
BUB1 Inhibition Overcomes Radio- and Chemoradiation Resistance in Lung Cancer


 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. BUB1 Expression in Lung Cancer
2.2. Immunohistological Analysis of BUB1 Expression in Lung Tumor Microarrays
2.3. Cell Culture
2.4. Drug Treatment and Radiation
2.5. Cell Survival Assay
2.6. Clonogenic Cell Survival Assay
2.7. DNA Damage Assay
2.8. Quantitative Real-Time PCR
2.9. Statistical Analysis
3. Results
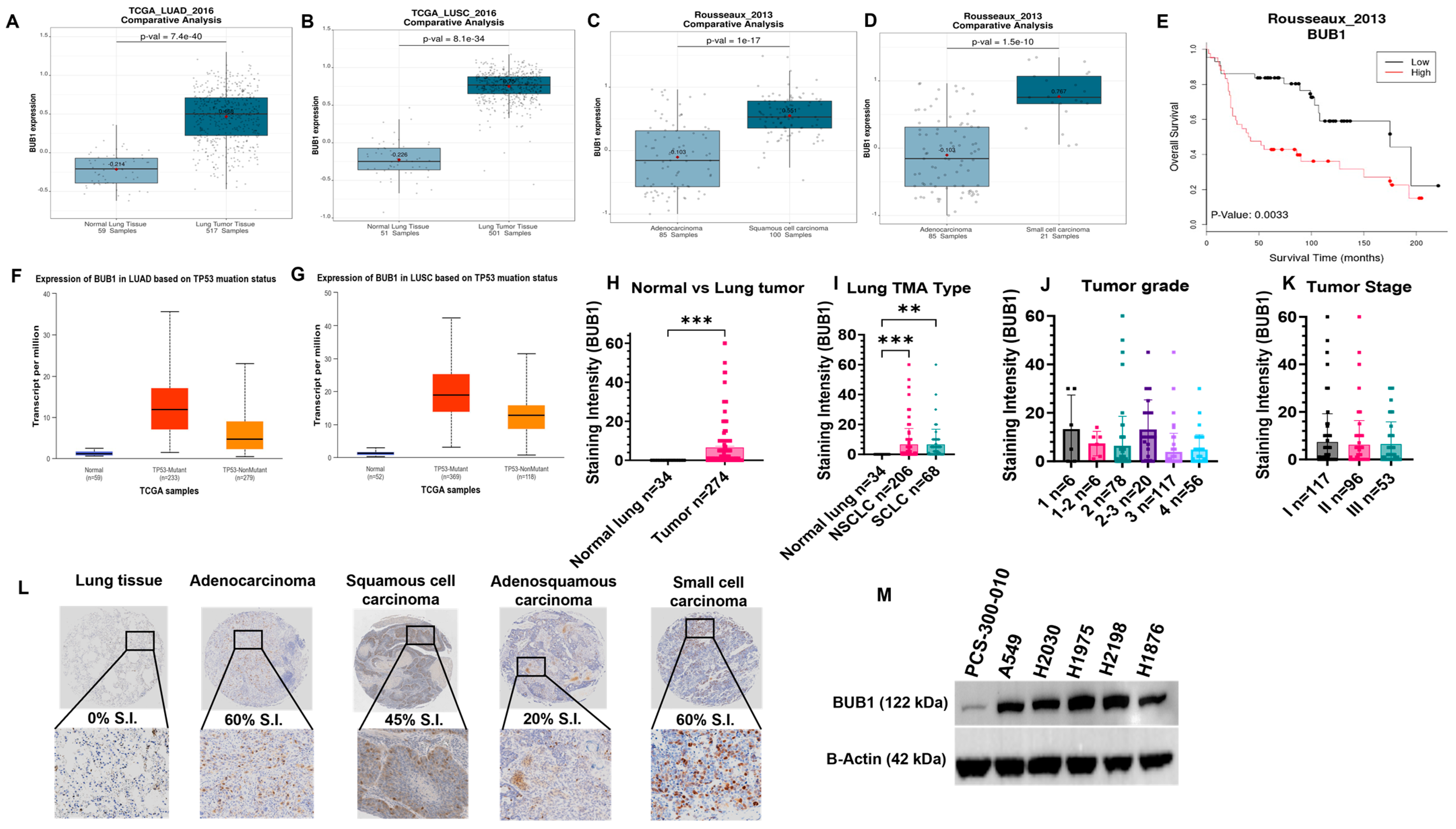
3.1. BUB1 Is Overexpressed in LUAD, LUSC, and SCLC, and Is Correlated with Poorer Survival
3.2. BUB1 Protein Is Overexpressed in Lung Tumor Tissues
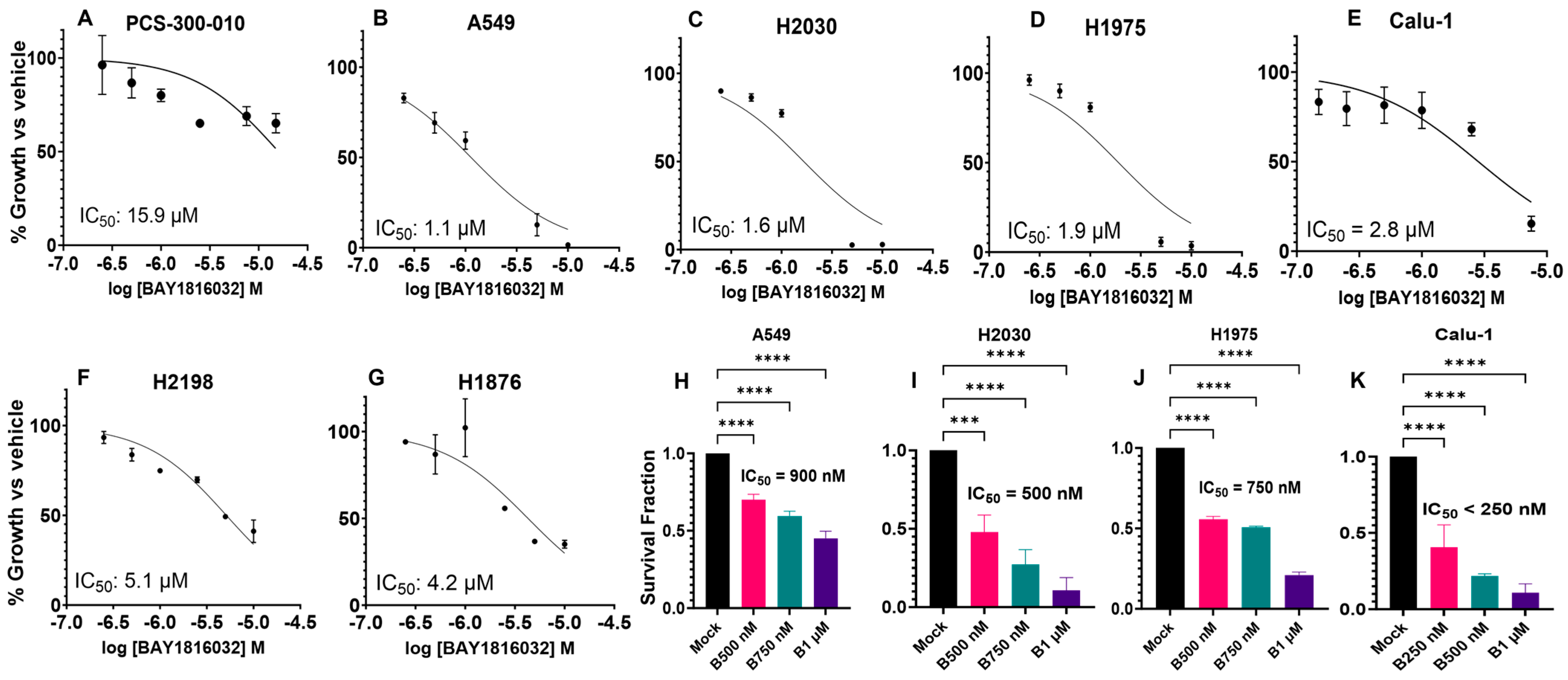
3.3. BUB1 Inhibition Suppresses Cell Proliferation and Clonogenic Capacity
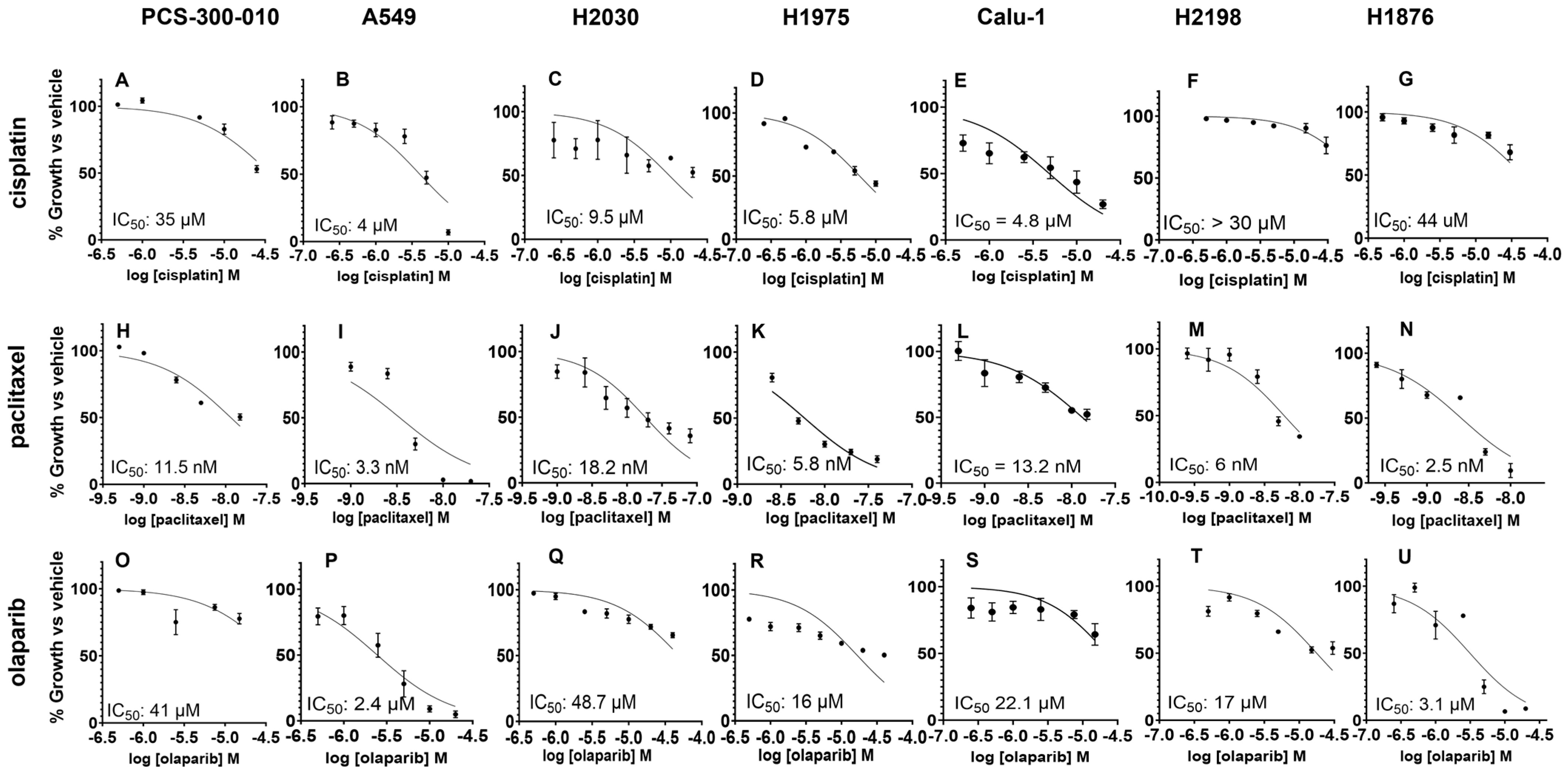
3.4. The Sequence and Doses of BUB1 Inhibitor Determine Sensitivity to Paclitaxel, Cisplatin, and Olaparib
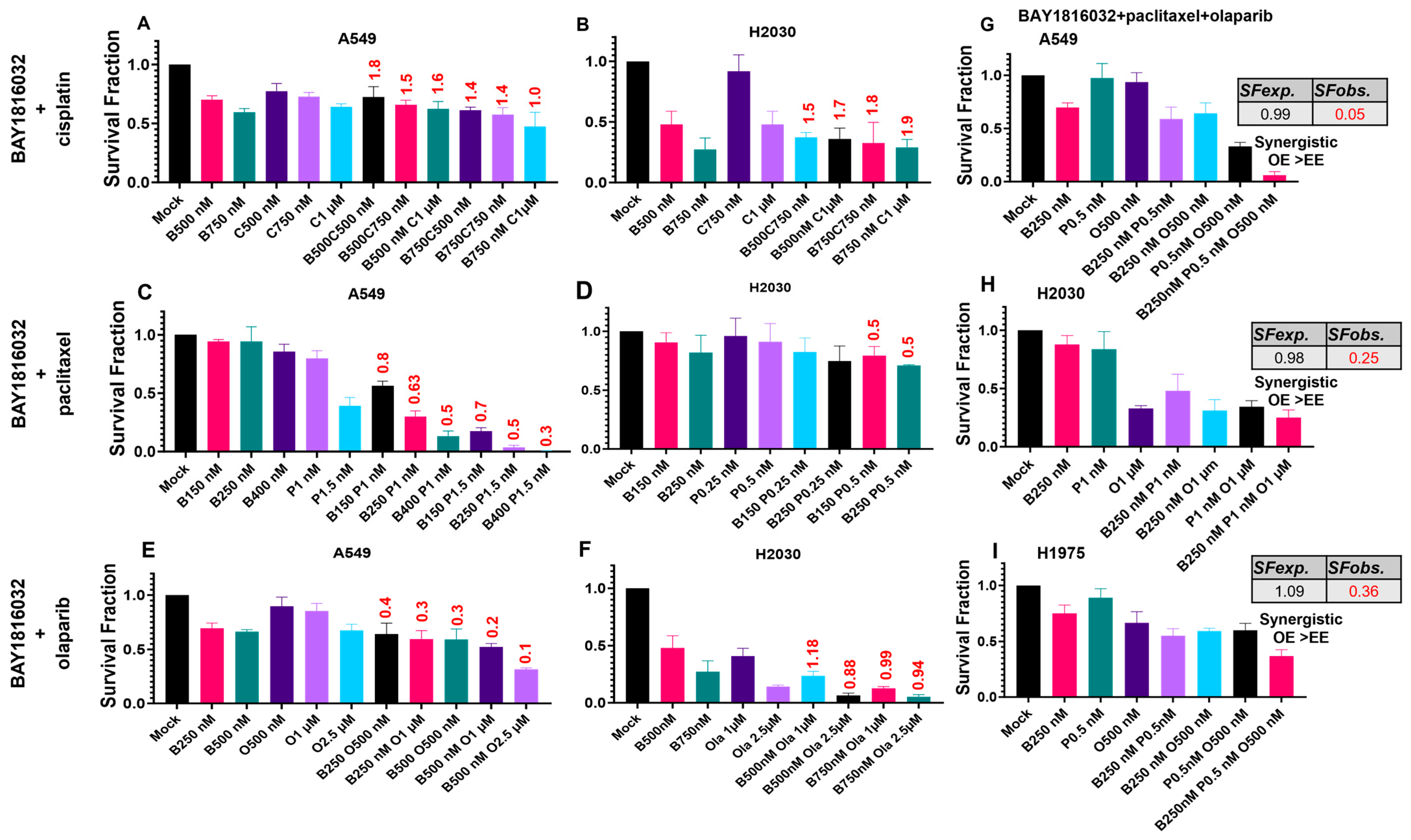
3.5. BUB1 Inhibition Chemosensitizes NSCLC in Double and Triple Drug Combinations
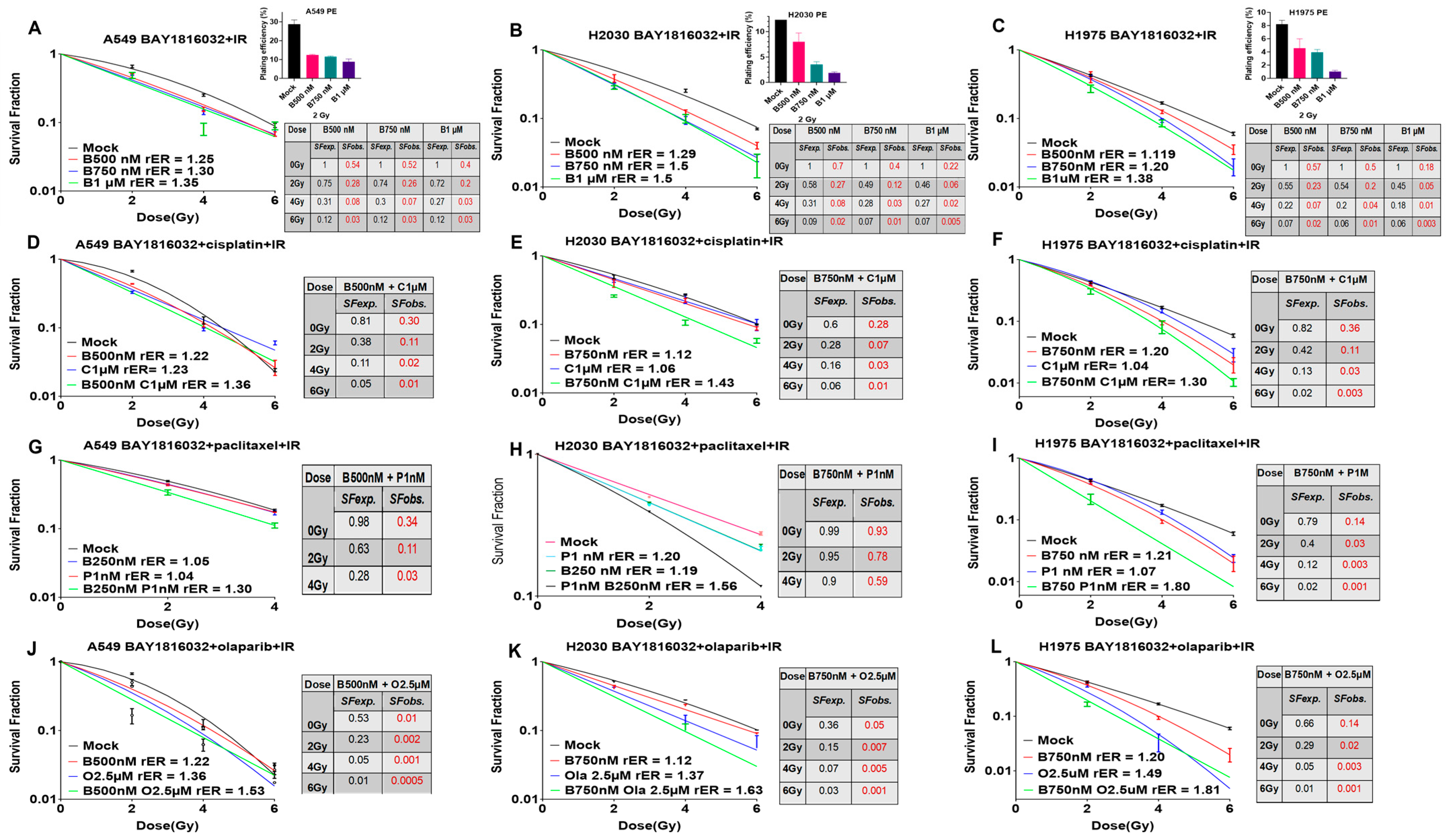
3.6. BUB1 Inhibition Sensitizes NSCLC to Radiation
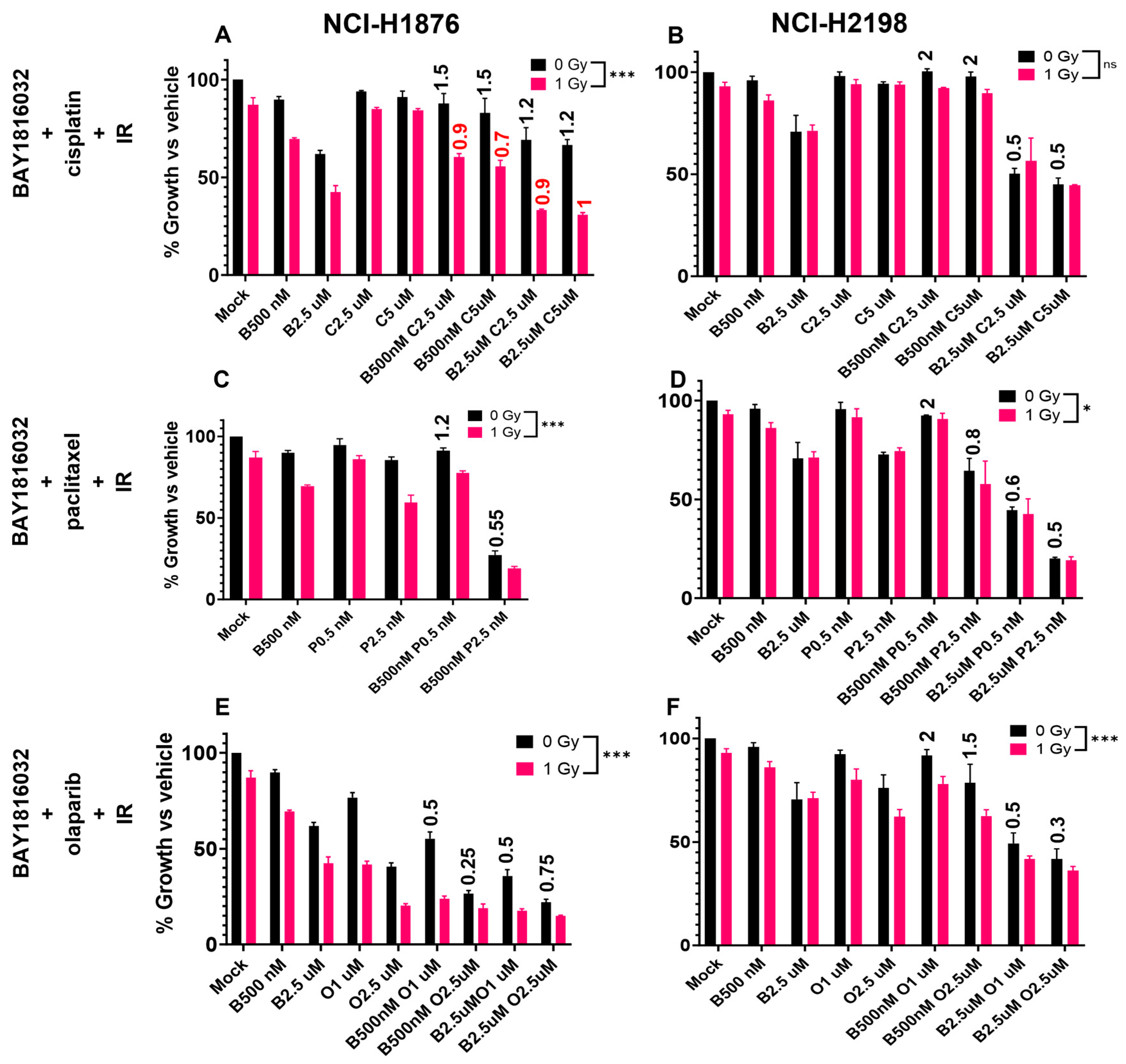
3.7. BUB1 Inhibition Sensitizes NSCLC and SCLC to Chemoradiation
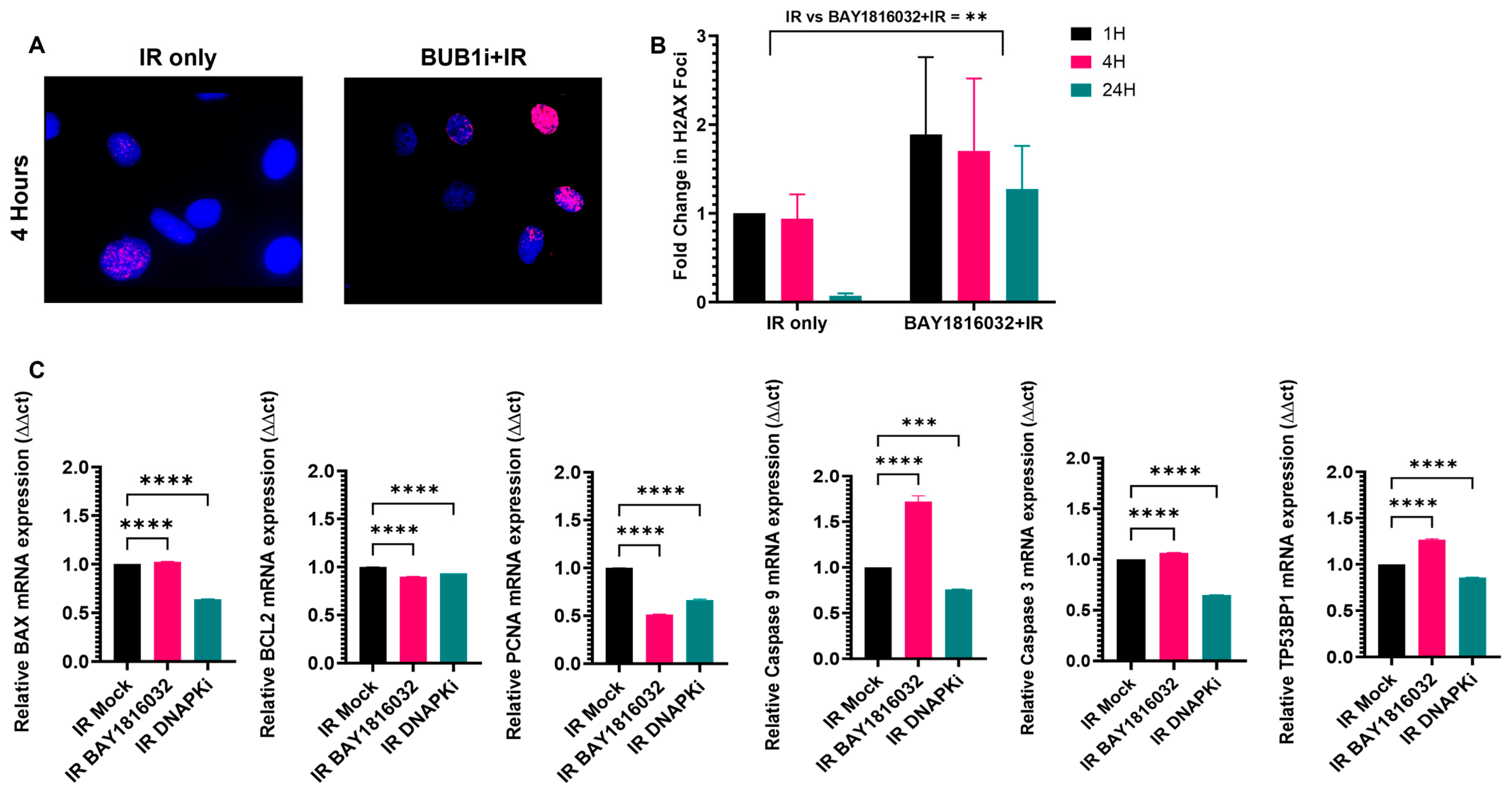
3.8. BUB1 Inhibition Delays DNA Double-Strand Break Repair and Elicits Pro-Apoptotic and Anti-Proliferative Responses
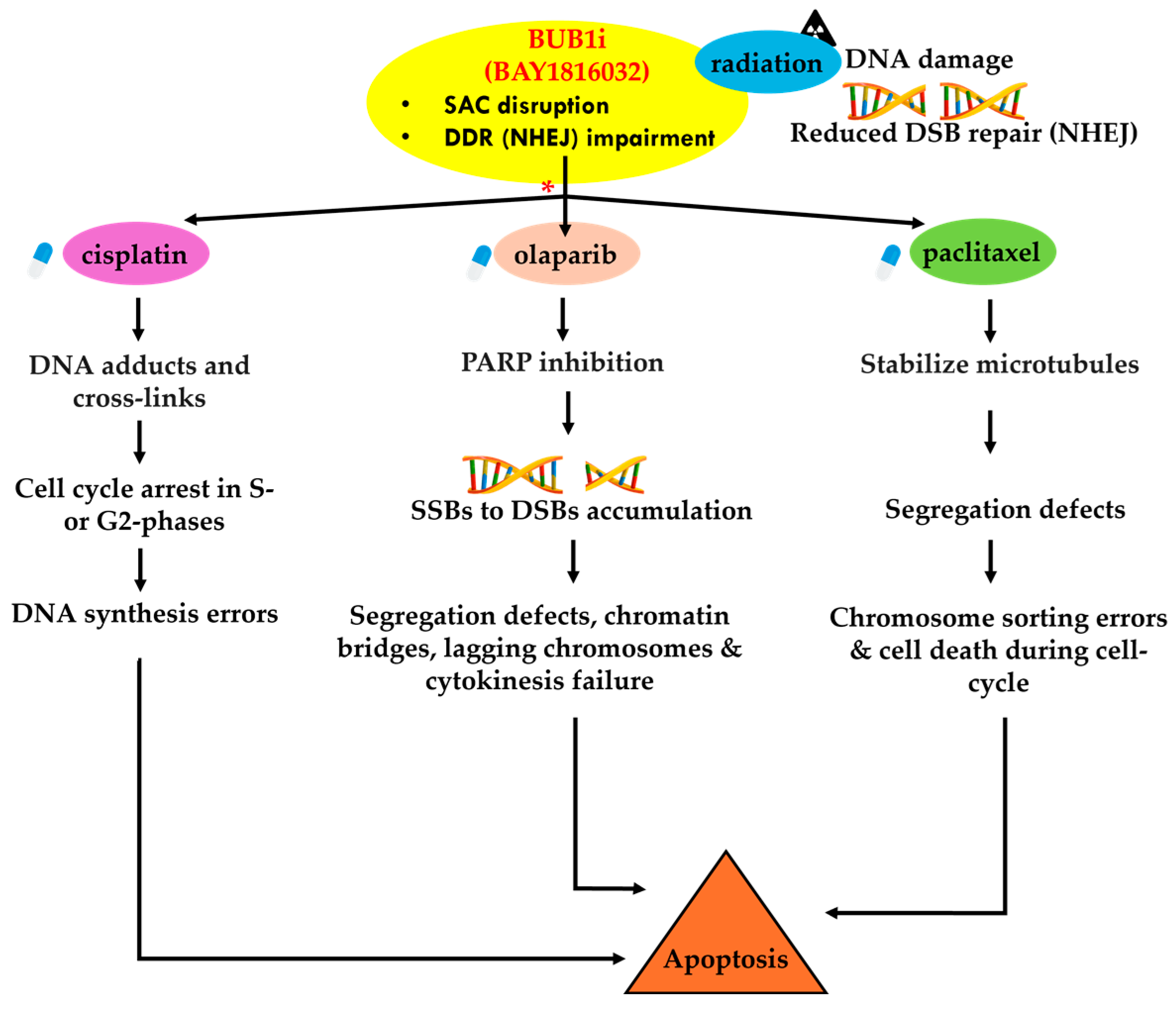
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, A.; Akter, Z.; Modareszadeh, P.; Modareszadeh, P.; Berisha, E.; Alemi, P.S.; Chacon Castro, M.D.C.; Deese, A.R.; Zhang, L. Current Landscape of Therapeutic Resistance in Lung Cancer and Promising Strategies to Overcome Resistance. Cancers 2022, 14, 4562. [Google Scholar] [CrossRef] [PubMed]
- Gadgeel, S.M. Role of Chemotherapy and Targeted Therapy in Early-Stage Non-Small Cell Lung Cancer. Am. Soc. Clin. Oncol. Educ. Book 2017, 37, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Nagasaka, M.; Gadgeel, S.M. Role of chemotherapy and targeted therapy in early-stage non-small cell lung cancer. Expert. Rev. Anticancer. Ther. 2018, 18, 63–70. [Google Scholar] [CrossRef]
- Oronsky, B.; Reid, T.R.; Oronsky, A.; Carter, C.A. What’s New in SCLC? A Review. Neoplasia 2017, 19, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Guberina, M.; Pöttgen, C.; Guberina, N.; Hoffmann, C.; Wiesweg, M.; Richlitzki, C.; Metzenmacher, M.; Aigner, C.; Bölükbas, S.; Gauler, T.; et al. Long-Term Survival in Patients with Oligometastatic Non-Small Cell Lung Cancer by a Multimodality Treatment—Comparison with Stage III Disease. Cancers 2024, 16, 1174. [Google Scholar] [CrossRef]
- Sepesi, B.; Jones, D.R.; Meyers, B.F.; Chaft, J.E.; Sholl, L.M.; Shyr, Y.; Kelly, K.; Lin, J.; Bunn, P.A.; Minna, J.D.; et al. LCMC LEADER neoadjuvant screening trial: LCMC4 evaluation of actionable drivers in early-stage lung cancers. J. Clin. Oncol. 2022, 40, TPS8596. [Google Scholar] [CrossRef]
- Deboever, N.; Eisenberg, M.; Chidi, A.; Sepesi, B. The role of immunotherapy and targeted therapy in the multimodal therapy for resectable lung cancer. J. Surg. Oncol. 2023, 127, 275–281. [Google Scholar] [CrossRef]
- Tricard, J.; Filaire, M.; Verge, R.; Pages, P.B.; Brichon, P.Y.; Loundou, A.; Boyer, L.; Thomas, P.A. Multimodality therapy for lung cancer invading the chest wall: A study of the French EPITHOR database. Lung Cancer 2023, 181, 107224. [Google Scholar] [CrossRef]
- Marchetti, F.; Venkatachalam, S. The multiple roles of Bub1 in chromosome segregation during mitosis and meiosis. Cell Cycle 2010, 9, 58–63. [Google Scholar] [CrossRef]
- Yang, C.; Wang, H.; Xu, Y.; Brinkman, K.L.; Ishiyama, H.; Wong, S.T.; Xu, B. The kinetochore protein Bub1 participates in the DNA damage response. DNA Repair 2012, 11, 185–191. [Google Scholar] [CrossRef]
- Hein, J.; Boichuk, S.; Wu, J.; Cheng, Y.; Freire, R.; Jat, P.S.; Roberts, T.M.; Gjoerup, O.V. Simian virus 40 large T antigen disrupts genome integrity and activates a DNA damage response via Bub1 binding. J. Virol. 2009, 83, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Long, Z.; Wu, T.; Tian, Q.; Carlson, L.A.; Wang, W.; Wu, G. Expression and prognosis analyses of BUB1, BUB1B and BUB3 in human sarcoma. Aging 2021, 13, 12395–12409. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Liao, Y.; Wang, M.; Wang, Y.; Wang, K.; Guo, J.; Wu, P.; Zhong, B.; Guo, T.; Wu, C. BUB1 drives the occurrence and development of bladder cancer by mediating the STAT3 signaling pathway. J. Exp. Clin. Cancer Res. 2021, 40, 378. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Yuan, Y.; Kuang, H.; Tang, B.; Zhang, H.; Zhang, M. BUB1B (BUB1 Mitotic Checkpoint Serine/Threonine Kinase B) promotes lung adenocarcinoma by interacting with Zinc Finger Protein ZNF143 and regulating glycolysis. Bioengineered 2022, 13, 2471–2485. [Google Scholar] [CrossRef]
- Nyati, S.; Schinske-Sebolt, K.; Pitchiaya, S.; Chekhovskiy, K.; Chator, A.; Chaudhry, N.; Dosch, J.; Van Dort, M.E.; Varambally, S.; Kumar-Sinha, C.; et al. The kinase activity of the Ser/Thr kinase BUB1 promotes TGF-β signaling. Sci. Signal 2015, 8, ra1. [Google Scholar] [CrossRef]
- Nyati, S.; Gregg, B.S.; Xu, J.; Young, G.; Kimmel, L.; Nyati, M.K.; Ray, D.; Speers, C.; Rehemtulla, A. TGFBR2 mediated phosphorylation of BUB1 at Ser-318 is required for transforming growth factor-β signaling. Neoplasia 2020, 22, 163–178. [Google Scholar] [CrossRef]
- Chen, R.; Wang, Z.; Lu, T.; Liu, Y.; Ji, Y.; Yu, Y.; Tou, F.; Guo, S. Budding uninhibited by benzimidazoles 1 overexpression is associated with poor prognosis and malignant phenotype: A promising therapeutic target for lung adenocarcinoma. Thorac. Cancer 2023, 14, 893–912. [Google Scholar] [CrossRef]
- Siemeister, G.; Mengel, A.; Fernández-Montalván, A.E.; Bone, W.; Schröder, J.; Zitzmann-Kolbe, S.; Briem, H.; Prechtl, S.; Holton, S.J.; Mönning, U.; et al. Inhibition of BUB1 Kinase by BAY 1816032 Sensitizes Tumor Cells toward Taxanes, ATR, and PARP Inhibitors In Vitro and In Vivo. Clin. Cancer Res. 2019, 25, 1404–1414. [Google Scholar] [CrossRef]
- Sriramulu, S.; Thoidingjam, S.; Chen, W.-M.; Hassan, O.; Siddiqui, F.; Brown, S.L.; Movsas, B.; Green, M.D.; Davis, A.J.; Speers, C.; et al. BUB1 regulates non-homologous end joining pathway to mediate radioresistance in triple-negative breast cancer. J. Exp. Clin. Cancer Res. 2024, 43, 163. [Google Scholar] [CrossRef]
- Cai, L.; Lin, S.; Girard, L.; Zhou, Y.; Yang, L.; Ci, B.; Zhou, Q.; Luo, D.; Yao, B.; Tang, H.; et al. LCE: An open web portal to explore gene expression and clinical associations in lung cancer. Oncogene 2019, 38, 2551–2564. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekar, D.S.; Karthikeyan, S.K.; Korla, P.K.; Patel, H.; Shovon, A.R.; Athar, M.; Netto, G.J.; Qin, Z.S.; Kumar, S.; Manne, U.; et al. UALCAN: An update to the integrated cancer data analysis platform. Neoplasia 2022, 25, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Li, X.; Gao, F.; de Groot, J.F.; Koul, D.; Yung, W.K.A. PARP-mediated PARylation of MGMT is critical to promote repair of temozolomide-induced O6-methylguanine DNA damage in glioblastoma. Neuro-Oncol. 2021, 23, 920–931. [Google Scholar] [CrossRef]
- Foucquier, J.; Guedj, M. Analysis of drug combinations: Current methodological landscape. Pharmacol. Res. Perspect. 2015, 3, e00149. [Google Scholar] [CrossRef]
- Gadgeel, S.; Rodriguez-Abreu, D.; Speranza, G.; Esteban, E.; Felip, E.; Domine, M.; Hui, R.; Hochmair, M.J.; Clingan, P.; Powell, S.F.; et al. Updated Analysis From KEYNOTE-189: Pembrolizumab or Placebo Plus Pemetrexed and Platinum for Previously Untreated Metastatic Nonsquamous Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2020, 38, 1505–1517. [Google Scholar] [CrossRef]
- Chen, X.; Wang, L.; Su, X.; Luo, S.Y.; Tang, X.; Huang, Y. Identification of potential target genes and crucial pathways in small cell lung cancer based on bioinformatic strategy and human samples. PLoS ONE 2020, 15, e0242194. [Google Scholar] [CrossRef]
- Wang, L.; Yang, X.; An, N.; Liu, J. Bioinformatics analysis of BUB1 expression and gene regulation network in lung adenocarcinoma. Transl. Cancer Res. 2020, 9, 4820–4833. [Google Scholar] [CrossRef]
- Wang, X.; Simon, R. Identification of potential synthetic lethal genes to p53 using a computational biology approach. BMC Med. Genom. 2013, 6, 30. [Google Scholar] [CrossRef]
- Li, X.; Wang, T.; Li, M.; Bao, X.; Ma, T.; Yang, C.; Wu, H.; Li, H. Comprehensive analysis of BUBs gene family in lung adenocarcinoma with immunological analysis. Aging 2023, 15, 810–829. [Google Scholar] [CrossRef]
- Sriramulu, S.; Thoidingjam, S.; Brown, S.L.; Siddiqui, F.; Movsas, B.; Nyati, S. Molecular targets that sensitize cancer to radiation killing: From the bench to the bedside. Biomed. Pharmacother. 2023, 158, 114126. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., III; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Luna-Maldonado, F.; Andonegui-Elguera, M.A.; Díaz-Chávez, J.; Herrera, L.A. Mitotic and DNA Damage Response Proteins: Maintaining the Genome Stability and Working for the Common Good. Front. Cell Dev. Biol. 2021, 9, 700162. [Google Scholar] [CrossRef] [PubMed]
- Sriramulu, S.; Thoidingjam, S.; Siddiqui, F.; Brown, S.L.; Movsas, B.; Walker, E.; Nyati, S. BUB1 Inhibition Sensitizes TNBC Cell Lines to Chemotherapy and Radiotherapy. Biomolecules 2024, 14, 625. [Google Scholar] [CrossRef]
- Panier, S.; Boulton, S.J. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2014, 15, 7–18. [Google Scholar] [CrossRef]
- Huang, Z.; Wang, S.; Wei, H.; Chen, H.; Shen, R.; Lin, R.; Wang, X.; Lan, W.; Lin, R.; Lin, J. Inhibition of BUB1 suppresses tumorigenesis of osteosarcoma via blocking of PI3K/Akt and ERK pathways. J. Cell Mol. Med. 2021, 25, 8442–8453. [Google Scholar] [CrossRef]
- Ji, W.; Weng, X.; Xu, D.; Cai, S.; Lou, H.; Ding, L. Non-small cell lung cancer cells with deficiencies in homologous recombination genes are sensitive to PARP inhibitors. Biochem. Biophys. Res. Commun. 2020, 522, 121–126. [Google Scholar] [CrossRef]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Tchounwou, P.B.; Dasari, S.; Noubissi, F.K.; Ray, P.; Kumar, S. Advances in Our Understanding of the Molecular Mechanisms of Action of Cisplatin in Cancer Therapy. J. Exp. Pharmacol. 2021, 13, 303–328. [Google Scholar] [CrossRef]
- O’Dwyer, P.J.; Johnson, S.W.; Hamilton, T.C. Cisplatin and its analogues. In Cancer: Principles and Practice of Oncology; DeVita, V.T., Hellman, S., Rosenberg, S.A., Eds.; Lippincott-Raven: Philadelphia, PA, USA, 1997; pp. 467–483. [Google Scholar]
- Schiff, P.B.; Fant, J.; Horwitz, S.B. Promotion of microtubule assembly in vitro by taxol. Nature 1979, 277, 665–667. [Google Scholar] [CrossRef] [PubMed]
- Schiff, P.B.; Horwitz, S.B. Taxol stabilizes microtubules in mouse fibroblast cells. Proc. Natl. Acad. Sci. USA 1980, 77, 1561–1565. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, S.B. Taxol (paclitaxel): Mechanisms of action. Ann. Oncol. 1994, 5 (Suppl. 6), S3–S6. [Google Scholar] [PubMed]
- Orr, G.A.; Verdier-Pinard, P.; McDaid, H.; Horwitz, S.B. Mechanisms of Taxol resistance related to microtubules. Oncogene 2003, 22, 7280–7295. [Google Scholar] [CrossRef]
- Elango, R.; Vishnubalaji, R.; Shaath, H.; Alajez, N.M. Transcriptional alterations of protein coding and noncoding RNAs in triple negative breast cancer in response to DNA methyltransferases inhibition. Cancer Cell Int. 2021, 21, 515. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thoidingjam, S.; Sriramulu, S.; Hassan, O.; Brown, S.L.; Siddiqui, F.; Movsas, B.; Gadgeel, S.; Nyati, S. BUB1 Inhibition Overcomes Radio- and Chemoradiation Resistance in Lung Cancer. Cancers 2024, 16, 3291. https://doi.org/10.3390/cancers16193291
Thoidingjam S, Sriramulu S, Hassan O, Brown SL, Siddiqui F, Movsas B, Gadgeel S, Nyati S. BUB1 Inhibition Overcomes Radio- and Chemoradiation Resistance in Lung Cancer. Cancers. 2024; 16(19):3291. https://doi.org/10.3390/cancers16193291
Chicago/Turabian StyleThoidingjam, Shivani, Sushmitha Sriramulu, Oudai Hassan, Stephen L. Brown, Farzan Siddiqui, Benjamin Movsas, Shirish Gadgeel, and Shyam Nyati. 2024. "BUB1 Inhibition Overcomes Radio- and Chemoradiation Resistance in Lung Cancer" Cancers 16, no. 19: 3291. https://doi.org/10.3390/cancers16193291
APA StyleThoidingjam, S., Sriramulu, S., Hassan, O., Brown, S. L., Siddiqui, F., Movsas, B., Gadgeel, S., & Nyati, S. (2024). BUB1 Inhibition Overcomes Radio- and Chemoradiation Resistance in Lung Cancer. Cancers, 16(19), 3291. https://doi.org/10.3390/cancers16193291