
GABA(A) Receptor Activation Drives GABARAP–Nix Mediated Autophagy to Radiation-Sensitize Primary and Brain-Metastatic Lung Adenocarcinoma Tumors

 , , , ,
, , , , 
 ,
,  ,
,  add
Show full author list
add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Maintenance
2.2. Reverse Transcriptase PCR
2.3. Immunohistochemistry
2.4. AM-101 and Pen3-ortho Stapled-Peptide Preparation
2.5. Electrophysiology
2.6. Mitochondrial Depolarization
2.7. Preparation of Lung Cancer Chips
2.8. Imaging of Lung Cancer Chips
2.9. Immunoblotting
2.10. Immunofluorescence Staining and Confocal Imaging
2.11. Clonogenic and In Vitro Viability Assays
2.12. Irradiation of NSCLC Cells
2.13. In Vitro Cell Survival Assay with Stapled-Peptide and AM-101
2.14. Mouse Experiments
2.14.1. Subcutaneous Xenograft Studies
2.14.2. Intracranial Xenograft Studies
2.15. Data Analysis and Statistics
3. Results
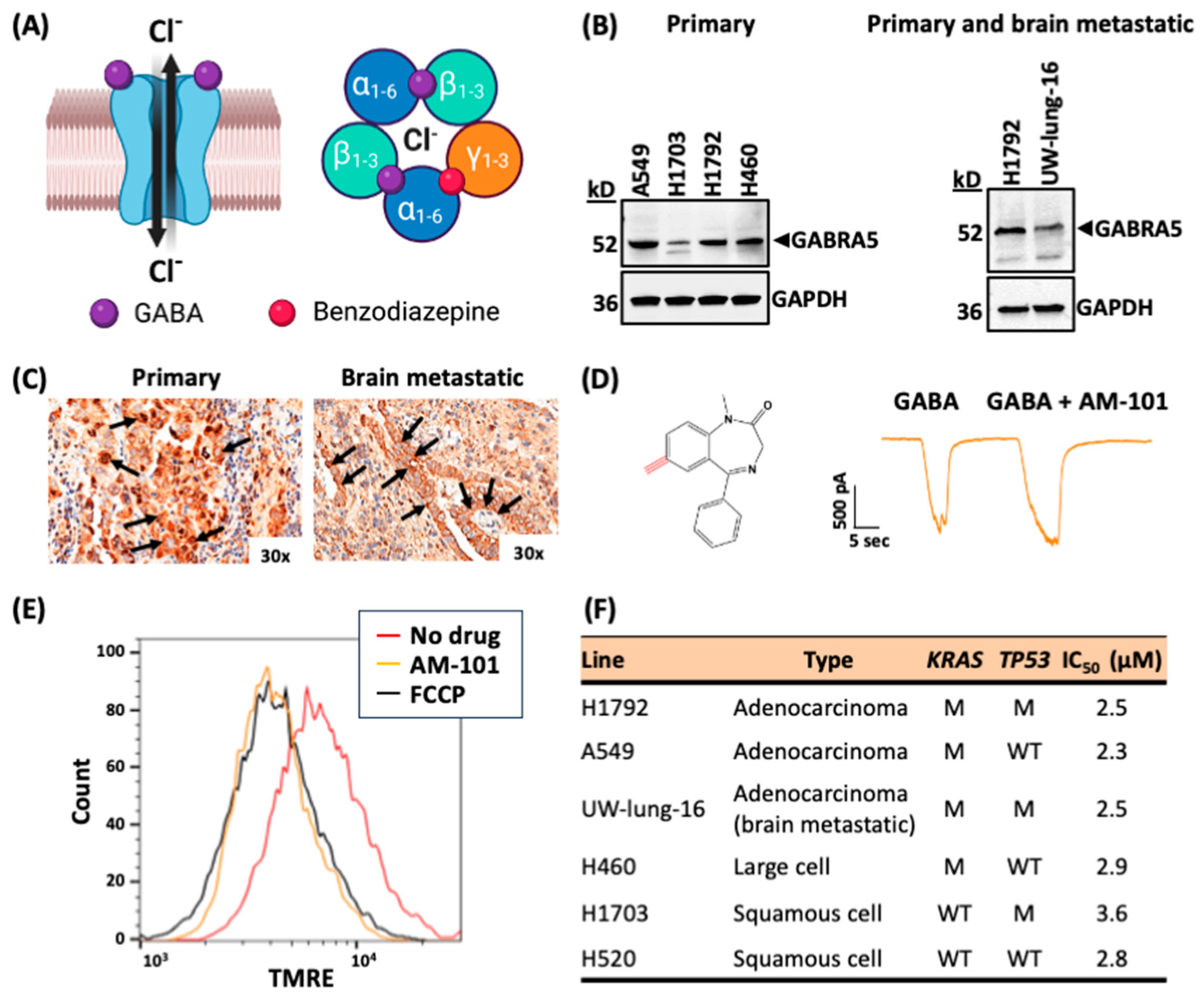
3.1. Lung Adenocarcinomas Express GABA(A) Receptor Subunits
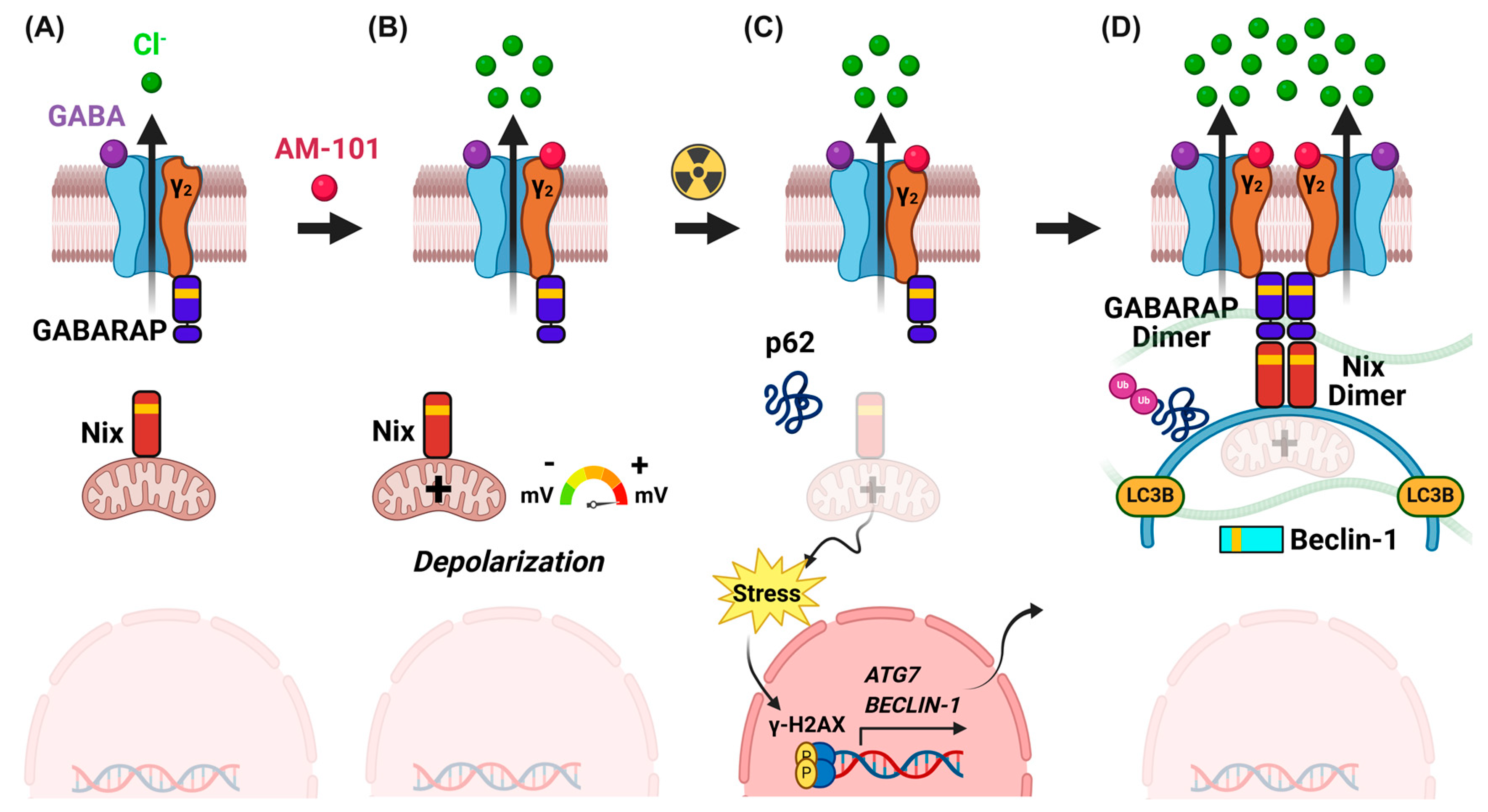
3.2. Activation of GABA(A) Receptors Is Depolarizing and Triggers Cell Death
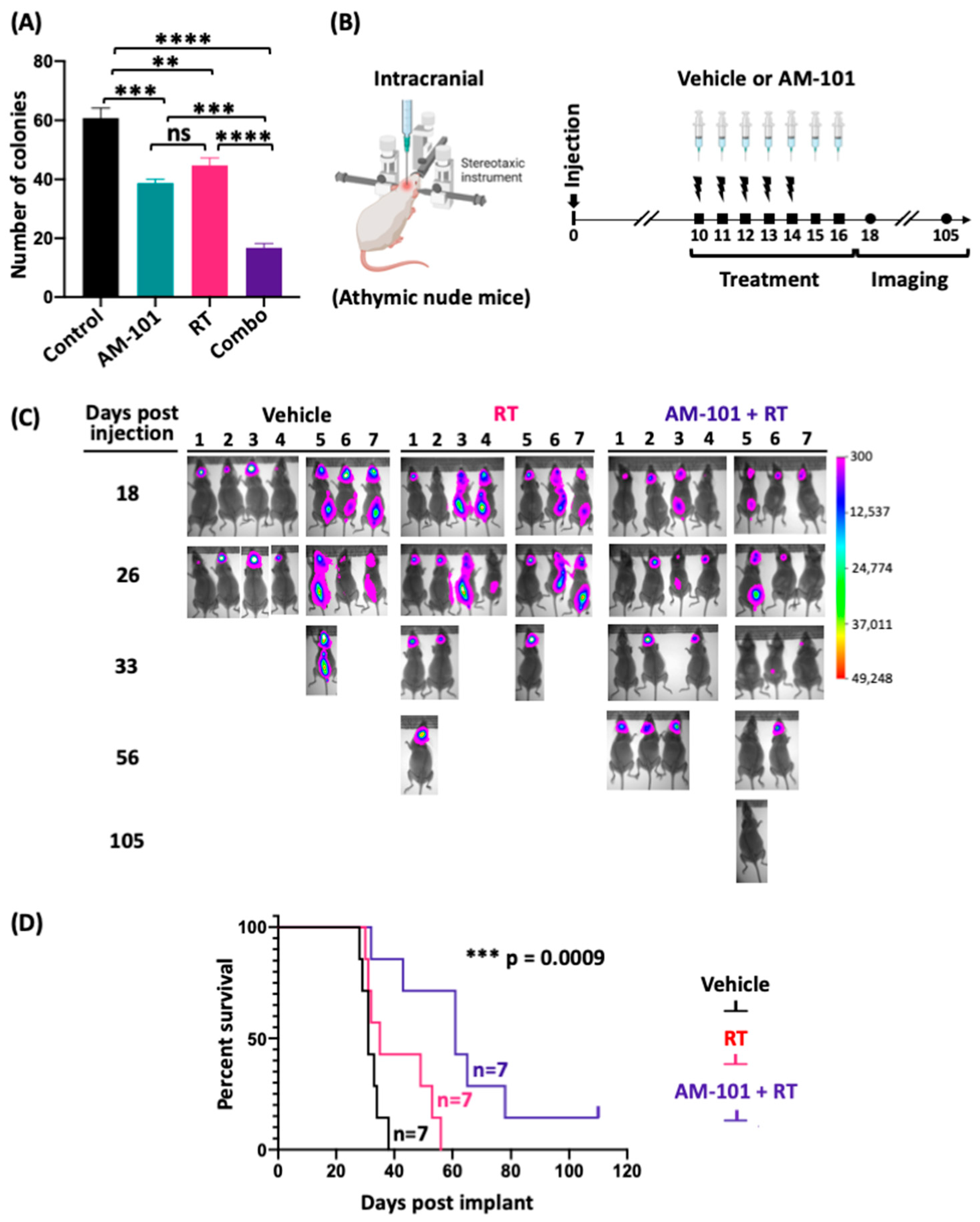
3.3. GABA(A) Receptor Activation Potentiates Radiation
3.4. Increased Survival of Mice Bearing a Lung Brain-Metastatic Tumor
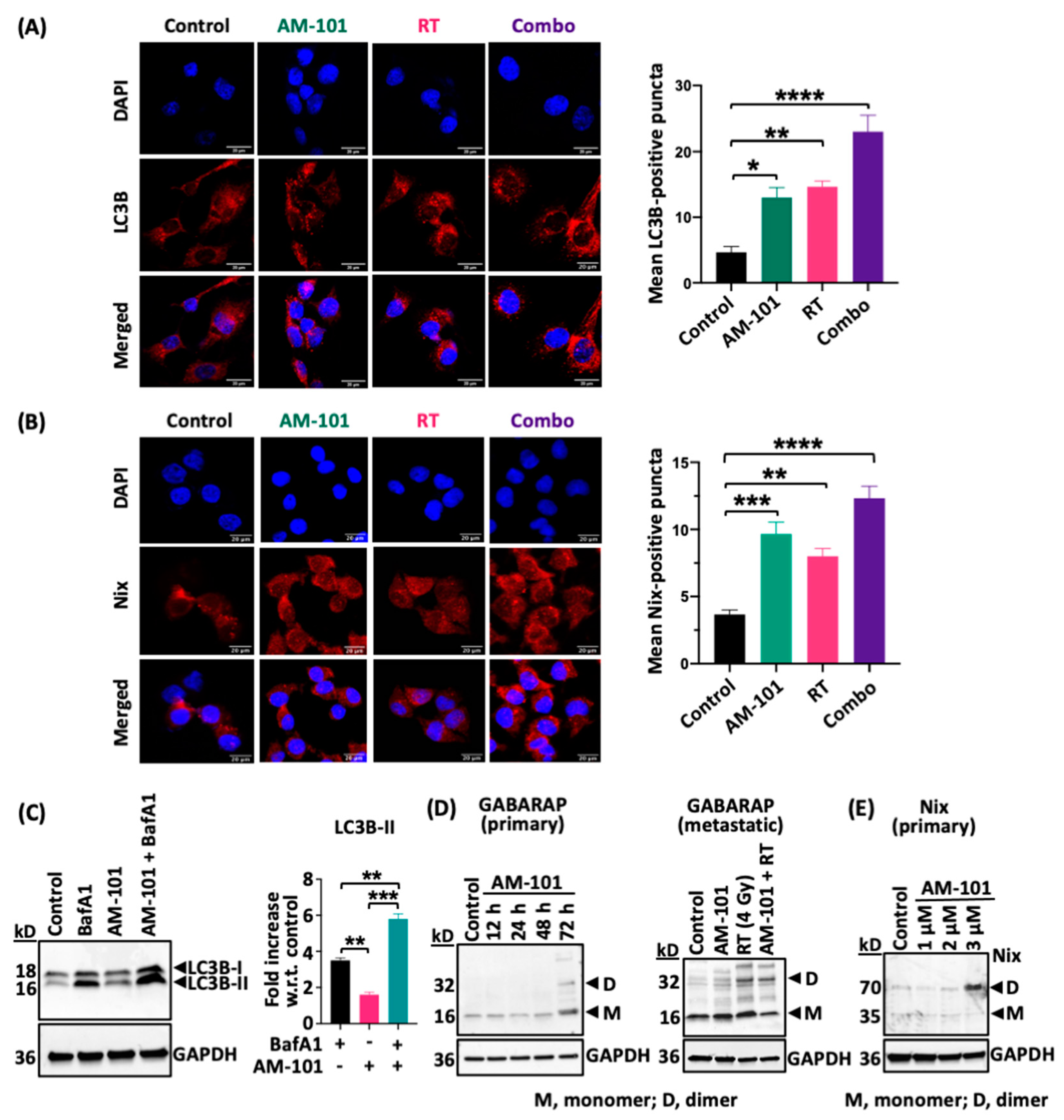
3.5. GABA(A) Receptor Activation Enhances Autophagic Puncta and GABARAP and Nix Multimerization
3.6. Enhanced Levels of Autophagy Biomarkers in Cells and Tumors
3.7. AM-101 Cytotoxicity Is Inhibited by Abrogating the GABARAP–Nix Axis
3.8. Contribution of γ-H2AX to Radiation Sensitization
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, J.; Fruh, M.; Papachristofilou, A.; Bubendorf, L.; Hauptle, P.; Jost, L.; Zippelius, A.; Rothschild, S.I. Resection of isolated brain metastases in non-small cell lung cancer (NSCLC) patients—Evaluation of outcome and prognostic factors: A retrospective multicenter study. PLoS ONE 2021, 16, e0253601. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, J.B.; Hansen, H.H.; Hansen, M.; Dombernowsky, P. Brain metastases in adenocarcinoma of the lung: Frequency, risk groups, and prognosis. J. Clin. Oncol. 1988, 6, 1474–1480. [Google Scholar] [CrossRef]
- Waqar, S.N.; Samson, P.P.; Robinson, C.G.; Bradley, J.; Devarakonda, S.; Du, L.; Govindan, R.; Gao, F.; Puri, V.; Morgensztern, D. Non-small-cell lung cancer with brain metastasis at presentation. Clin. Lung Cancer 2018, 19, e373–e379. [Google Scholar] [CrossRef] [PubMed]
- Baschnagel, A.M.; Elnaggar, J.H.; VanBeek, H.J.; Kromke, A.C.; Skiba, J.H.; Kaushik, S.; Abel, L.; Clark, P.A.; Longhurst, C.A.; Nickel, K.P.; et al. ATR inhibitor M6620 (VX-970) enhances the effect of radiation in non-small cell lung cancer brain metastasis patient-derived xenografts. Mol. Cancer Ther. 2021, 20, 2129–2139. [Google Scholar] [CrossRef]
- Sperduto, P.W.; Yang, T.J.; Beal, K.; Pan, H.; Brown, P.D.; Bangdiwala, A.; Shanley, R.; Yeh, N.; Gaspar, L.E.; Braunstein, S.; et al. Estimating survival in patients with lung cancer and brain metastases: An update of the graded prognostic assessment for lung cancer using molecular markers (Lung-molGPA). JAMA Oncol. 2017, 3, 827–831. [Google Scholar] [CrossRef]
- Shepherd, F.A.; Fossella, F.V.; Lynch, T.; Armand, J.P.; Rigas, J.R.; Kris, M.G. Docetaxel (Taxotere) shows survival and quality-of-life benefits in the second-line treatment of non-small cell lung cancer: A review of two phase III trials. Semin. Oncol. 2001, 28, 4–9. [Google Scholar] [CrossRef]
- Amaravadi, R.K.; Lippincott-Schwartz, J.; Yin, X.M.; Weiss, W.A.; Takebe, N.; Timmer, W.; DiPaola, R.S.; Lotze, M.T.; White, E. Principles and current strategies for targeting autophagy for cancer treatment. Clin. Cancer Res. 2011, 17, 654–666. [Google Scholar] [CrossRef]
- Brown, H.; Chung, M.; Uffing, A.; Batistatou, N.; Tsang, T.; Doskocil, S.; Mao, W.; Willbold, D.; Bast, R.C., Jr.; Lu, Z.; et al. Structure-based design of stapled peptides that bind GABARAP and inhibit autophagy. J. Am. Chem. Soc. 2022, 144, 14687–14697. [Google Scholar] [CrossRef]
- Schlafli, A.M.; Adams, O.; Galvan, J.A.; Gugger, M.; Savic, S.; Bubendorf, L.; Schmid, R.A.; Becker, K.F.; Tschan, M.P.; Langer, R.; et al. Prognostic value of the autophagy markers LC3 and p62/SQSTM1 in early-stage non-small cell lung cancer. Oncotarget 2016, 7, 39544–39555. [Google Scholar] [CrossRef]
- Wang, X.; Du, Z.; Li, L.; Shi, M.; Yu, Y. Beclin 1 and p62 expression in non-small cell lung cancer: Relation with malignant behaviors and clinical outcome. Int. J. Clin. Exp. Pathol. 2015, 8, 10644–10652. [Google Scholar] [PubMed]
- Debnath, J.; Gammoh, N.; Ryan, K.M. Autophagy and autophagy-related pathways in cancer. Nat. Rev. Mol. Cell Biol. 2023, 24, 560–575. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Su, L.; Xiao, Z.; Liu, X.; Liu, X. Methyl jasmonate induces apoptosis and pro-apoptotic autophagy via the ROS pathway in human non-small cell lung cancer. Am. J. Cancer Res. 2016, 6, 187–199. [Google Scholar] [PubMed]
- Cao, C.; Subhawong, T.; Albert, J.M.; Kim, K.W.; Geng, L.; Sekhar, K.R.; Gi, Y.J.; Lu, B. Inhibition of mammalian target of rapamycin or apoptotic pathway induces autophagy and radiosensitizes PTEN null prostate cancer cells. Cancer Res. 2006, 66, 10040–10047. [Google Scholar] [CrossRef]
- Chaurasia, M.; Bhatt, A.N.; Das, A.; Dwarakanath, B.S.; Sharma, K. Radiation-induced autophagy: Mechanisms and consequences. Free Radic. Res. 2016, 50, 273–290. [Google Scholar] [CrossRef]
- Gao, P.; Hao, F.; Dong, X.; Qiu, Y. The role of autophagy and Beclin-1 in radiotherapy-induced apoptosis in thyroid carcinoma cells. Int. J. Clin. Exp. Pathol. 2019, 12, 885–892. [Google Scholar]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef]
- Senft, D.; Ronai, Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef]
- Rao, R.; Shah, S.; Bhattacharya, D.; Toukam, D.K.; Caceres, R.; Pomeranz Krummel, D.A.; Sengupta, S. Ligand-gated ion channels as targets for treatment and management of cancers. Front. Physiol. 2022, 13, 839437. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Gawali, V.S.; Kallay, L.; Toukam, D.K.; Koehler, A.; Stambrook, P.; Krummel, D.P.; Sengupta, S. Therapeutically leveraging GABA(A) receptors in cancer. Exp. Biol. Med. 2021, 246, 2128–2135. [Google Scholar] [CrossRef]
- Kallay, L.; Keskin, H.; Ross, A.; Rupji, M.; Moody, O.A.; Wang, X.; Li, G.; Ahmed, T.; Rashid, F.; Stephen, M.R.; et al. Modulating native GABA(A) receptors in medulloblastoma with positive allosteric benzodiazepine-derivatives induces cell death. J. Neurooncol. 2019, 142, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Pomeranz Krummel, D.A.; Nasti, T.H.; Kaluzova, M.; Kallay, L.; Bhattacharya, D.; Melms, J.C.; Izar, B.; Xu, M.; Burnham, A.; Ahmed, T.; et al. Melanoma cell ntrinsic GABA(A) receptor enhancement potentiates radiation and immune checkpoint inhibitor response by promoting direct and T cell-mediated antitumor activity. Int. J. Radiat. Oncol. Biol. Phys. 2021, 109, 1040–1053. [Google Scholar] [CrossRef] [PubMed]
- Biel, T.G.; Rao, V.A. Mitochondrial dysfunction activates lysosomal-dependent mitophagy selectively in cancer cells. Oncotarget 2018, 9, 995–1011. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, M.; Hirata, N.; Tanaka, T.; Suizu, F.; Nakajima, H.; Chiorini, J.A. Autophagy as a modulator of cell death machinery. Cell Death Dis. 2020, 11, 517. [Google Scholar] [CrossRef]
- Kneussel, M.; Haverkamp, S.; Fuhrmann, J.C.; Wang, H.; Wassle, H.; Olsen, R.W.; Betz, H. The gamma-aminobutyric acid type A receptor (GABAAR)-associated protein GABARAP interacts with gephyrin but is not involved in receptor anchoring at the synapse. Proc. Natl. Acad. Sci. USA 2000, 97, 8594–8599. [Google Scholar] [CrossRef]
- Chen, L.; Wang, H.; Vicini, S.; Olsen, R.W. The gamma-aminobutyric acid type A (GABAA) receptor-associated protein (GABARAP) promotes GABAA receptor clustering and modulates the channel kinetics. Proc. Natl. Acad. Sci. USA 2000, 97, 11557–11562. [Google Scholar] [CrossRef]
- Ye, J.; Zou, G.; Zhu, R.; Kong, C.; Miao, C.; Zhang, M.; Li, J.; Xiong, W.; Wang, C. Structural basis of GABARAP-mediated GABA(A) receptor trafficking and functions on GABAergic synaptic transmission. Nat. Commun. 2021, 12, 297. [Google Scholar] [CrossRef]
- Irwin, B.W.J.; Wanjura, C.C.; Molnar, D.; Rutter, M.J.; Payne, M.C.; Chau, P.L. GABA receptor associated protein changes the electrostatic environment around the GABA type A receptor. Proteins 2022, 90, 476–484. [Google Scholar] [CrossRef]
- Schwarten, M.; Mohrluder, J.; Ma, P.; Stoldt, M.; Thielmann, Y.; Stangler, T.; Hersch, N.; Hoffmann, B.; Merkel, R.; Willbold, D. Nix directly binds to GABARAP: A possible crosstalk between apoptosis and autophagy. Autophagy 2009, 5, 690–698. [Google Scholar] [CrossRef]
- Nieto-Torres, J.L.; Leidal, A.M.; Debnath, J.; Hansen, M. Beyond autophagy: The expanding roles of ATG8 proteins. Trends Biochem. Sci. 2021, 46, 673–686. [Google Scholar] [CrossRef]
- Olsen, R.W.; Sieghart, W. GABA A receptors: Subtypes provide diversity of function and pharmacology. Neuropharmacology 2009, 56, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Baschnagel, A.M.; Kaushik, S.; Durmaz, A.; Goldstein, S.; Ong, I.M.; Abel, L.; Clark, P.A.; Gurel, Z.; Leal, T.; Buehler, D.; et al. Development and characterization of patient-derived xenografts from non-small cell lung cancer brain metastases. Sci. Rep. 2021, 11, 2520. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Zhang, W.J.; Liu, R.Y.; McKernan, R.M.; Cook, J.M. Benzo-fused benzodiazepines employed as topological probes for the study of benzodiazepine receptor subtypes. Med. Chem. Res. 1996, 6, 384–391. [Google Scholar]
- Cox, E.D.; Diaz-Arauzo, H.; Huang, Q.; Reddy, M.S.; Ma, C.; Harris, B.; McKernan, R.; Skolnick, P.; Cook, J.M. Synthesis and evaluation of analogues of the partial agonist 6-(propyloxy)-4-(methoxymethyl)-beta-carboline-3-carboxylic acid ethyl ester (6-PBC) and the full agonist 6-(benzyloxy)-4-(methoxymethyl)-beta-carboline-3-carboxylic acid ethyl ester (Zk 93423) at wild type and recombinant GABAA receptors. J. Med. Chem. 1998, 41, 2537–2552. [Google Scholar] [CrossRef]
- Dhir, A.; Bruun, D.A.; Guignet, M.; Tsai, Y.H.; Gonzalez, E.; Calsbeek, J.; Vu, J.; Saito, N.; Tancredi, D.J.; Harvey, D.J.; et al. Allopregnanolone and perampanel as adjuncts to midazolam for treating diisopropylfluorophosphate-induced status epilepticus in rats. Ann. N. Y. Acad. Sci. 2020, 1480, 183–206. [Google Scholar] [CrossRef]
- Sano, E.; Mori, C.; Matsuoka, N.; Ozaki, Y.; Yagi, K.; Wada, A.; Tashima, K.; Yamasaki, S.; Tanabe, K.; Yano, K.; et al. Tetrafluoroethylene-propylene elastomer for fabrication of microfluidic organs-on-chips resistant to drug absorption. Micromachines 2019, 10, 793. [Google Scholar] [CrossRef]
- Thacker, V.V.; Dhar, N.; Sharma, K.; Barrile, R.; Karalis, K.; McKinney, J.D. A lung-on-chip model of early Mycobacterium tuberculosis infection reveals an essential role for alveolar epithelial cells in controlling bacterial growth. Elife 2020, 9, e59961. [Google Scholar] [CrossRef]
- Hassell, B.A.; Goyal, G.; Lee, E.; Sontheimer-Phelps, A.; Levy, O.; Chen, C.S.; Ingber, D.E. Human organ chip models recapitulate orthotopic lung cancer growth, therapeutic responses, and tumor dormancy in vitro. Cell Rep. 2017, 21, 508–516. [Google Scholar] [CrossRef]
- Huh, D.; Matthews, B.D.; Mammoto, A.; Montoya-Zavala, M.; Hsin, H.Y.; Ingber, D.E. Reconstituting organ-level lung functions on a chip. Science 2010, 328, 1662–1668. [Google Scholar] [CrossRef]
- Jain, A.; Barrile, R.; van der Meer, A.D.; Mammoto, A.; Mammoto, T.; De Ceunynck, K.; Aisiku, O.; Otieno, M.A.; Louden, C.S.; Hamilton, G.A.; et al. Primary Human lung alveolus-on-a-chip model of intravascular thrombosis for assessment of therapeutics. Clin. Pharmacol. Ther. 2018, 103, 332–340. [Google Scholar] [CrossRef]
- Chang, S.H.; Minai-Tehrani, A.; Shin, J.Y.; Park, S.; Kim, J.E.; Yu, K.N.; Hong, S.H.; Hong, C.M.; Lee, K.H.; Beck, G.R., Jr.; et al. Beclin1-induced autophagy abrogates radioresistance of lung cancer cells by suppressing osteopontin. J. Radiat. Res. 2012, 53, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, D.; Chaudhuri, S.; Singh, M.K.; Chaudhuri, S. T11TS inhibits Angiopoietin-1/Tie-2 signaling, EGFR activation and Raf/MEK/ERK pathway in brain endothelial cells restraining angiogenesis in glioma model. Exp. Mol. Pathol. 2015, 98, 455–466. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Weeraratne, S.D.; Sun, H.; Phallen, J.; Rallapalli, S.K.; Teider, N.; Kosaras, B.; Amani, V.; Pierre-Francois, J.; Tang, Y.; et al. alpha5-GABAA receptors negatively regulate MYC-amplified medulloblastoma growth. Acta Neuropathol. 2014, 127, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Dingley, S.; Chapman, K.A.; Falk, M.J. Fluorescence-activated cell sorting analysis of mitochondrial content, membrane potential, and matrix oxidant burden in human lymphoblastoid cell lines. Methods Mol. Biol. 2012, 837, 231–239. [Google Scholar] [CrossRef]
- Kondratskyi, A.; Kondratska, K.; Skryma, R.; Klionsky, D.J.; Prevarskaya, N. Ion channels in the regulation of autophagy. Autophagy 2018, 14, 3–21. [Google Scholar] [CrossRef]
- Ruedi-Bettschen, D.; Rowlett, J.K.; Rallapalli, S.; Clayton, T.; Cook, J.M.; Platt, D.M. Modulation of alpha5 subunit-containing GABAA receptors alters alcohol drinking by rhesus monkeys. Alcohol. Clin. Exp. Res. 2013, 37, 624–634. [Google Scholar] [CrossRef]
- Shultz, L.D.; Lang, P.A.; Christianson, S.W.; Gott, B.; Lyons, B.; Umeda, S.; Leiter, E.; Hesselton, R.; Wagar, E.J.; Leif, J.H.; et al. NOD/LtSz-Rag1null mice: An immunodeficient and radioresistant model for engraftment of human hematolymphoid cells, HIV infection, and adoptive transfer of NOD mouse diabetogenic T cells. J. Immunol. 2000, 164, 2496–2507. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhao, L.; Liu, L.; Gao, P.; Tian, W.; Wang, X.; Jin, H.; Xu, H.; Chen, Q. Beclin 1 cleavage by caspase-3 inactivates autophagy and promotes apoptosis. Protein Cell 2010, 1, 468–477. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Shen, X.L.; Zhang, B.; Liang, R.; Cheng, W.H.; Xu, W.; Luo, Y.; Zhao, C.; Huang, K. Central role of Nix in the autophagic response to ochratoxin A. Food Chem. Toxicol. 2014, 69, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.W.; Chang, C.S.; Leil, T.A.; Olsen, R.W. C-terminal modification is required for GABARAP-mediated GABA(A) receptor trafficking. J. Neurosci. 2007, 27, 6655–6663. [Google Scholar] [CrossRef] [PubMed]
- Marinkovic, M.; Sprung, M.; Novak, I. Dimerization of mitophagy receptor BNIP3L/NIX is essential for recruitment of autophagic machinery. Autophagy 2021, 17, 1232–1243. [Google Scholar] [CrossRef] [PubMed]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Lohr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010, 11, 45–51. [Google Scholar] [CrossRef]
- Noda, N.N.; Satoo, K.; Fujioka, Y.; Kumeta, H.; Ogura, K.; Nakatogawa, H.; Ohsumi, Y.; Inagaki, F. Structural basis of Atg8 activation by a homodimeric E1, Atg7. Mol. Cell 2011, 44, 462–475. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Schulman, B.A. Dynamic regulation of macroautophagy by distinctive ubiquitin-like proteins. Nat. Struct. Mol. Biol. 2014, 21, 336–345. [Google Scholar] [CrossRef]
- Collier, J.J.; Suomi, F.; Olahova, M.; McWilliams, T.G.; Taylor, R.W. Emerging roles of ATG7 in human health and disease. EMBO Mol. Med. 2021, 13, e14824. [Google Scholar] [CrossRef]
- Walczak, M.; Martens, S. Dissecting the role of the Atg12-Atg5-Atg16 complex during autophagosome formation. Autophagy 2013, 9, 424–425. [Google Scholar] [CrossRef]
- Karras, P.; Riveiro-Falkenbach, E.; Canon, E.; Tejedo, C.; Calvo, T.G.; Martinez-Herranz, R.; Alonso-Curbelo, D.; Cifdaloz, M.; Perez-Guijarro, E.; Gomez-Lopez, G.; et al. p62/SQSTM1 fuels melanoma progression by opposing mrna decay of a selective set of pro-metastatic factors. Cancer Cell 2019, 35, 46–63.e10. [Google Scholar] [CrossRef]
- Kageyama, S.; Gudmundsson, S.R.; Sou, Y.S.; Ichimura, Y.; Tamura, N.; Kazuno, S.; Ueno, T.; Miura, Y.; Noshiro, D.; Abe, M.; et al. p62/SQSTM1-droplet serves as a platform for autophagosome formation and anti-oxidative stress response. Nat. Commun. 2021, 12, 16. [Google Scholar] [CrossRef]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Kinner, A.; Wu, W.; Staudt, C.; Iliakis, G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008, 36, 5678–5694. [Google Scholar] [CrossRef] [PubMed]
- Vessoni, A.T.; Filippi-Chiela, E.C.; Menck, C.F.; Lenz, G. Autophagy and genomic integrity. Cell Death Differ. 2013, 20, 1444–1454. [Google Scholar] [CrossRef] [PubMed]
- Fernando, R.N.; Eleuteri, B.; Abdelhady, S.; Nussenzweig, A.; Andang, M.; Ernfors, P. Cell cycle restriction by histone H2AX limits proliferation of adult neural stem cells. Proc. Natl. Acad. Sci. USA 2011, 108, 5837–5842. [Google Scholar] [CrossRef]
- Lyamzaev, K.G.; Tokarchuk, A.V.; Panteleeva, A.A.; Mulkidjanian, A.Y.; Skulachev, V.P.; Chernyak, B.V. Induction of autophagy by depolarization of mitochondria. Autophagy 2018, 14, 921–924. [Google Scholar] [CrossRef]
- Grunwald, D.S.; Otto, N.M.; Park, J.M.; Song, D.; Kim, D.H. GABARAPs and LC3s have opposite roles in regulating ULK1 for autophagy induction. Autophagy 2020, 16, 600–614. [Google Scholar] [CrossRef]
- Bavro, V.N.; Sola, M.; Bracher, A.; Kneussel, M.; Betz, H.; Weissenhorn, W. Crystal structure of the GABA(A)-receptor-associated protein, GABARAP. EMBO Rep. 2002, 3, 183–189. [Google Scholar] [CrossRef]
- Cook, J.L.; Re, R.N.; deHaro, D.L.; Abadie, J.M.; Peters, M.; Alam, J. The trafficking protein GABARAP binds to and enhances plasma membrane expression and function of the angiotensin II type 1 receptor. Circ. Res. 2008, 102, 1539–1547. [Google Scholar] [CrossRef]
- Chen, Z.W.; Chang, C.S.; Leil, T.A.; Olcese, R.; Olsen, R.W. GABAA receptor-associated protein regulates GABAA receptor cell-surface number in Xenopus laevis oocytes. Mol. Pharmacol. 2005, 68, 152–159. [Google Scholar] [CrossRef]
- Daido, S.; Yamamoto, A.; Fujiwara, K.; Sawaya, R.; Kondo, S.; Kondo, Y. Inhibition of the DNA-dependent protein kinase catalytic subunit radiosensitizes malignant glioma cells by inducing autophagy. Cancer Res. 2005, 65, 4368–4375. [Google Scholar] [CrossRef]
- Zhang, Z.; Jin, F.; Lian, X.; Li, M.; Wang, G.; Lan, B.; He, H.; Liu, G.D.; Wu, Y.; Sun, G.; et al. Genistein promotes ionizing radiation-induced cell death by reducing cytoplasmic Bcl-xL levels in non-small cell lung cancer. Sci. Rep. 2018, 8, 328. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Pilarczyk, M.; Fazel-Najafabadi, M.; Kouril, M.; Shamsaei, B.; Vasiliauskas, J.; Niu, W.; Mahi, N.; Zhang, L.; Clark, N.A.; Ren, Y.; et al. Connecting omics signatures and revealing biological mechanisms with iLINCS. Nat. Commun. 2022, 13, 4678. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhattacharya, D.; Barrile, R.; Toukam, D.K.; Gawali, V.S.; Kallay, L.; Ahmed, T.; Brown, H.; Rezvanian, S.; Karve, A.; Desai, P.B.; et al. GABA(A) Receptor Activation Drives GABARAP–Nix Mediated Autophagy to Radiation-Sensitize Primary and Brain-Metastatic Lung Adenocarcinoma Tumors. Cancers 2024, 16, 3167. https://doi.org/10.3390/cancers16183167
Bhattacharya D, Barrile R, Toukam DK, Gawali VS, Kallay L, Ahmed T, Brown H, Rezvanian S, Karve A, Desai PB, et al. GABA(A) Receptor Activation Drives GABARAP–Nix Mediated Autophagy to Radiation-Sensitize Primary and Brain-Metastatic Lung Adenocarcinoma Tumors. Cancers. 2024; 16(18):3167. https://doi.org/10.3390/cancers16183167
Chicago/Turabian StyleBhattacharya, Debanjan, Riccardo Barrile, Donatien Kamdem Toukam, Vaibhavkumar S. Gawali, Laura Kallay, Taukir Ahmed, Hawley Brown, Sepideh Rezvanian, Aniruddha Karve, Pankaj B. Desai, and et al. 2024. "GABA(A) Receptor Activation Drives GABARAP–Nix Mediated Autophagy to Radiation-Sensitize Primary and Brain-Metastatic Lung Adenocarcinoma Tumors" Cancers 16, no. 18: 3167. https://doi.org/10.3390/cancers16183167
APA StyleBhattacharya, D., Barrile, R., Toukam, D. K., Gawali, V. S., Kallay, L., Ahmed, T., Brown, H., Rezvanian, S., Karve, A., Desai, P. B., Medvedovic, M., Wang, K., Ionascu, D., Harun, N., Vallabhapurapu, S., Wang, C., Qi, X., Baschnagel, A. M., Kritzer, J. A., ... Sengupta, S. (2024). GABA(A) Receptor Activation Drives GABARAP–Nix Mediated Autophagy to Radiation-Sensitize Primary and Brain-Metastatic Lung Adenocarcinoma Tumors. Cancers, 16(18), 3167. https://doi.org/10.3390/cancers16183167