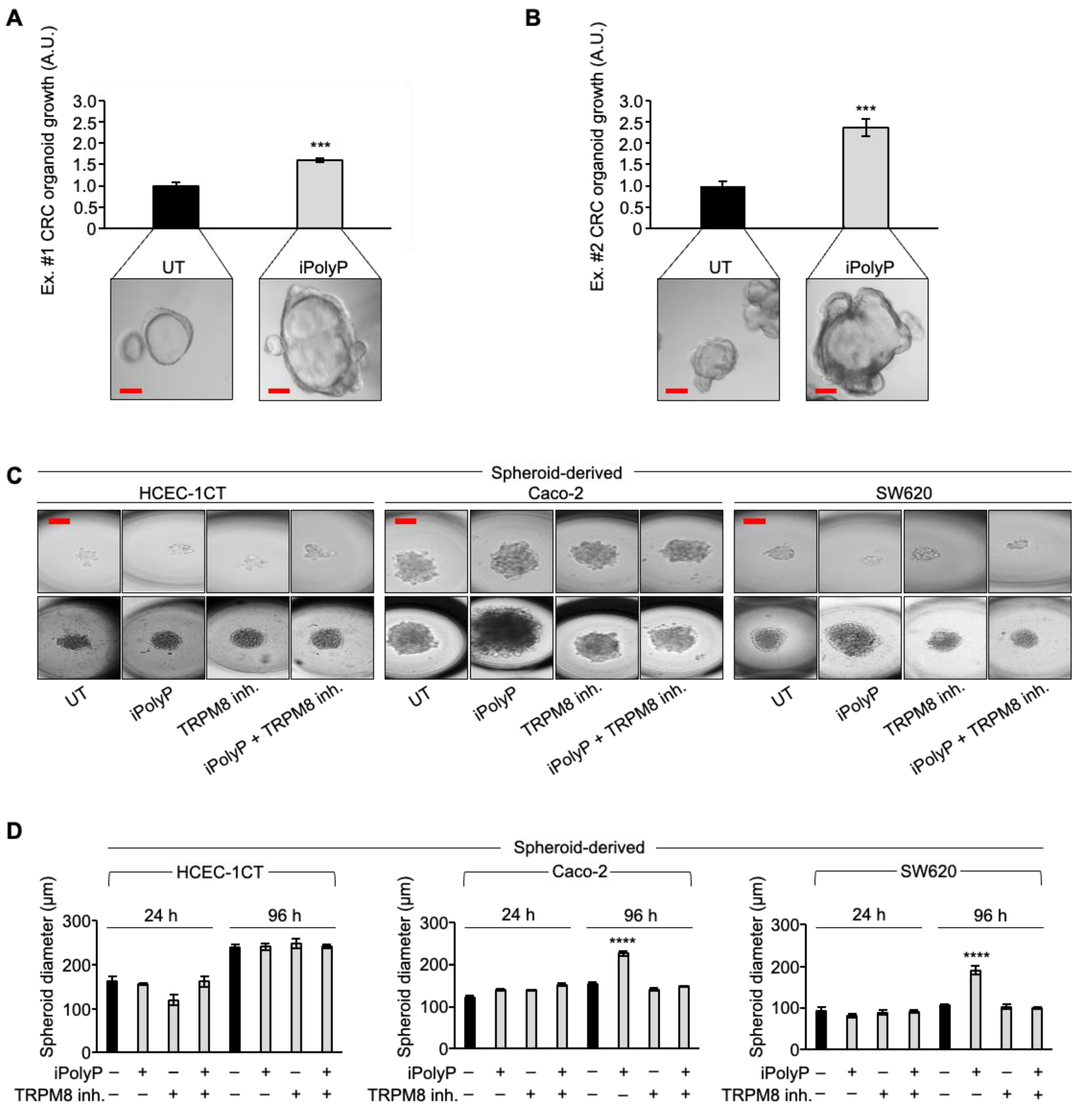
3.3. iPolyP Encourages CRC Patient-Derived Organoids’ Growth and Caco-2- and SW620 Cells-Derived Spheroids’ Formation
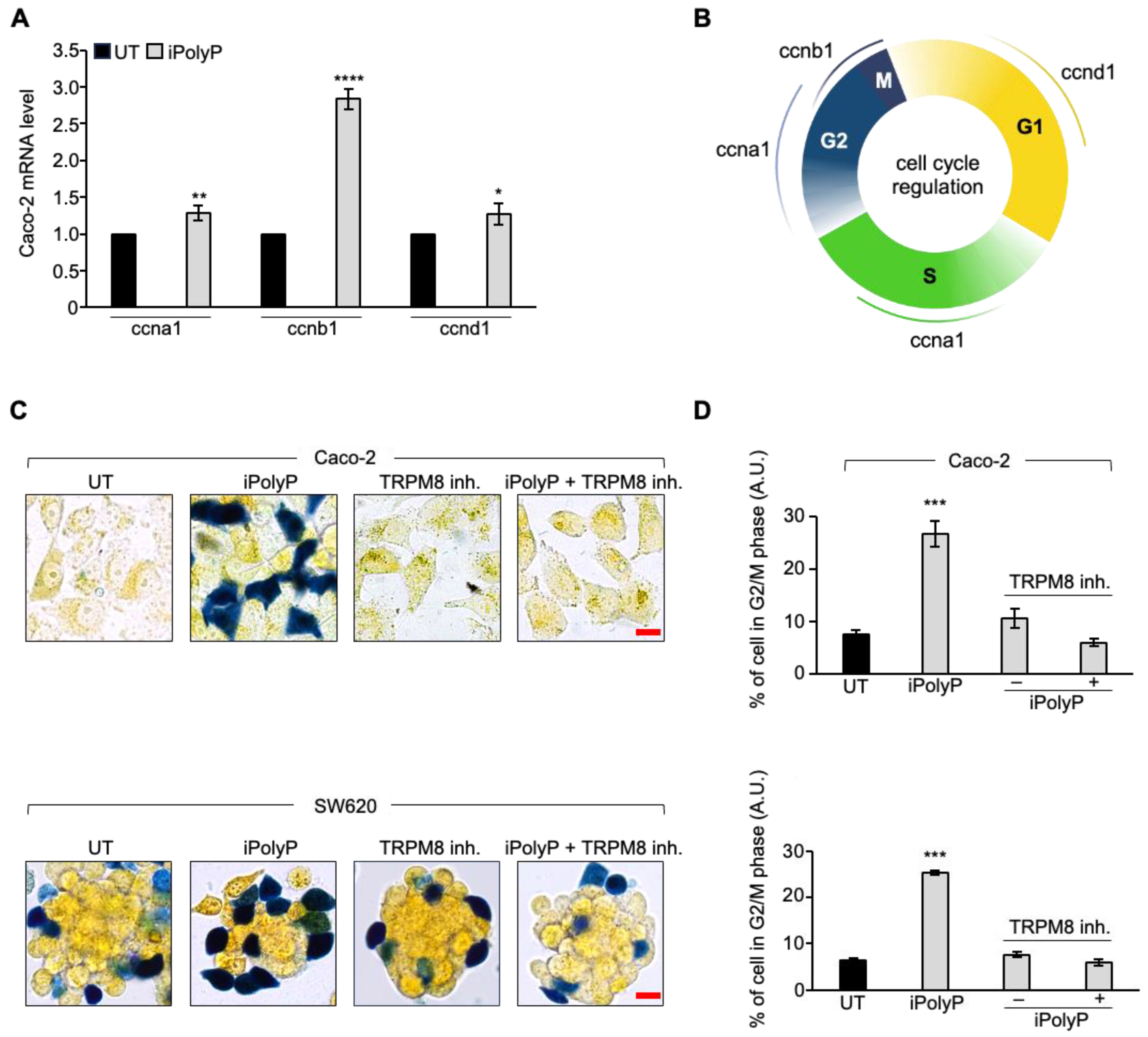
3.4. iPolyP Induces ccnb1 Expression in the Caco-2 Cell Line and Drives the Cells into M Phase via the TRPM8 Receptor
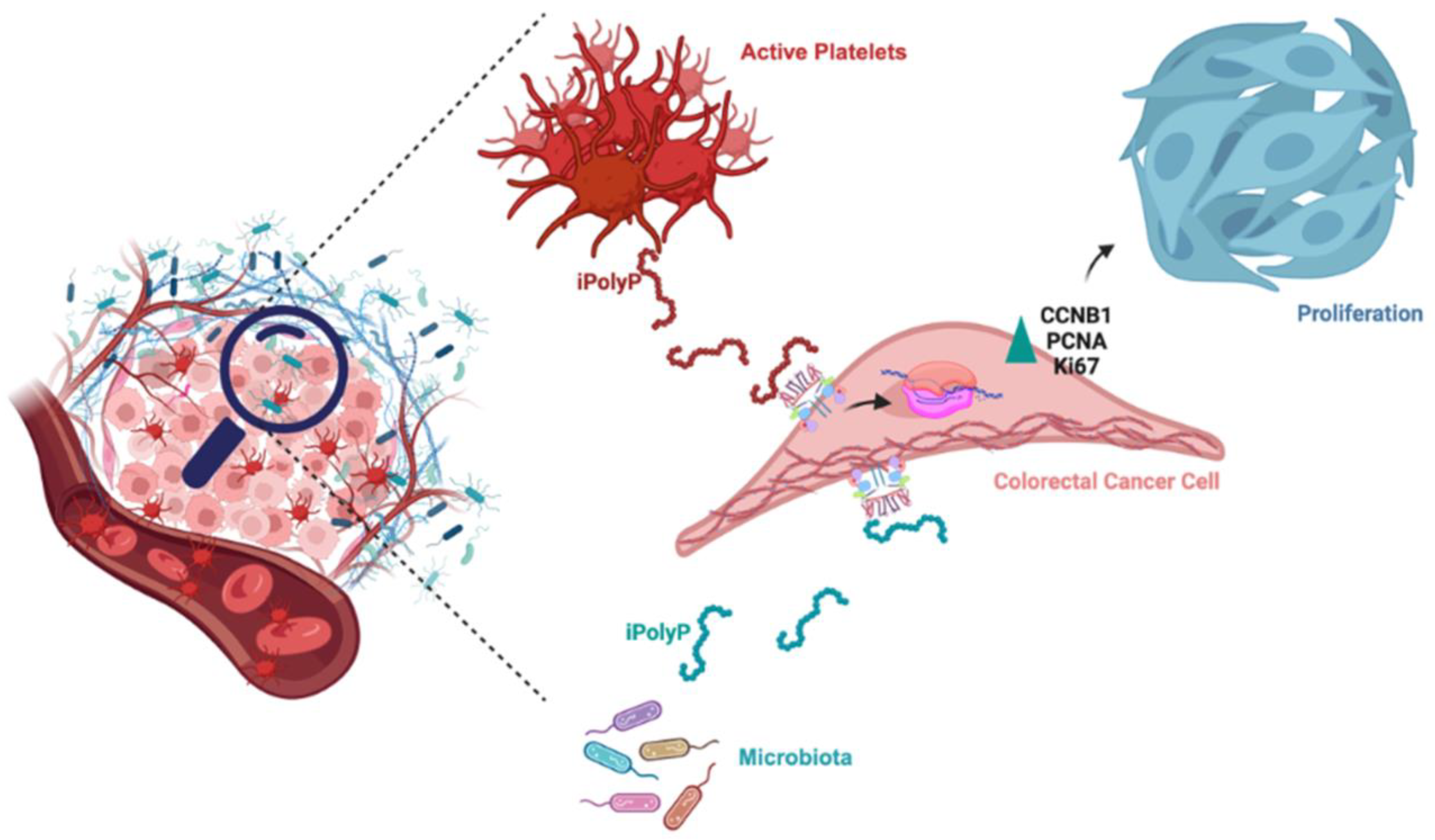
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Jasperson, K.W.; Tuohy, T.M.; Neklason, D.W.; Burt, R.W. Hereditary and Familial Colon Cancer. Gastroenterology 2010, 136, 2044–2058. [Google Scholar] [CrossRef] [PubMed]
- Keum, N.; Giovannucci, E. Global Burden of Colorectal Cancer: Emerging Trends, Risk Factors and Prevention Strategies. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 713–732. [Google Scholar] [CrossRef] [PubMed]
- Fearon, E.R. Molecular Genetics of Colorectal Cancer. Annu. Rev. Pathol. 2011, 6, 479–507. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.; Greten, F.R. The Inflammatory Pathogenesis of Colorectal Cancer. Nat. Rev. Immunol. 2021, 21, 653–667. [Google Scholar] [CrossRef]
- Kim, J.; Lee, H.K. Potential Role of the Gut Microbiome In Colorectal Cancer Progression. Front. Immunol. 2021, 12, 807648. [Google Scholar] [CrossRef]
- Kudryavtseva, A.V.; Lipatova, A.V.; Zaretsky, A.R.; Moskalev, A.A.; Fedorova, M.S.; Rasskazova, A.S.; Shibukhova, G.A.; Snezhkina, A.V.; Kaprin, A.D.; Alekseev, B.Y.; et al. Important molecular genetic markers of colorectal cancer. Oncotarget 2016, 7, 53959–53983. [Google Scholar] [CrossRef]
- Chakradhar, S. Colorectal cancer: 5 big questions. Nature 2015, 521, S16. [Google Scholar] [CrossRef]
- Hassanian, S.M.; Avan, A.; Ardeshirylajimi, A. Inorganic polyphosphate: A key modulator of inflammation. J. Thromb. Haemost. 2017, 15, 213–218. [Google Scholar] [CrossRef]
- Kulakovskaya, T.; Pavlov, E.; Dedkova, E.N. Inorganic Polyphosphate in Eukaryotic Cells, 1st ed.; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Kulakovskaya, E.V.; Zemskova, M.Y.; Kulakovskaya, T.V. Inorganic Polyphosphate and Cancer. Biochemistry 2018, 83, 961–968. [Google Scholar] [CrossRef]
- Kornberg, A. Inorganic polyphosphate: Toward making a forgotten polymer unforgettable. J. Bacteriol. 1995, 177, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Pisoni, R.L.; Lindley, E.R. Incorporation of [32P] orthophosphate into long chains of inorganic polyphosphate within lysosomes of human fibroblasts. J. Biol. Chem. 1992, 267, 3626–3631. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, F.A.; Lea, C.R.; Oldfield, E.; Docampo, R. Human platelet dense granules contain polyphosphate and are similar to acidocalcisomes of bacteria and unicellular eukaryotes. J. Biol. Chem. 2004, 279, 44250–44257. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Sanchez, D.; Hernandez-Ruiz, L.; Ruiz, F.A.; Docampo, R. Polyphosphate is a novel pro-Inflammatory regulator of mast cells and is located in acidocalcisomes. J. Biol. Chem. 2012, 287, 28435–28444. [Google Scholar] [CrossRef]
- Müller, W.E.G.; Wang, S.; Neufurth, M.; Kokkinopoulou, M.; Feng, Q.; Schröder, H.C.; Wang, X. Polyphosphate as a donor of high-energy phosphate for the synthesis of ADP and ATP. J. Cell Sci. 2017, 130, 2747–2756. [Google Scholar] [CrossRef]
- Bae, J.-S.; Lee, W.; Rezaie, A.R. Polyphosphate elicits pro-inflammatory responses that are counteracted By activated protein C in both cellular and animal models. J. Thromb. Haemost. 2012, 10, 1145–1151. [Google Scholar] [CrossRef]
- Müller, F.; Mutch, N.J.; Schenk, W.A.; Smith, S.A.; Esterl, L.; Spronk, H.M.; Schmidbauer, S.; Gahl, W.A.; Morrissey, J.H.; Renné, T. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell 2009, 139, 1143–1156. [Google Scholar] [CrossRef] [PubMed]
- Hassanian, S.M.; Dinarvand, P.; Smith, S.A.; Rezaie, A.R. Inorganic polyphosphate elicits pro-inflammatory responses through activation of the mammalian target of rapamycin complexes 1 and 2 in vascular endothelial cells. J. Thromb. Haemost. 2015, 13, 860–871. [Google Scholar] [CrossRef]
- Semeraro, F.; Ammollo, C.T.; Morrissey, J.H.; Dale, G.L.; Friese, P.; Esmon, N.L.; Esmon, C.T. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood 2011, 118, 1952–1961. [Google Scholar] [CrossRef]
- Wat, J.M.; Foley, J.H.; Krisinger, M.J.; Ocariza, L.M.; Lei, V.; Wasney, G.A.; Lameignere, E.; Strynadka, N.C.; Smith, S.A.; Morrissey, J.H.; et al. Polyphosphate suppresses complement via the terminal pathway. Blood 2014, 123, 768–776. [Google Scholar] [CrossRef]
- Wijeyewickrema, L.C.; Lameignere, E.; Hor, L.; Duncan, R.C.; Shiba, T.; Travers, R.J.; Kapopara, P.R.; Lei, V.; Smith, S.A.; Kim, H.; et al. Polyphosphate is a novel cofactor for regulation of complement by a serpin, C1 inhibitor. Blood 2016, 128, 1766–1776. [Google Scholar] [CrossRef] [PubMed]
- Galasso, A.; Zollo, M. The Nm23-H1-h-Prune complex in cellular physiology: A tip of the iceberg’ protein network perspective. Mol. Cell Biochem. 2009, 329, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Tammenkoski, M.; Koivula, K.; Cusanelli, E.; Zollo, M.; Steegborn, C.; Baykov, A.A.; Lahti, R. Human metastasis regulator protein H-prune is a short-chain exopolyphosphatase. Biochemistry 2008, 47, 9707–9713. [Google Scholar] [CrossRef]
- Wang, L.; Fraley, C.D.; Faridi, J.; Kornberg, A.; Roth, R.A. Inorganic polyphosphate stimulates mammalian TOR, a kinase involved in the proliferation of mammary cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11249–12254. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Yamamoto, S.; Yamaguchi, K.; Sato, M.; Kaneko, Y.; Goto, S.; Goto, Y.; Narita, I. Inorganic polyphosphate potentiates lipopolysaccharide-induced macrophage inflammatory response. J. Biol. Chem. 2020, 295, 4014–4023. [Google Scholar] [CrossRef] [PubMed]
- Dinarvand, P.; Hassanian, S.M.; Qureshi, S.H.; Manithody, C.; Eissenberg, J.C.; Yang, L.; Rezaie, A.R. Polyphosphate amplifies proinflammatory responses of nuclear proteins through interaction with receptor. for advanced glycation end products and P2Y1 purinergic receptor. Blood 2014, 123, 935–945. [Google Scholar] [CrossRef]
- Zakharian, E.; Thyagarajan, B.; French, R.J.; Pavlov, E.; Rohacs, T. Inorganic polyphosphate modulates TRPM8 Channels. PLoS ONE 2009, 4, e5404. [Google Scholar] [CrossRef]
- Mollace, A.; Coluccio, M.L.; Donato, G.; Mollace, V.; Malara, N. Cross-talks in colon cancer between RAGE/AGEs axis and inflammation/immunotherapy. Oncotarget 2021, 12, 1281–1295. [Google Scholar] [CrossRef]
- Hassanian, S.M.; Ardeshirylajimi, A.; Dinarvand, P.; Rezaie, A.R. Inorganic polyphosphate promotes cyclin D1 synthesis through activation of mTOR/Wnt/β-catenin signaling in endothelial cells. J. Thromb. Haemost. 2016, 14, 2261–2273. [Google Scholar] [CrossRef]
- Dillard, C.; Borde, C.; Mohammad, A.; Puchois, V.; Jourdren, L.; Larsen, A.K.; Sabbah, M.; Maréchal, V.; Escargueil, A.E.; Pramil, E. Expression Pattern of Purinergic Signaling Components in Colorectal Cancer Cells and Differential Cellular Outcomes Induced by Extracellular ATP and Adenosine. Int. J. Mol. Sci. 2021, 22, 11472. [Google Scholar] [CrossRef]
- Pagano, P.; Romano, B.; Cicia, D.; Iannotti, F.A.; Venneri, T.; Lucariello, G.; Nanì, M.F.; Cattaneo, F.; De Cicco, P.; D’Armiento, M.; et al. TRPM8 indicates poor prognosis in colorectal cancer patients and its pharmacological targeting reduces tumour growth in mice by inhibiting Wnt/β-catenin signalling. Br. J. Pharmacol. 2023, 180, 235–251. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Zhang, F.; Feng, S.; Butay, K.J.; Borgnia, M.J.; Im, W.; Lee, S.-Y. Activation mechanism of the mouse cold-sensing TRPM8 channel by cooling agonist and PIP2. Science 2022, 378, eadd1268. [Google Scholar] [CrossRef]
- Tsutsumi, K.; Tippayamontri, T.; Hayashi, M.; Matsuda, N.; Goto, Y. The dynamic relationship between inorganic polyphosphate and adenosine triphosphate in human non-small cell lung cancer H1299 cells. FEBS Open Bio. 2024, 14, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Ogunwobi, O.O.; Mahmood, F.; Akingboye, A. Biomarkers in Colorectal Cancer: Current Research and Future Prospects. Int. J. Mol. Sci. 2020, 21, 5311. [Google Scholar] [CrossRef]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernández-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.S.; Karuniawati, H.; Jairoun, A.A.; Urbi, Z.; Ooi, D.J.; John, A.; Lim, Y.C.; Kibria, K.M.K.; Mohiuddin, A.K.M.; Ming, L.C.; et al. Colorectal Cancer: A Review of Carcinogenesis, Global Epidemiology, Current Challenges, Risk Factors, Preventive and Treatment Strategies. Cancers 2022, 14, 1732. [Google Scholar] [CrossRef]
- Hibberd, A.A.; Lyra, A.; Ouwehand, A.C.; Rolny, P.; Lindegren, H.; Cedgård, L.; Wettergren, Y. Intestinal microbiota is altered in patients with colon cancer and modified by probiotic intervention. BMJ Open Gastroenterol. 2017, 4, e000145. [Google Scholar] [CrossRef] [PubMed]
- Zackular, J.P.; Baxter, N.T.; Iverson, K.D.; Sadler, W.D.; Petrosino, J.F.; Chen, G.Y.; Schloss, P.D. The gut microbiome modulates colon tumorigenesis. mBio 2013, 4, e00692-13. [Google Scholar] [CrossRef]
- Karaca, C.; Karaman, E.D.; Leblebici, A.; Kurter, H.; Ellidokuz, H.; Koc, A.; Ellidokuz, E.B.; Isik, Z.; Basbinar, Y. New treatment alternatives for primary and metastatic colorectal cancer by an integrated transcriptome and network analyses. Sci. Rep. 2024, 14, 8762. [Google Scholar] [CrossRef]
- Wu, Z.; Zuo, M.; Zeng, L.; Cui, K.; Liu, B.; Yan, C.; Chen, L.; Dong, J.; Shangguan, F.; Hu, W.; et al. OMA1 reprograms metabolism under hypoxia to promote colorectal cancer development. EMBO Rep. 2021, 22, e50827. [Google Scholar] [CrossRef]
- Rao, N.N.; Gómez-García, M.R.; Kornberg, A. Inorganic polyphosphate: Essential for growth and survival. Annu. Rev. Biochem. 2009, 78, 605–647. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, A.; Rao, N.N.; Ault-Riché, D. Inorganic polyphosphate: A molecule of many functions. Annu. Rev. Biochem. 1999, 68, 89–125. [Google Scholar] [CrossRef] [PubMed]
- Kumble, K.D.; Kornberg, A. Inorganic polyphosphate in mammalian cells and tissues. J. Biol. Chem. 1995, 70, 5818–5822. [Google Scholar] [CrossRef]
- Ahn, K.; Kornberg, A. Polyphosphate kinase from Escherichia coli. Purification and demonstration of a phosphoenzyme intermediate. J. Biol. Chem. 1990, 265, 11734–11739. [Google Scholar] [CrossRef]
- Schröder, H.C.; Kurz, L.; Müller, W.E.; Lorenz, B. Polyphosphate in bone. Biochemistry 2000, 65, 296–303. [Google Scholar]
- Docampo, R. Polyphosphate: A target for thrombosis attenuation. Blood 2014, 124, 3177–3178. [Google Scholar] [CrossRef]
- Dulal, S.; Keku, T.O. Gut microbiome and colorectal adenomas. Cancer J. 2014, 20, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Flemer, B.; Lynch, C.B.; Brown, J.M.R.; Jeffery, I.B.; Ryan, F.J.; Claesson, M.J.; O’Riordain, M.; Shanahan, F.; O’Toole, P.W.l. Tumour-associated and non-tumour-associated microbiota in colorectal cancer. Gut 2017, 66, 633–643. [Google Scholar] [CrossRef]
- Candon, H.L.; Allan, B.J.; Fraley, C.D.; Gaynor, E.C. Polyphosphate kinase 1 is a pathogenesis determinant in Campylobacter jejuni. J. Bacteriol. 2007, 189, 8099–8108. [Google Scholar] [CrossRef]
- Jenal, U.; Hengge-Aronis, R. Regulation by proteolysis in bacterial cells. Curr. Opin. Microbiol. 2003, 6, 163–172. [Google Scholar] [CrossRef]
- Bowlin, M.Q.; Gray, M.J. Inorganic Polyphosphate in Host and Microbe Biology. Trends Microbiol. 2021, 29, 1013–1023. [Google Scholar] [CrossRef] [PubMed]
- Boyineni, J.; Sredni, S.T.; Margaryan, N.V.; Demirkhanyan, L.; Tye, M.; Johnson, R.; Gonzalez-Nilo, F.; Hendrix, M.J.C.; Pavlov, E.; Soares, M.B.; et al. Inorganic polyphosphate as an energy source in Tumorigenesis. Oncotarget 2020, 11, 4613–4624. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Age | Sex | Diagnosis | TNM | Grading |
|---|---|---|---|---|---|
| 1 | 58 | f | CRC | T1N0MX | G3 |
| 2 | 58 | m | CRC | T1N0MX | G2 |
| 3 | 58 | f | CRC | T1N0MX | G2 |
| 4 | 81 | f | CRC | T2N0MX | G3 |
| 5 | 72 | m | CRC | T2N0MX | G2 |
| 6 | 78 | f | CRC | T2N0MX | G3 |
| 7 | 76 | m | CRC | T2N0MX | G3 |
| 8 | 82 | m | CRC | T2N0MX | G2 |
| 9 | 85 | m | CRC | T2N0MX | G2 |
| 10 | 73 | f | CRC | T2N0MX | G3 |
| 11 | 82 | f | CRC | T2N0MX | G2 |
| 12 | 81 | m | CRC | T2N1aMX | G2 |
| 13 | 70 | m | CRC | T2N1aMX | G3 |
| 14 | 60 | f | CRC | T2N1aM1 | G3 |
| 15 | 71 | m | CRC | T3N0MX | G2 |
| 16 | 75 | m | CRC | T3N0MX | G2 |
| 17 | 59 | f | CRC | T3N0MX | G3 |
| 18 | 69 | m | CRC | T3N0MX | G3 |
| 19 | 89 | m | CRC | T3N0MX | G3 |
| 20 | 72 | f | CRC | T3N0MX | G3 |
| 21 | 59 | f | CRC | T3N0MX | G3 |
| 22 | 77 | f | CRC | T3N0MX | G3 |
| 23 | 70 | m | CRC | T3N0MX | G2 |
| 24 | 65 | m | CRC | T3N0MX | G3 |
| 25 | 43 | f | CRC | T3N0MX | G2 |
| 26 | 76 | m | CRC | T3N0MX | G2 |
| 27 | 65 | f | CRC | T3N0MX | G2 |
| 28 | 74 | m | CRC | T3N0MX | G2 |
| 29 | 69 | f | CRC | T3N0MX | G3 |
| 30 | 71 | f | CRC | T3N0MX | G2 |
| 31 | 70 | f | CRC | T3N0M1b | G2 |
| 32 | 88 | m | CRC | T3N1aMX | G2 |
| 33 | 74 | m | CRC | T3N1aMX | G2 |
| 34 | 80 | f | CRC | T3N1aMX | G3 |
| 35 | 74 | m | CRC | T3N1aMX | G3 |
| 36 | 70 | f | CRC | T3N1aMX | G3 |
| 37 | 67 | f | CRC | T3N1aMX | G3 |
| 38 | 78 | m | CRC | T3N1bMX | G2/G3 |
| 39 | 79 | m | CRC | T3N1bMX | G3 |
| 40 | 77 | m | CRC | T3N1bMX | G3 |
| 41 | 83 | m | CRC | T3N2aMX | G3 |
| 42 | 76 | m | CRC | T4aN0MX | G3 |
| 43 | 56 | m | CRC | T4aN1MX | G3 |
| 44 | 35 | m | CRC | T4aN1bMX | G3 |
| 45 | 86 | m | CRC | T4aN2bMX | G2 |
| 46 | 93 | f | CRC | T4aN2bM1c | G3 |
| 47 | 81 | f | CRC | T4bN0MX | G2 |
| 48 | 71 | f | CRC | T4bN0MX | G2 |
| 49 | 53 | f | CRC | T4bN0MX | G2 |
| 50 | 74 | f | CRC | T4bN0MX | G2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arrè, V.; Balestra, F.; Scialpi, R.; Dituri, F.; Donghia, R.; Coletta, S.; Stabile, D.; Bianco, A.; Vincenti, L.; Fedele, S.; et al. Inorganic Polyphosphate Promotes Colorectal Cancer Growth via TRPM8 Receptor Signaling Pathway. Cancers 2024, 16, 3326. https://doi.org/10.3390/cancers16193326
Arrè V, Balestra F, Scialpi R, Dituri F, Donghia R, Coletta S, Stabile D, Bianco A, Vincenti L, Fedele S, et al. Inorganic Polyphosphate Promotes Colorectal Cancer Growth via TRPM8 Receptor Signaling Pathway. Cancers. 2024; 16(19):3326. https://doi.org/10.3390/cancers16193326
Chicago/Turabian StyleArrè, Valentina, Francesco Balestra, Rosanna Scialpi, Francesco Dituri, Rossella Donghia, Sergio Coletta, Dolores Stabile, Antonia Bianco, Leonardo Vincenti, Salvatore Fedele, and et al. 2024. "Inorganic Polyphosphate Promotes Colorectal Cancer Growth via TRPM8 Receptor Signaling Pathway" Cancers 16, no. 19: 3326. https://doi.org/10.3390/cancers16193326
APA StyleArrè, V., Balestra, F., Scialpi, R., Dituri, F., Donghia, R., Coletta, S., Stabile, D., Bianco, A., Vincenti, L., Fedele, S., Shen, C., Pettinato, G., Scavo, M. P., Giannelli, G., & Negro, R. (2024). Inorganic Polyphosphate Promotes Colorectal Cancer Growth via TRPM8 Receptor Signaling Pathway. Cancers, 16(19), 3326. https://doi.org/10.3390/cancers16193326