
Xeroderma Pigmentosum Type C Primary Skin Fibroblasts Overexpress HGF and Promote Squamous Cell Carcinoma Invasion in the Absence of Genotoxic Stress


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Scratch Assay
2.3. Contractile Property Measurements and Invasion Assays in Organotypic Skin Cultures
2.4. Histology and Immunostaining
2.5. Protein Extraction and Analyses
2.6. RNA Extraction and Quantitative RT-PCR
2.7. Spheroid Assay
2.8. HGF Enzyme-Linked Immunoassay
2.9. Transduction of XP-C Fibroblasts and Fluorescence-Activated Cell Sorting
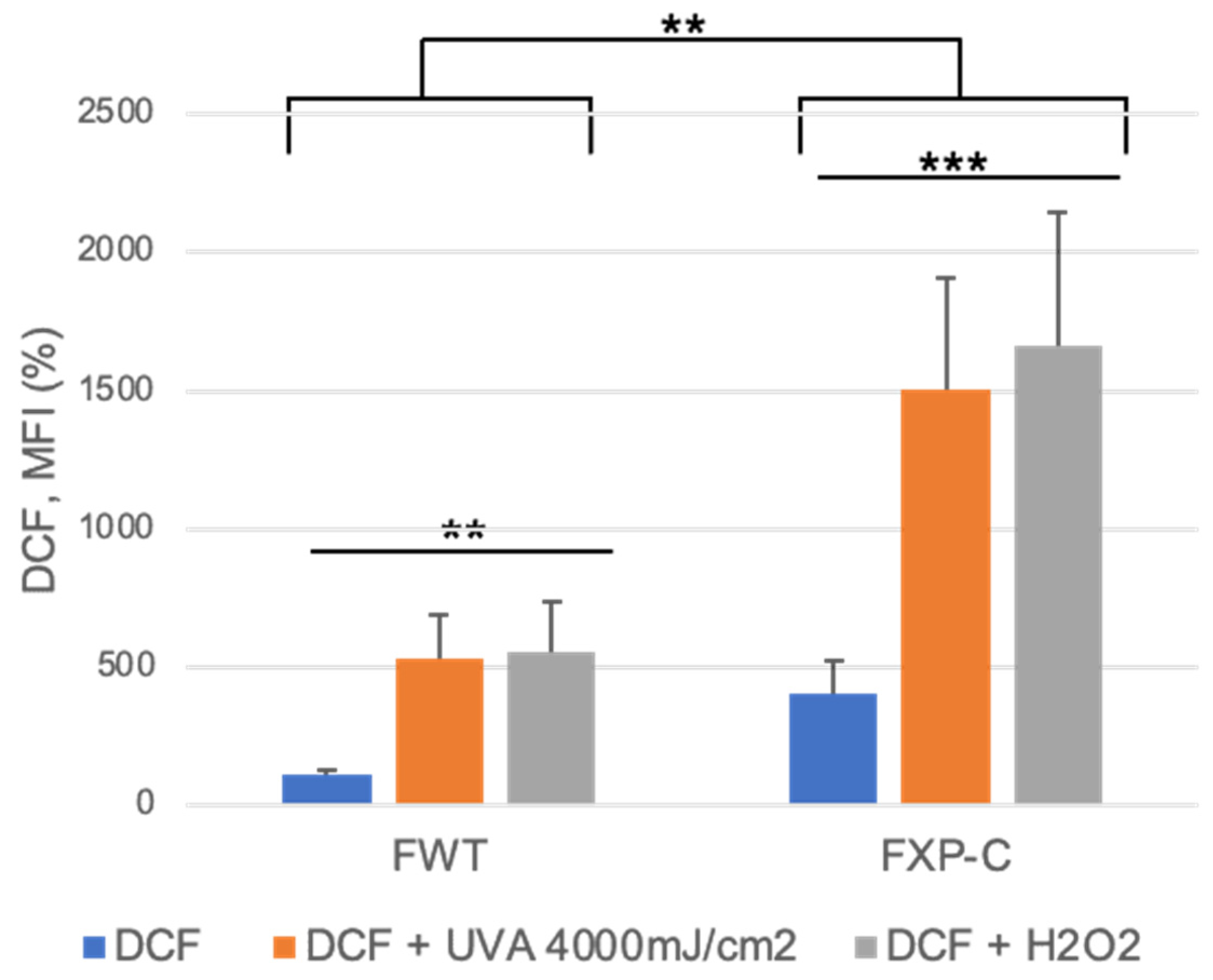
2.10. Quantitation of ROS Accumulation
2.11. Animals and Experimental Protocol
2.12. Statistical Analysis
3. Results
3.1. XP-C Primary Fibroblasts Promote Invasiveness of SCC Ex Vivo
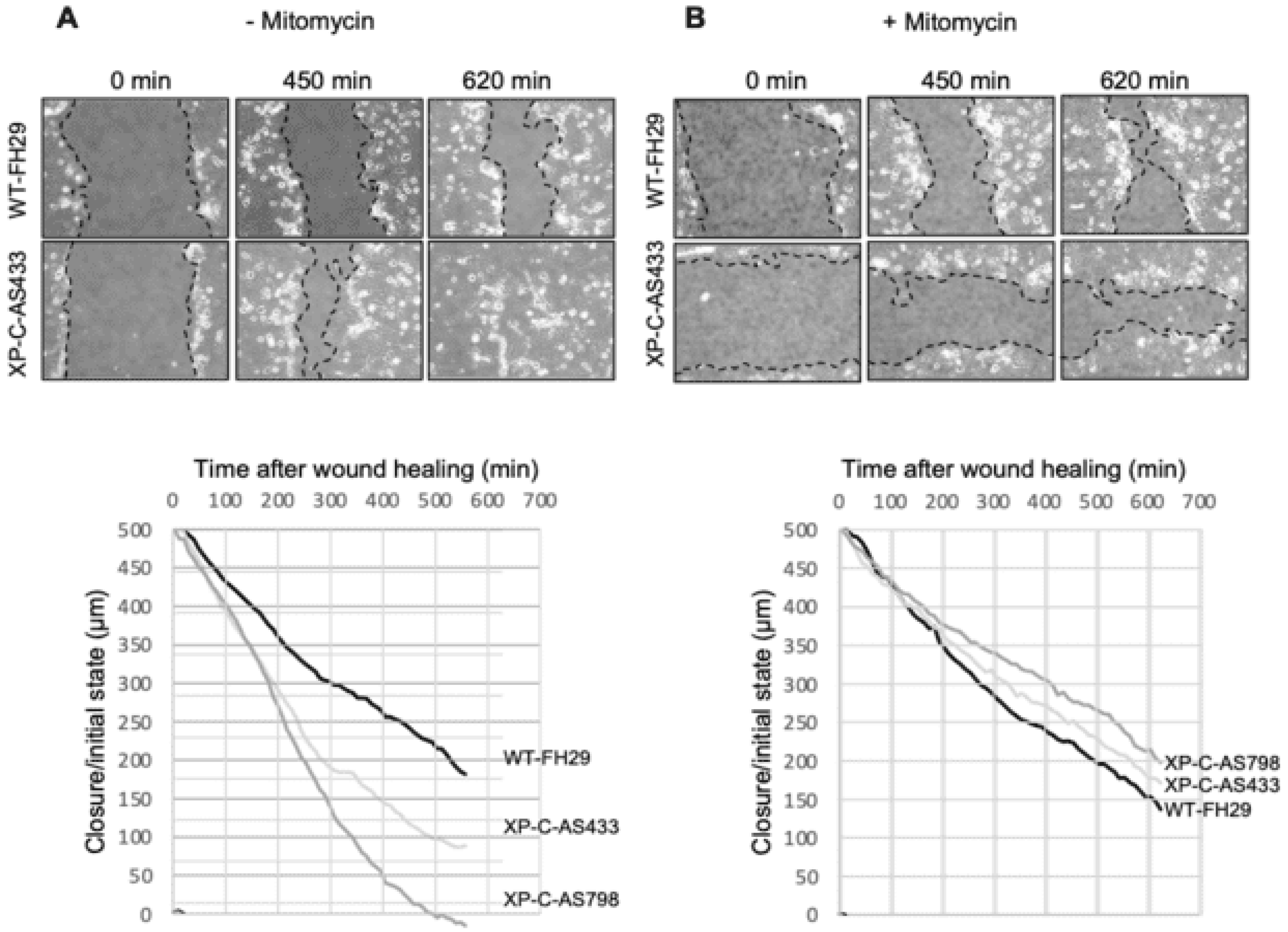
3.2. XP-C Fibroblasts Accelerate Scratch Closure of SCC Cells
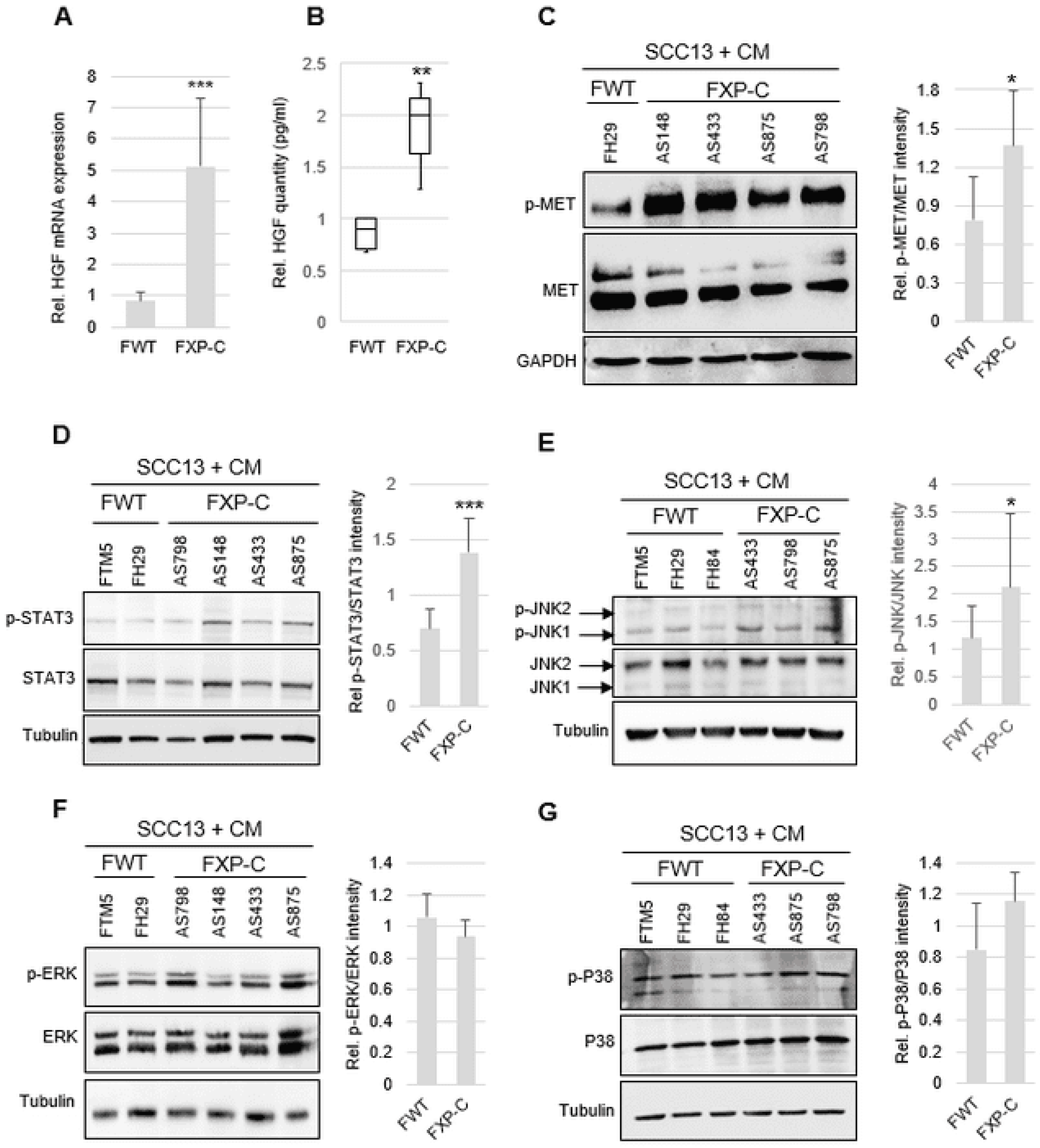
3.3. XP-C Fibroblasts Overexpress HGF in a Cell-Autonomous Manner
3.4. HGF/SF Overexpression in XP-C Fibroblasts Activates cMET and Downstream STAT3 and JNK Signaling Pathways
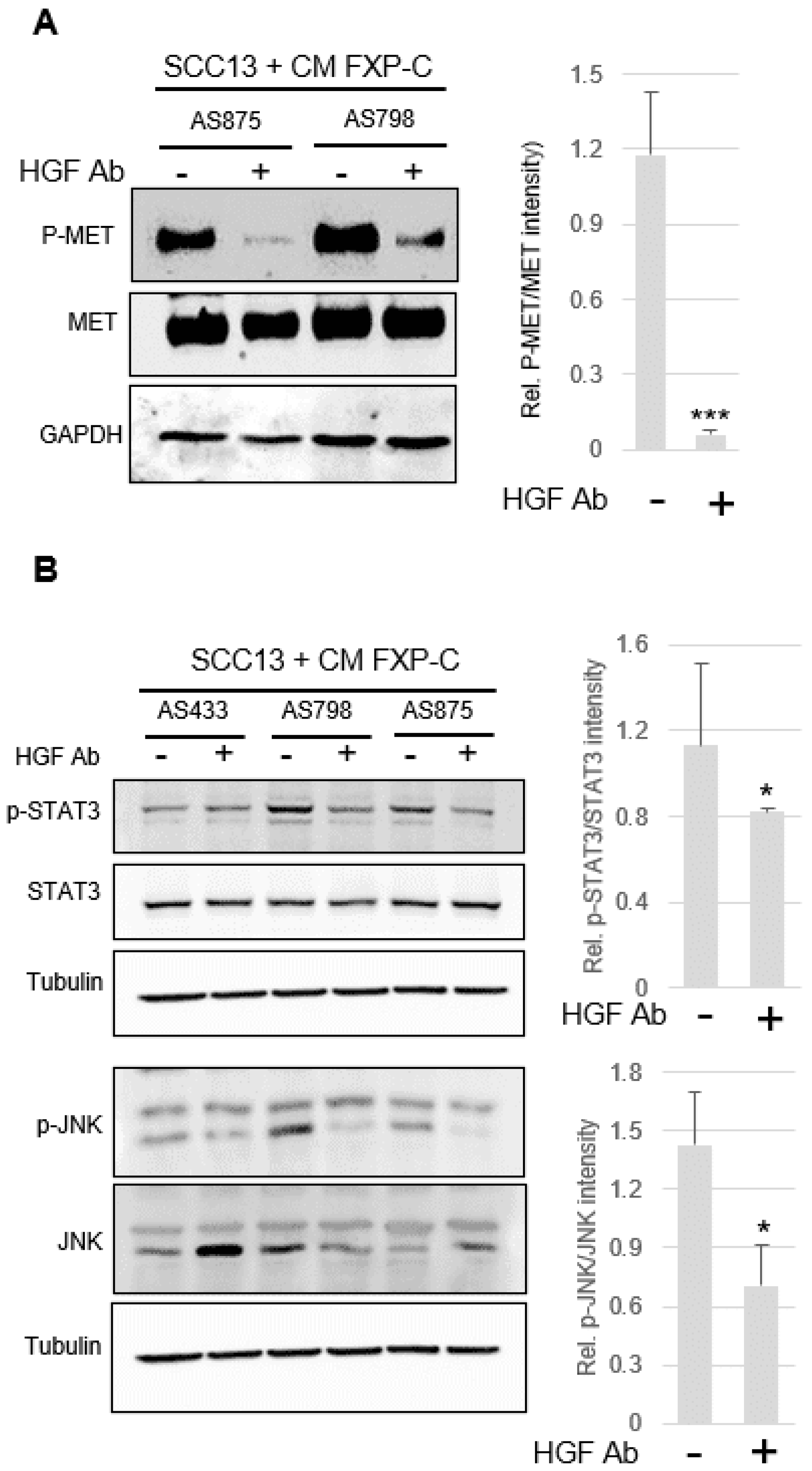
3.5. Inhibition of cMet Activation Counteracts Increased Invasiveness of SCC Cells Due to XP-C Fibroblasts
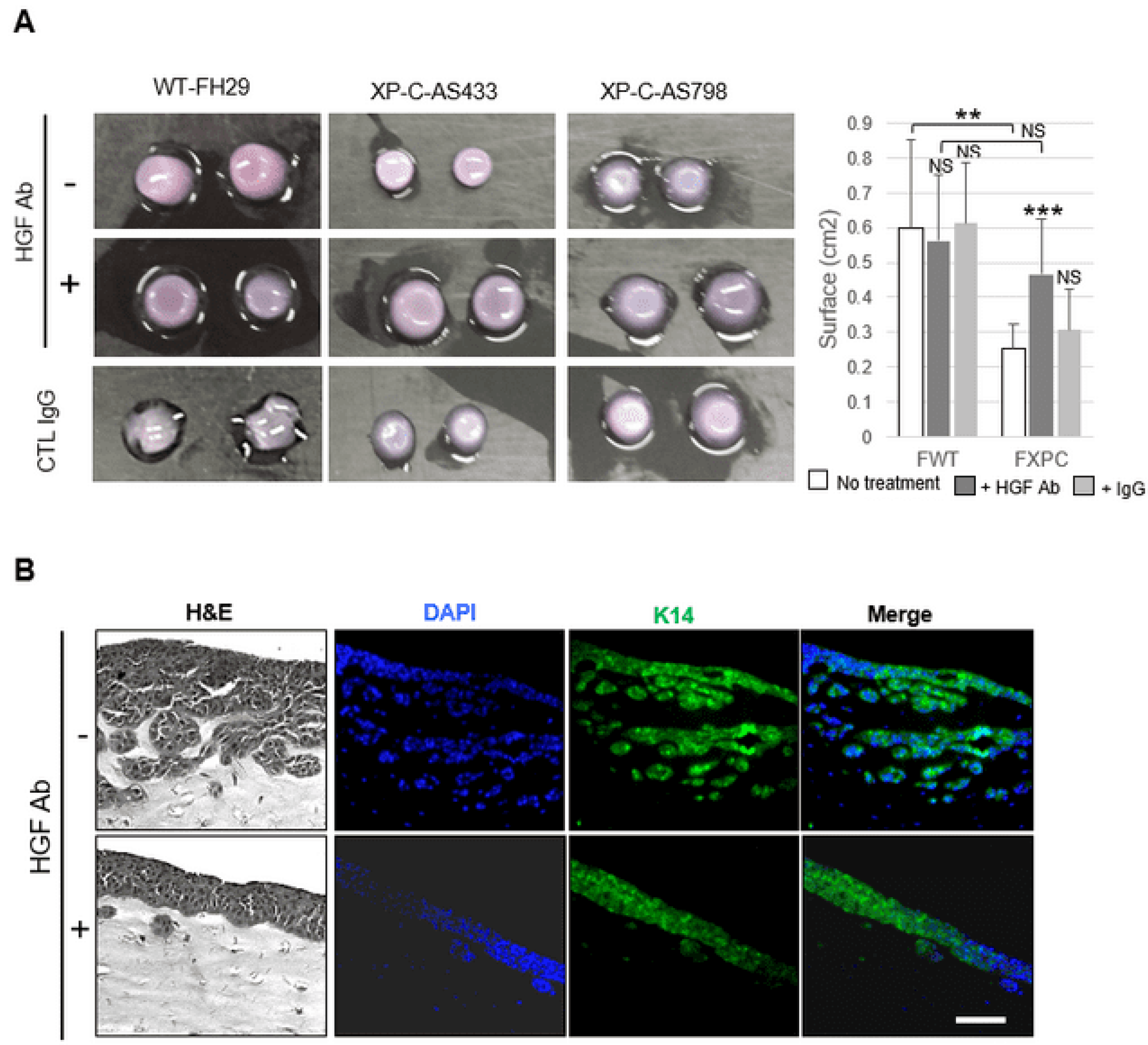
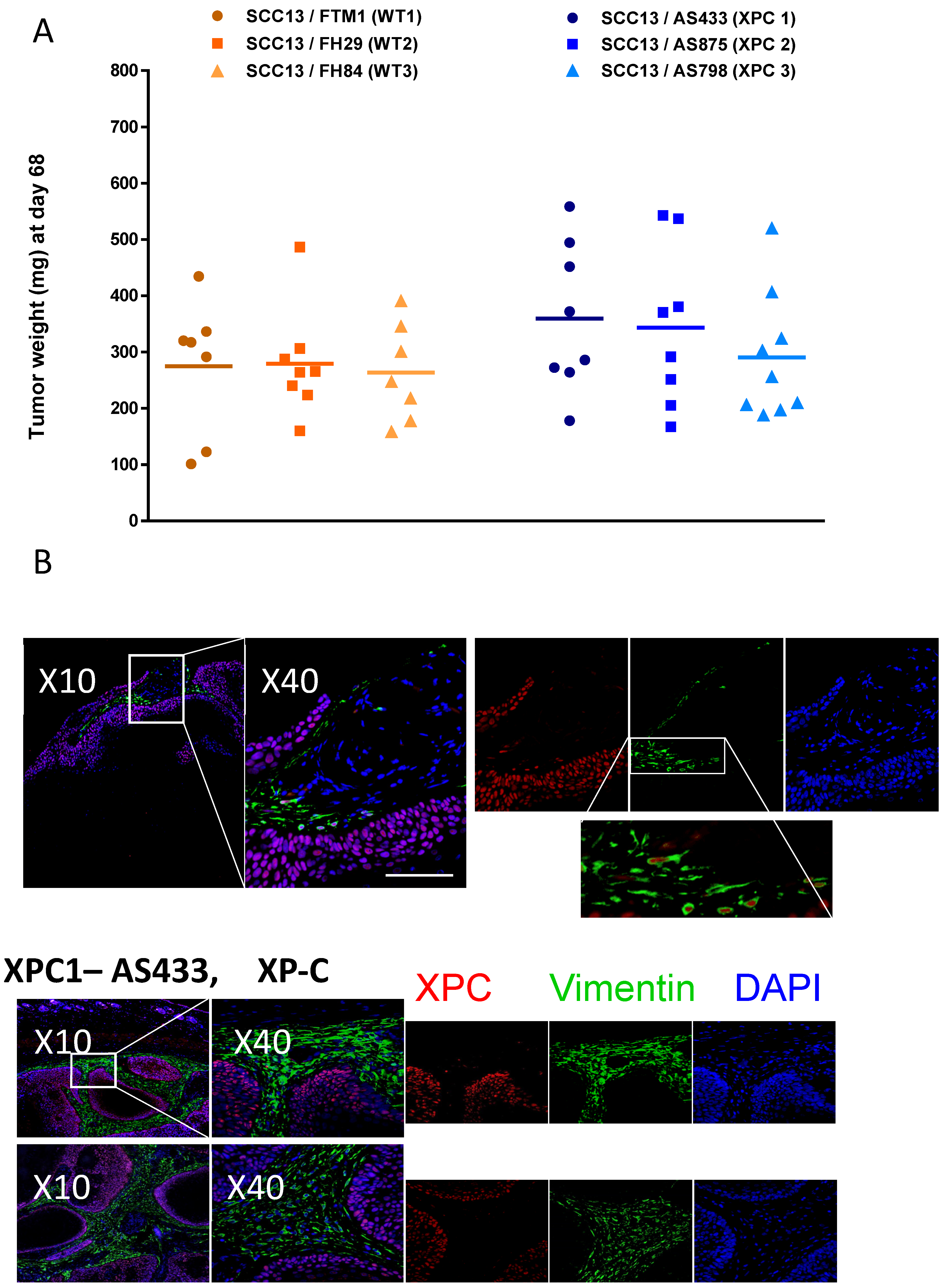
3.6. XP-C Fibroblasts Promote Cancer Cell Invasion In Vivo
4. Discussion
4.1. In Vivo Relevance
4.2. Molecular Mechanisms
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Abbreviations | Meaning |
| a-SMA | alpha smooth muscle actin |
| anti-K14 | anti-keratin 14 |
| CAFs | Carcinoma-Associated Fibroblasts |
| CM | conditioned culture medium supernatant |
| c-Met | mesenchymal–epithelial transition factor |
| CPDs | cyclobutane pyrimidine dimers |
| ECM | extracellular matrix |
| EDTA | ethylenediaminetetraacetic acid |
| EGF | epidermal growth factor |
| ERKs | extracellular signal-regulated kinase |
| FACS | fluorescence-activated cell sorting |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| GDF15 | growth/differentiation factor 15 |
| GFP | Green Fluorescent Protein |
| HGF | Hepatocyte Growth Factor |
| HBSS | sickle cell anemia medium |
| JNKs | c-Jun NH2-terminal kinases |
| K-WT | wild type keratinocytes |
| LIF1 | leukemia inhibitory factor 1 |
| MMC | mitomycin C |
| MMP1 | matrix metalloproteinase 1 |
| mRNA | messenger ribonucleic acid |
| NER | Nucleotide Excision Repair |
| NK | natural killer |
| NSG mouse | NOD SCID gamma mouse |
| PBS | phosphate-buffered saline |
| PPIA | peptidylprolyl isomerase A |
| ROS | reactive oxygen species |
| RPLO1 | apolipoprotein L1 |
| RT-PCR | reverse transcription quantitative real-time PCR |
| SCC | squamous cell carcinoma |
| SDF1a | stromal cell-derived factor 1 |
| SF | Scatter Factor |
| TGFβ | transforming growth factor beta |
| TFIIH | transcription factor II H |
| STAT3 | signal transducer and activator of transcription 3 |
| UVR | ultraviolet radiation |
| XP-C | xeroderma pigmentosum type C |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Bradford, P.T.; Goldstein, A.M.; Tamura, D.; Khan, S.G.; Ueda, T.; Boyle, J.; Oh, K.S.; Imoto, K.; Inui, H.; Moriwaki, S.; et al. Cancer and neurologic degeneration in xeroderma pigmentosum: Long term follow-up characterises the role of DNA repair. J. Med. Genet. 2011, 48, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Farkas, G.; Kocsis, Z.S.; Szekely, G.; Dobozi, M.; Kenessey, I.; Polgar, C.; Juranyi, Z. Smoking, chromosomal aberrations, and cancer incidence in healthy subjects. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2021, 867, 503373. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.H.; Seo, J.Y.; Choi, H.R.; Lee, M.K.; Youn, C.S.; Rhie, G.; Cho, K.H.; Kim, K.H.; Park, K.C.; Eun, H.C. Modulation of skin collagen metabolism in aged and photoaged human skin in vivo. J. Investig. Dermatol. 2001, 117, 1218–1224. [Google Scholar] [CrossRef]
- de Feraudy, S.; Boubakour-Azzouz, I.; Fraitag, S.; Berneburg, M.; Chan, L.; Chew, K.; Clericuzio, C.L.; Cunningham, B.; Tope, W.D.; Cleaver, J.E. Diagnosing xeroderma pigmentosum group C by immunohistochemistry. Am. J. Dermatopathol. 2010, 32, 109–117. [Google Scholar] [CrossRef]
- Giglia-Mari, G.; Sarasin, A. TP53 mutations in human skin cancers. Hum. Mutat. 2003, 21, 217–228. [Google Scholar] [CrossRef]
- Dong, Z.; Birrer, M.; Watts, R.; Matrisian, L.; Colburn, N. Blocking of tumor promoter-induced AP-1 activity inhibits induced transformation in JB6 mouse epidermal cells. Proc. Natl. Acad. Sci. USA 1994, 91, 609–613. [Google Scholar] [CrossRef]
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat. Cell Biol. 2007, 9, 1392–1400. [Google Scholar] [CrossRef]
- Bernerd, F.; Asselineau, D.; Vioux, C.; Chevallier-Lagente, O.; Bouadjar, B.; Sarasin, A.; Magnaldo, T. Clues to epidermal cancer proneness revealed by reconstruction of DNA repair-deficient xeroderma pigmentosum skin in vitro. Proc. Natl. Acad. Sci. USA 2001, 98, 7817–7822. [Google Scholar] [CrossRef]
- Goncalves-Maia, M.; Gache, Y.; Basante, M.; Cosson, E.; Salavagione, E.; Muller, M.; Bernerd, F.; Avril, M.F.; Schaub, S.; Sarasin, A.; et al. NK Cell and Fibroblast-Mediated Regulation of Skin Squamous Cell Carcinoma Invasion by CLEC2A Is Compromised in Xeroderma Pigmentosum. J. Investig. Dermatol. 2020, 140, 1723–1732. [Google Scholar] [CrossRef]
- Rheinwald, J.G.; Beckett, M.A. Tumorigenic keratinocyte lines requiring anchorage and fibroblast support cultured from human squamous cell carcinomas. Cancer Res. 1981, 41, 1657–1663. [Google Scholar] [PubMed]
- Warrick, E.; Garcia, M.; Chagnoleau, C.; Chevallier, O.; Bergoglio, V.; Sartori, D.; Mavilio, F.; Angulo, J.F.; Avril, M.F.; Sarasin, A.; et al. Preclinical corrective gene transfer in xeroderma pigmentosum human skin stem cells. Mol. Ther. 2012, 20, 798–807. [Google Scholar] [CrossRef]
- Albrengues, J.; Meneguzzi, G.; Gaggioli, C. Analysis of collective invasion of carcinoma cells in a 3D organotypic model. Methods Mol. Biol. 2013, 961, 243–252. [Google Scholar]
- Nystrom, M.L.; Thomas, G.J.; Stone, M.; Mackenzie, I.C.; Hart, I.R.; Marshall, J.F. Development of a quantitative method to analyse tumour cell invasion in organotypic culture. J. Pathol. 2005, 205, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Villaronga, M.A.; Teijeiro, S.A.; Hermida-Prado, F.; Garzon-Arango, M.; Sanz-Moreno, V.; Garcia-Pedrero, J.M. Analysis of Invasive Activity of CAF Spheroids into Three Dimensional (3D) Collagen Matrices. Methods Mol. Biol. 2018, 1731, 145–154. [Google Scholar] [PubMed]
- Bergoglio, V.; Larcher, F.; Chevallier-Lagente, O.; Bernheim, A.; Danos, O.; Sarasin, A.; Rio, M.D.; Magnaldo, T. Safe selection of genetically manipulated human primary keratinocytes with very high growth potential using CD24. Mol. Ther. 2007, 15, 2186–2193. [Google Scholar] [CrossRef] [PubMed]
- Kuzet, S.E.; Gaggioli, C. Fibroblast activation in cancer: When seed fertilizes soil. Cell Tissue Res. 2016, 365, 607–619. [Google Scholar] [CrossRef]
- Orimo, A.; Weinberg, R.A. Heterogeneity of stromal fibroblasts in tumors. Cancer Biol. Ther. 2007, 6, 618–619. [Google Scholar] [CrossRef]
- Sappino, A.P.; Skalli, O.; Jackson, B.; Schurch, W.; Gabbiani, G. Smooth-muscle differentiation in stromal cells of malignant and non-malignant breast tissues. Int. J. Cancer 1988, 41, 707–712. [Google Scholar] [CrossRef]
- Birchmeier, C.; Birchmeier, W.; Gherardi, E.; Vande Woude, G.F. Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 2003, 4, 915–925. [Google Scholar] [CrossRef]
- Matsumoto, K.; Nakamura, T.; Sakai, K.; Nakamura, T. Hepatocyte growth factor and Met in tumor biology and therapeutic approach with NK4. Proteomics 2008, 8, 3360–3370. [Google Scholar] [CrossRef] [PubMed]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signalling: Principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Paulsson, J.; Micke, P. Prognostic relevance of cancer-associated fibroblasts in human cancer. Semin. Cancer Biol. 2014, 25, 61–68. [Google Scholar] [CrossRef]
- Naldini, L.; Vigna, E.; Narsimhan, R.P.; Gaudino, G.; Zarnegar, R.; Michalopoulos, G.K.; Comoglio, P.M. Hepatocyte growth factor (HGF) stimulates the tyrosine kinase activity of the receptor encoded by the proto-oncogene c-MET. Oncogene 1991, 6, 501–504. [Google Scholar]
- De Bacco, F.; Luraghi, P.; Medico, E.; Reato, G.; Girolami, F.; Perera, T.; Gabriele, P.; Comoglio, P.M.; Boccaccio, C. Induction of MET by ionizing radiation and its role in radioresistance and invasive growth of cancer. J. Natl. Cancer Inst. 2011, 103, 645–661. [Google Scholar] [CrossRef]
- Frechet, M.; Warrick, E.; Vioux, C.; Chevallier, O.; Spatz, A.; Benhamou, S.; Sarasin, A.; Bernerd, F.; Magnaldo, T. Overexpression of matrix metalloproteinase 1 in dermal fibroblasts from DNA repair-deficient/cancer-prone xeroderma pigmentosum group C patients. Oncogene 2008, 27, 5223–5232. [Google Scholar] [CrossRef] [PubMed]
- Bidon, B.; Iltis, I.; Semer, M.; Nagy, Z.; Larnicol, A.; Cribier, A.; Benkirane, M.; Coin, F.; Egly, J.M.; Le May, N. XPC is an RNA polymerase II cofactor recruiting ATAC to promoters by interacting with E2F1. Nat. Commun. 2018, 9, 2610. [Google Scholar] [CrossRef]
- Fong, Y.W.; Inouye, C.; Yamaguchi, T.; Cattoglio, C.; Grubisic, I.; Tjian, R. A DNA repair complex functions as an Oct4/Sox2 coactivator in embryonic stem cells. Cell 2011, 147, 120–131. [Google Scholar] [CrossRef]
- Cattoglio, C.; Zhang, E.T.; Grubisic, I.; Chiba, K.; Fong, Y.W.; Tjian, R. Functional and mechanistic studies of XPC DNA-repair complex as transcriptional coactivator in embryonic stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, E2317–E2326. [Google Scholar] [CrossRef]
- Soufir, N.; Ged, C.; Bourillon, A.; Austerlitz, F.; Chemin, C.; Stary, A.; Armier, J.; Pham, D.; Khadir, K.; Roume, J.; et al. A prevalent mutation with founder effect in xeroderma pigmentosum group C from north Africa. J. Investig. Dermatol. 2010, 130, 1537–1542. [Google Scholar] [CrossRef]
- Ho, J.J.; Cattoglio, C.; McSwiggen, D.T.; Tjian, R.; Fong, Y.W. Regulation of DNA demethylation by the XPC DNA repair complex in somatic and pluripotent stem cells. Genes. Dev. 2017, 31, 830–844. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Su, X.; Li, Z.; Deng, L.; Liu, X.; Feng, X.; Peng, J. HGF/c-MET pathway in cancer: From molecular characterization to clinical evidence. Oncogene 2021, 40, 4625–4651. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-qaraghuli, S.; Gache, Y.; Goncalves-Maia, M.; Alcor, D.; Muzotte, E.; Mahfouf, W.; Rezvani, H.-R.; Magnaldo, T. Xeroderma Pigmentosum Type C Primary Skin Fibroblasts Overexpress HGF and Promote Squamous Cell Carcinoma Invasion in the Absence of Genotoxic Stress. Cancers 2024, 16, 3277. https://doi.org/10.3390/cancers16193277
Al-qaraghuli S, Gache Y, Goncalves-Maia M, Alcor D, Muzotte E, Mahfouf W, Rezvani H-R, Magnaldo T. Xeroderma Pigmentosum Type C Primary Skin Fibroblasts Overexpress HGF and Promote Squamous Cell Carcinoma Invasion in the Absence of Genotoxic Stress. Cancers. 2024; 16(19):3277. https://doi.org/10.3390/cancers16193277
Chicago/Turabian StyleAl-qaraghuli, Sahar, Yannick Gache, Maria Goncalves-Maia, Damien Alcor, Elodie Muzotte, Walid Mahfouf, Hamid-Reza Rezvani, and Thierry Magnaldo. 2024. "Xeroderma Pigmentosum Type C Primary Skin Fibroblasts Overexpress HGF and Promote Squamous Cell Carcinoma Invasion in the Absence of Genotoxic Stress" Cancers 16, no. 19: 3277. https://doi.org/10.3390/cancers16193277
APA StyleAl-qaraghuli, S., Gache, Y., Goncalves-Maia, M., Alcor, D., Muzotte, E., Mahfouf, W., Rezvani, H.-R., & Magnaldo, T. (2024). Xeroderma Pigmentosum Type C Primary Skin Fibroblasts Overexpress HGF and Promote Squamous Cell Carcinoma Invasion in the Absence of Genotoxic Stress. Cancers, 16(19), 3277. https://doi.org/10.3390/cancers16193277