
Scoping Review: Methods and Applications of Spatial Transcriptomics in Tumor Research


Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Quantity of Deposited Samples and Studies
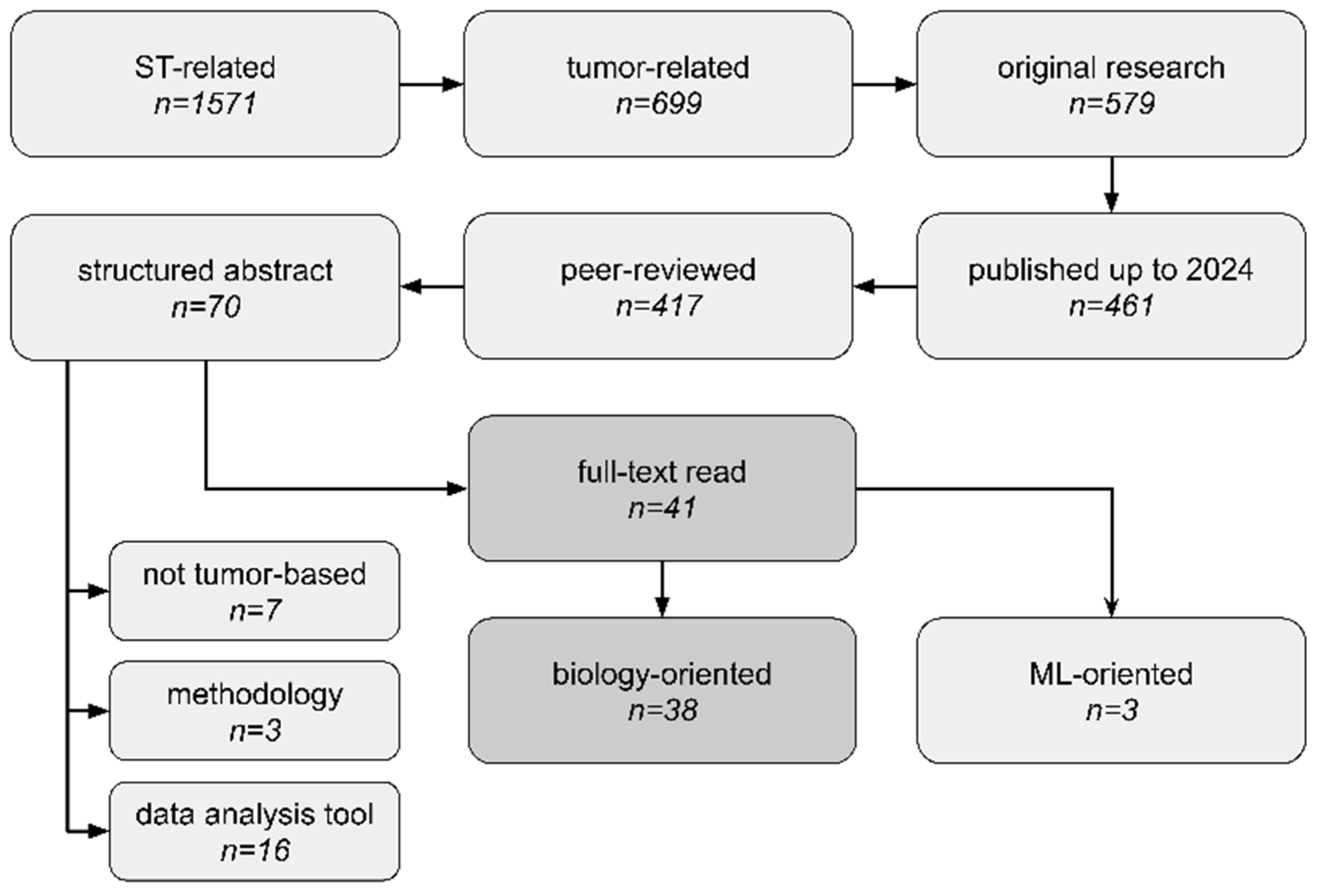
2.2. Article Search and Data Charting
2.2.1. Study Selection
2.2.2. Data Retrieval
2.3. Data Analysis
3. Results
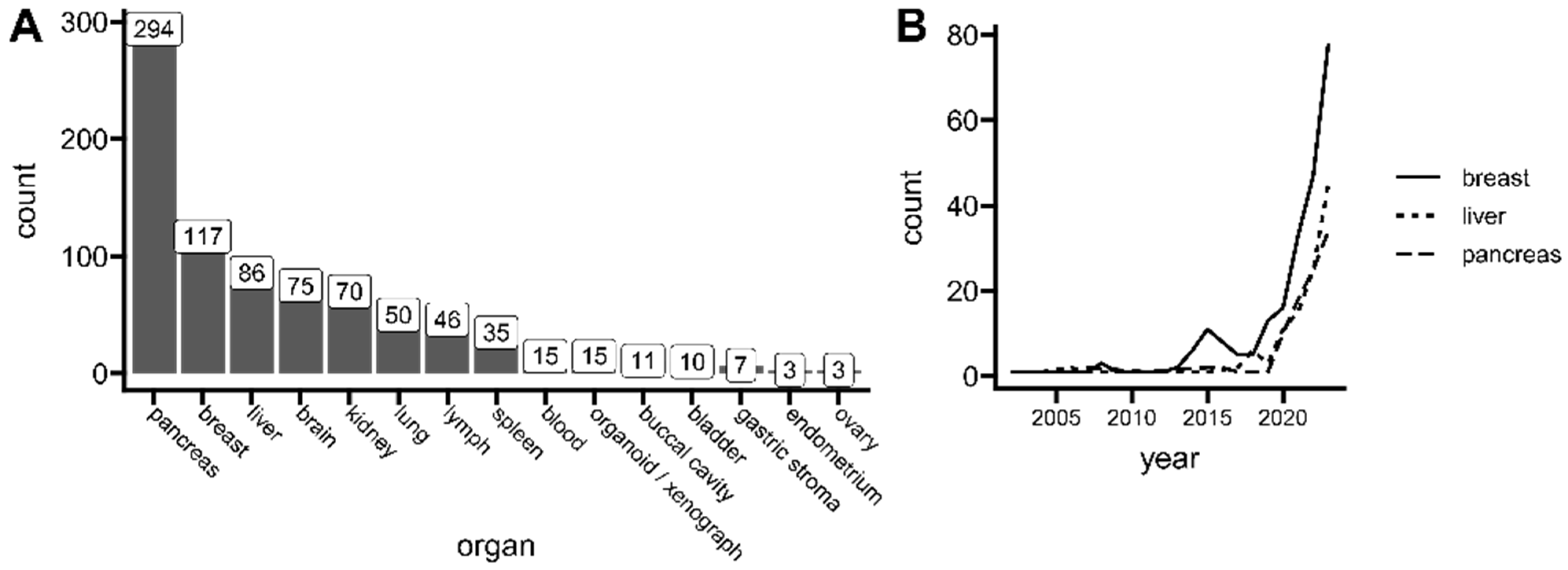
3.1. Tumors Types and Availability of Materials
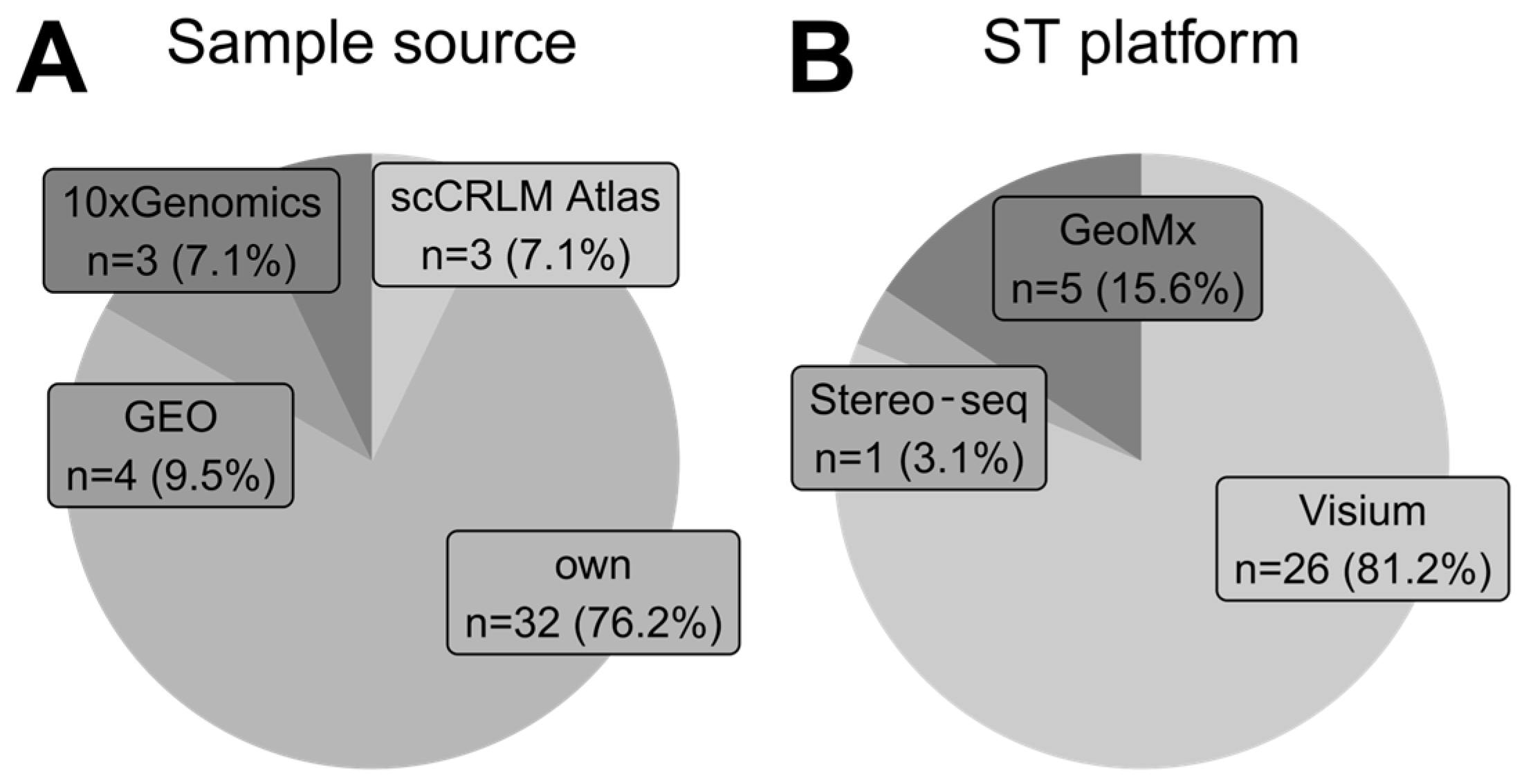
3.2. Sample Source and ST Platforms
3.3. Quality Control and Data Normalization
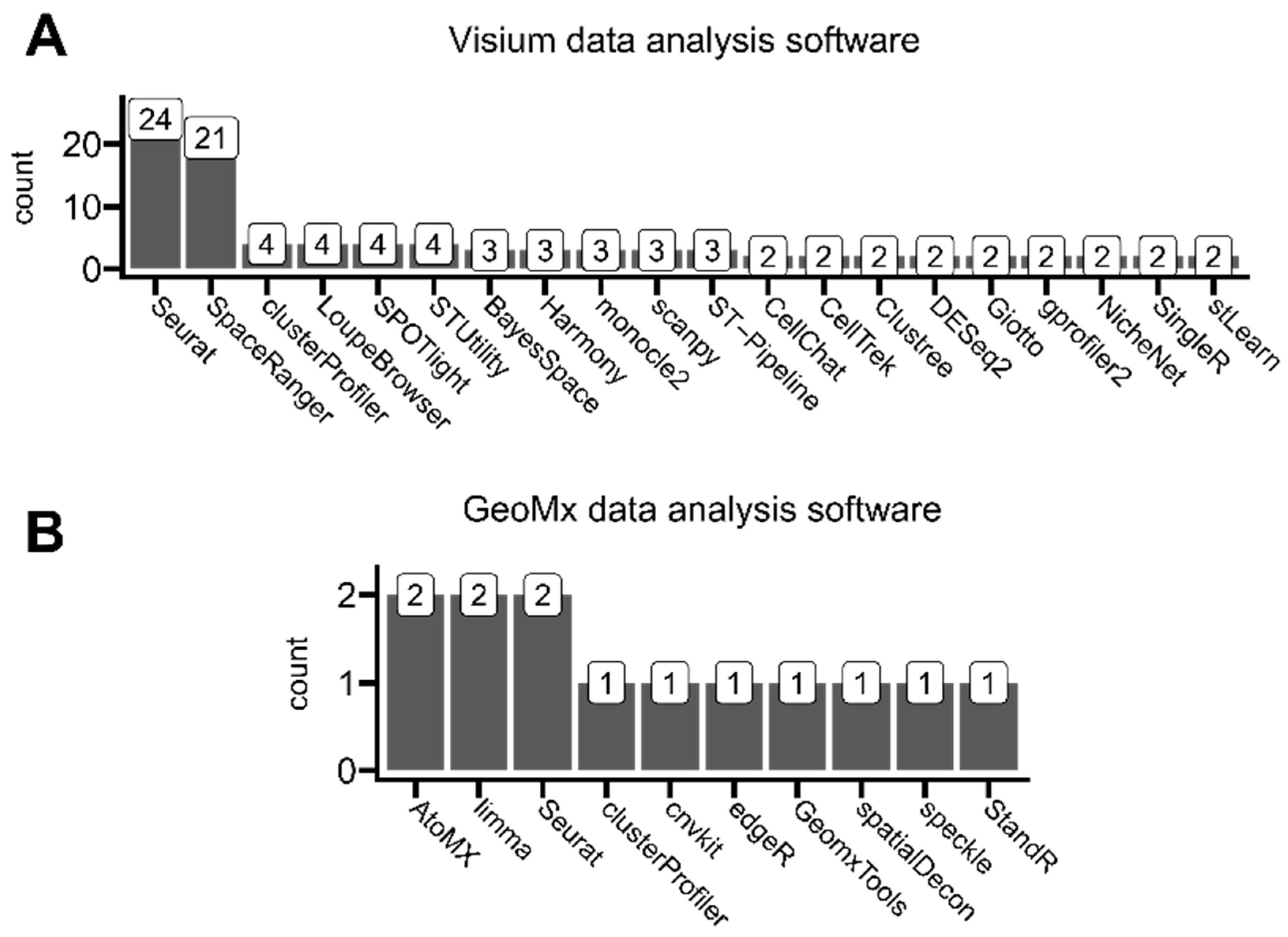
3.4. Application-Oriented Data Analysis Software
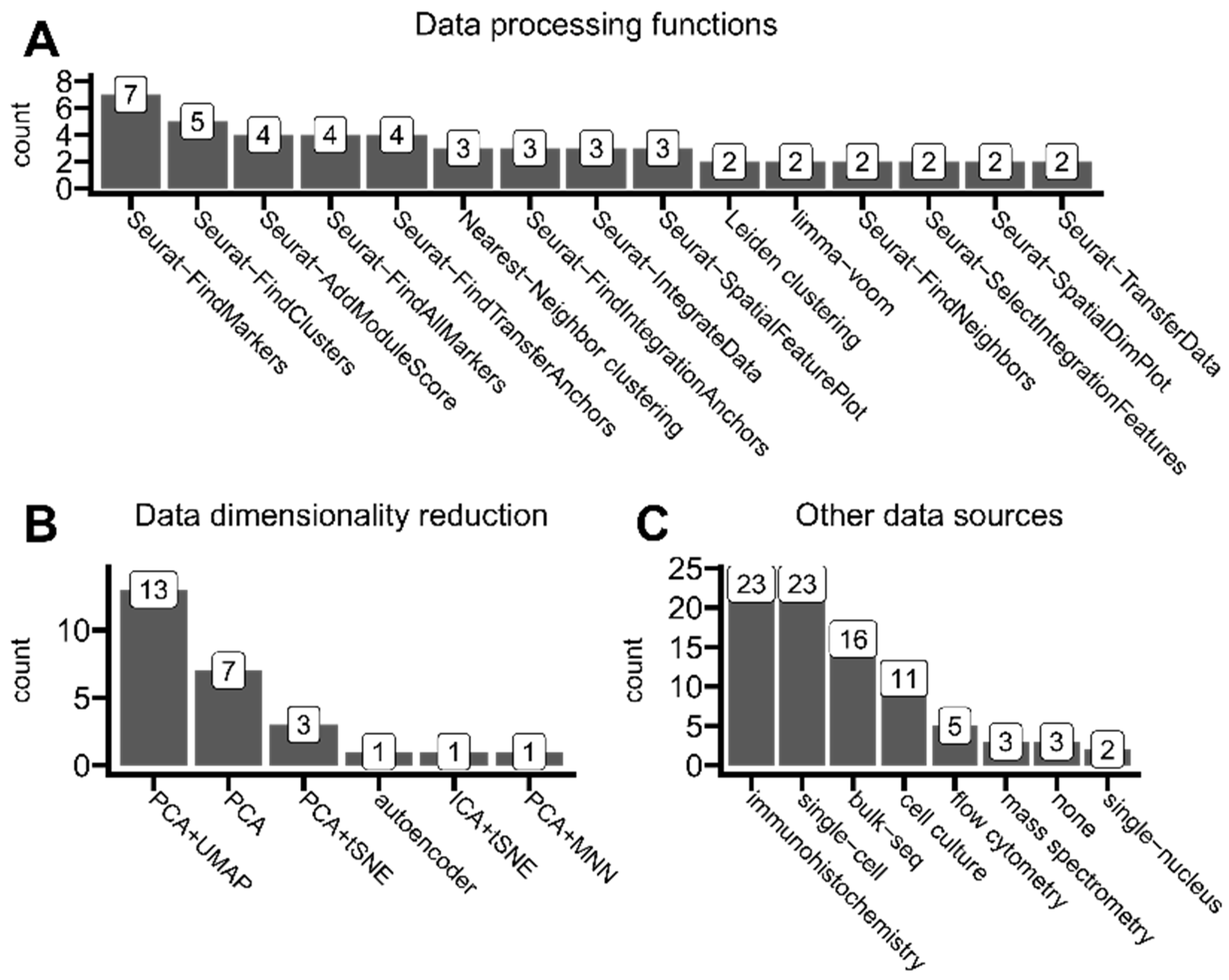
3.5. Data Integration and Dimensionality Reduction
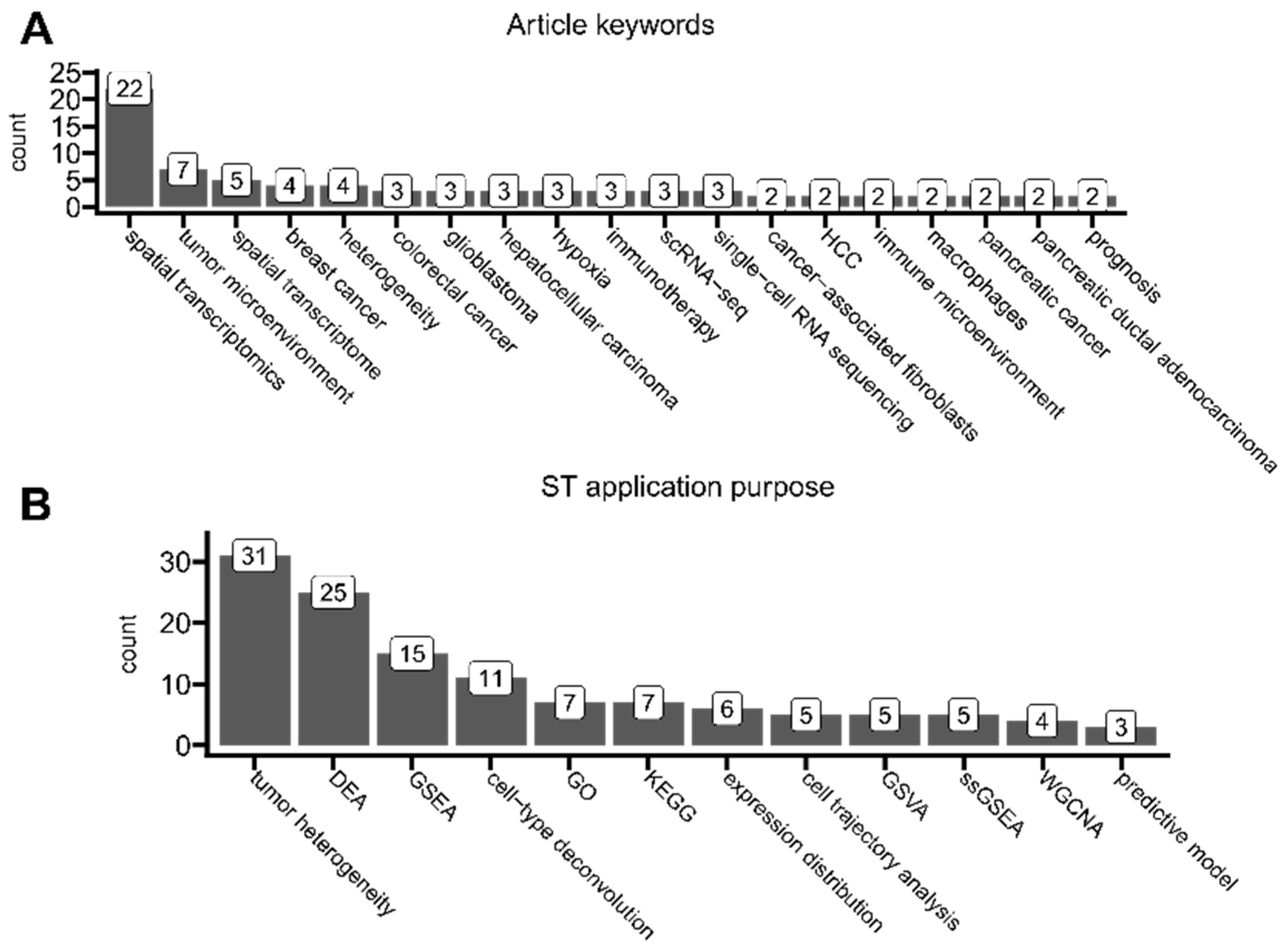
3.6. Semantic Analysis and Application Purposes
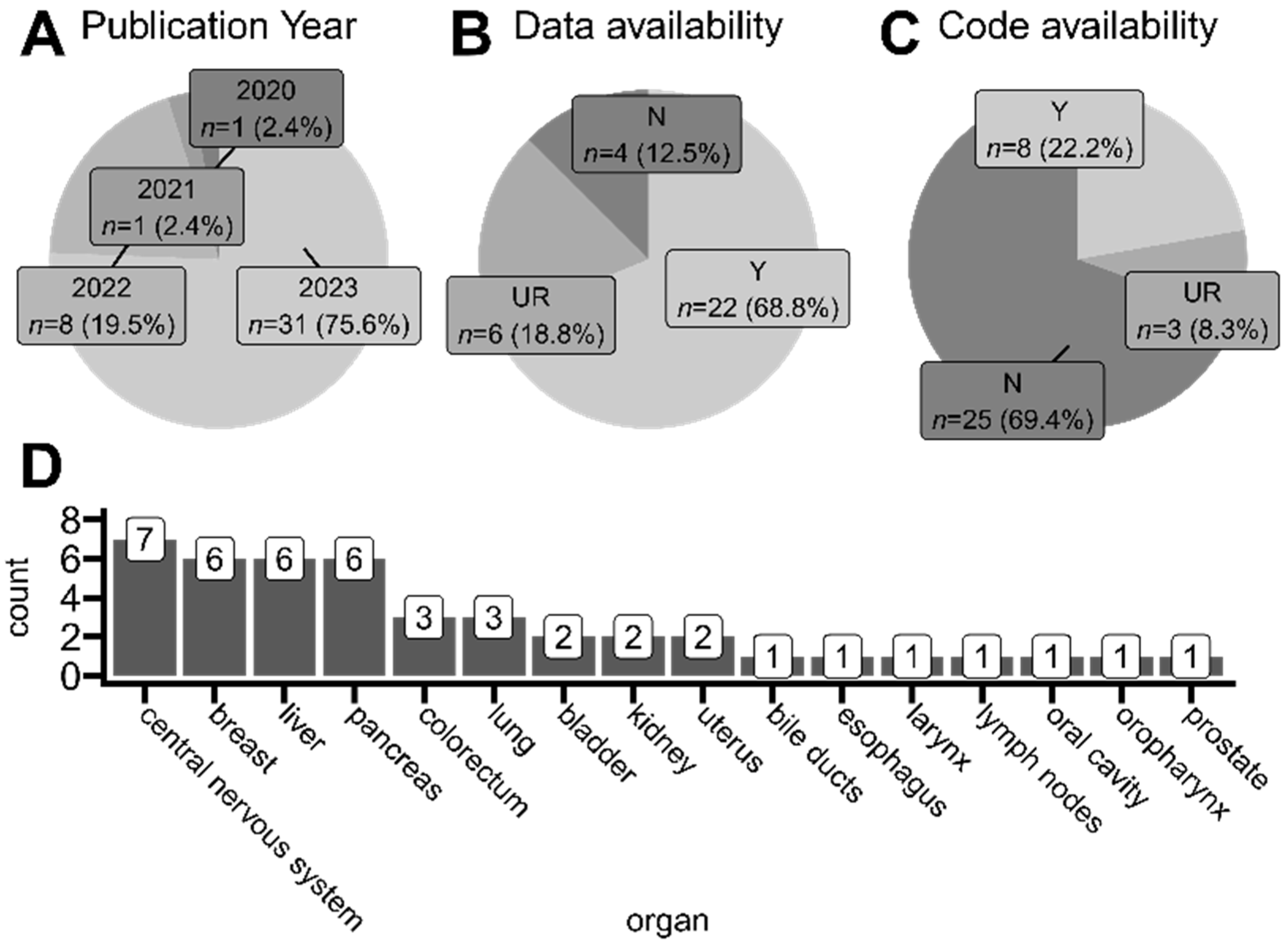
3.7. ST-Derived Biomedical Advancements
3.7.1. Central Nervous System
3.7.2. Breast
3.7.3. Liver
4. Discussion
5. Conclusions
- Neoplasm biology comprises a great share of ST-based research. The trend is highly increasing, with more and more articles published every year. This also concerns the development of novel data analysis methods and publicly available datasets;
- Visium (10x Genomics) was identified as the most popular NGS-based ST platform for new sample generation, alongside Seurat’s SCTransform normalization and integration method. Studies employing ST analyzes almost always use other data types, such as single-cell sequencing and immunohistochemistry, to support their findings;
- More attention should be paid to the direct application of single-cell data analysis methods in ST protocols;
- As all the analyses employing programming language-based tools are becoming more and more complex, authors should publish the analysis code far more often for higher transparency. Moreover, data processing protocols should be described in a fully exhaustive manner, making it possible to exactly reproduce the results;
- ST technology is predominantly applied to uncover tumor landscape composition and heterogeneity, determine tumor micro-environment characteristics, or identify cell–cell interactions, including spatial expression and co-occurrence patterns. The most common ST application purposes are differential expression analysis, gene set enrichment analysis, spatial cell-type deconvolution, and pathway analyses.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mazzarini, M.; Falchi, M.; Bani, D.; Migliaccio, A.R. Evolution and New Frontiers of Histology in Bio-medical Research. Microsc. Res. Tech. 2021, 84, 217–237. [Google Scholar] [CrossRef] [PubMed]
- Moses, L.; Pachter, L. Museum of Spatial Transcriptomics. Nat. Methods 2022, 19, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.M.; Asselin-Labat, M.-L.; Nguyen, Q.; Berthelet, J.; Tan, X.; Wimmer, V.C.; Merino, D.; Rogers, K.L.; Naik, S.H. Spatial Omics and Multiplexed Imaging to Explore Cancer Biology. Nat. Methods 2021, 18, 997–1012. [Google Scholar] [CrossRef] [PubMed]
- Asp, M.; Bergenstråhle, J.; Lundeberg, J. Spatially Resolved Transcriptomes-Next Generation Tools for Tissue Exploration. Bioessays 2020, 42, e1900221. [Google Scholar] [CrossRef]
- He, S.; Bhatt, R.; Brown, C.; Brown, E.A.; Buhr, D.L.; Chantranuvatana, K.; Danaher, P.; Dunaway, D.; Garrison, R.G.; Geiss, G.; et al. High-Plex Imaging of RNA and Proteins at Subcellular Resolution in Fixed Tissue by Spatial Molecular Imaging. Nat. Biotechnol. 2022, 40, 1794–1806. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.H.; Boettiger, A.N.; Moffitt, J.R.; Wang, S.; Zhuang, X. Spatially Resolved, Highly Multiplexed RNA Profiling in Single Cells. Science 2015, 348, aaa6090. [Google Scholar] [CrossRef]
- Lubeck, E.; Coskun, A.F.; Zhiyentayev, T.; Ahmad, M.; Cai, L. Single Cell in Situ RNA Profiling by Sequential Hybridization. Nat. Methods 2014, 11, 360–361. [Google Scholar] [CrossRef]
- Ke, R.; Mignardi, M.; Pacureanu, A.; Svedlund, J.; Botling, J.; Wählby, C.; Nilsson, M. In Situ Sequencing for RNA Analysis in Preserved Tissue and Cells. Nat. Methods 2013, 10, 857–860. [Google Scholar] [CrossRef]
- Lee, J.H.; Daugharthy, E.R.; Scheiman, J.; Kalhor, R.; Ferrante, T.C.; Terry, R.; Turczyk, B.M.; Yang, J.L.; Lee, H.S.; Aach, J.; et al. Fluorescent in Situ Sequencing (FISSEQ) of RNA for Gene Expression Profiling in Intact Cells and Tissues. Nat. Protoc. 2015, 10, 442–458. [Google Scholar] [CrossRef]
- Williams, C.G.; Lee, H.J.; Asatsuma, T.; Vento-Tormo, R.; Haque, A. An Introduction to Spatial Transcriptomics for Biomedical Research. Genome Med. 2022, 14, 68. [Google Scholar] [CrossRef]
- Merritt, C.R.; Ong, G.T.; Church, S.E.; Barker, K.; Danaher, P.; Geiss, G.; Hoang, M.; Jung, J.; Liang, Y.; McKay-Fleisch, J.; et al. Multiplex Digital Spatial Profiling of Proteins and RNA in Fixed Tissue. Nat. Biotechnol. 2020, 38, 586–599. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, M.; Deng, Y.; Su, G.; Enninful, A.; Guo, C.C.; Tebaldi, T.; Zhang, D.; Kim, D.; Bai, Z.; et al. High-Spatial-Resolution Multi-Omics Sequencing via Deterministic Barcoding in Tissue. Cell 2020, 183, 1665–1681.e18. [Google Scholar] [CrossRef]
- Ståhl, P.L.; Salmén, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and Analysis of Gene Expression in Tissue Sections by Spatial Transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Xia, K.; Sun, H.-X.; Li, J.; Li, J.; Zhao, Y.; Chen, L.; Qin, C.; Chen, R.; Chen, Z.; Liu, G.; et al. The Single-Cell Stereo-Seq Reveals Region-Specific Cell Subtypes and Transcriptome Profiling in Arabidopsis Leaves. Dev. Cell 2022, 57, 1299–1310.e4. [Google Scholar] [CrossRef]
- Rodriques, S.G.; Stickels, R.R.; Goeva, A.; Martin, C.A.; Murray, E.; Vanderburg, C.R.; Welch, J.; Chen, L.M.; Chen, F.; Macosko, E.Z. Slide-Seq: A Scalable Technology for Measuring Genome-Wide Expression at High Spatial Resolution. Science 2019, 363, 1463–1467. [Google Scholar] [CrossRef]
- Espina, V.; Wulfkuhle, J.D.; Calvert, V.S.; VanMeter, A.; Zhou, W.; Coukos, G.; Geho, D.H.; Petricoin, E.F.; Liotta, L.A. Laser-Capture Microdissection. Nat. Protoc. 2006, 1, 586–603. [Google Scholar] [CrossRef]
- Clément-Ziza, M.; Munnich, A.; Lyonnet, S.; Jaubert, F.; Besmond, C. Stabilization of RNA during Laser Capture Microdissection by Performing Experiments under Argon Atmosphere or Using Ethanol as a Solvent in Staining Solutions. RNA 2008, 14, 2698–2704. [Google Scholar] [CrossRef]
- Lee, Y.; Bogdanoff, D.; Wang, Y.; Hartoularos, G.C.; Woo, J.M.; Mowery, C.T.; Nisonoff, H.M.; Lee, D.S.; Sun, Y.; Lee, J.; et al. XYZeq: Spatially Resolved Single-Cell RNA Sequencing Reveals Expression Heterogeneity in the Tumor Microenvironment. Sci. Adv. 2021, 7, eabg4755. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Yang, Y.-C.; An, Z.-J.; Zhang, M.-H.; Fu, X.-H.; Huang, Z.-F.; Yuan, Y.; Hou, J. Advances in Spatial Transcriptomics and Related Data Analysis Strategies. J. Transl. Med. 2023, 21, 330. [Google Scholar] [CrossRef]
- Smith, K.D.; Prince, D.K.; MacDonald, J.W.; Bammler, T.K.; Akilesh, S. Challenges and Opportunities for the Clinical Translation of Spatial Transcriptomics Technologies. Glomerular Dis. 2024, 4, 49–63. [Google Scholar] [CrossRef]
- Cook, D.P.; Jensen, K.B.; Wise, K.; Roach, M.J.; Dezem, F.S.; Ryan, N.K.; Zamojski, M.; Vlachos, I.S.; Knott, S.R.V.; Butler, L.M.; et al. A Comparative Analysis of Imaging-Based Spatial Transcriptomics Platforms. bioRxiv 2023. [Google Scholar] [CrossRef]
- Cho, C.-S.; Xi, J.; Si, Y.; Park, S.-R.; Hsu, J.-E.; Kim, M.; Jun, G.; Kang, H.M.; Lee, J.H. Microscopic Examination of Spatial Transcriptome Using Seq-Scope. Cell 2021, 184, 3559–3572.e22. [Google Scholar] [CrossRef]
- Fang, S.; Chen, B.; Zhang, Y.; Sun, H.; Liu, L.; Liu, S.; Li, Y.; Xu, X. Computational Approaches and Challenges in Spatial Transcriptomics. Genom. Proteom. Bioinform. 2023, 21, 24–47. [Google Scholar] [CrossRef]
- Tricco, A.C.; Lillie, E.; Zarin, W.; O’Brien, K.K.; Colquhoun, H.; Levac, D.; Moher, D.; Peters, M.D.J.; Horsley, T.; Weeks, L.; et al. PRISMA Extension for Scoping Reviews (PRISMA-ScR): Checklist and Explanation. Ann. Intern. Med. 2018, 169, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; He, Y.; Zheng, J.; Wang, X.; Chen, S. Dissecting Order amidst Chaos of Programmed Cell Deaths: Construction of a Diagnostic Model for KIRC Using Transcriptomic Information in Blood-Derived Exosomes and Single-Cell Multi-Omics Data in Tumor Microenvironment. Front. Immunol. 2023, 14, 1130513. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Klein, C.; Caruso, S.; Maille, P.; Allende, D.S.; Mínguez, B.; Iavarone, M.; Ningarhari, M.; Casadei-Gardini, A.; Pedica, F.; et al. Artificial Intelligence-Based Pathology as a Biomarker of Sensitivity to Atezolizumab-Bevacizumab in Patients with Hepatocellular Carcinoma: A Multicentre Retrospective Study. Lancet Oncol. 2023, 24, 1411–1422. [Google Scholar] [CrossRef]
- Yoosuf, N.; Navarro, J.F.; Salmén, F.; Ståhl, P.L.; Daub, C.O. Identification and Transfer of Spatial Transcriptomics Signatures for Cancer Diagnosis. Breast Cancer Res. 2020, 22, 6. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Dawson, C. ggprism: A ‘ggplot2’ Extension Inspired by ‘GraphPad Prism’. R Package Version 1.0.5. Available online: https://github.com/csdaw/ggprism (accessed on 14 March 2024).
- Marx, V. Method of the Year: Spatially Resolved Transcriptomics. Nat. Methods 2021, 18, 9–14. [Google Scholar] [CrossRef]
- Heumos, L.; Schaar, A.C.; Lance, C.; Litinetskaya, A.; Drost, F.; Zappia, L.; Lücken, M.D.; Strobl, D.C.; Henao, J.; Curion, F.; et al. Best Practices for Single-Cell Analysis across Modalities. Nat. Rev. Genet. 2023, 24, 550–572. [Google Scholar] [CrossRef]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating Single-Cell Transcriptomic Data across Different Conditions, Technologies, and Species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Lytal, N.; Ran, D.; An, L. Normalization Methods on Single-Cell RNA-Seq Data: An Empirical Survey. Front. Genet. 2020, 11, 41. [Google Scholar] [CrossRef] [PubMed]
- Saiselet, M.; Rodrigues-Vitória, J.; Tourneur, A.; Craciun, L.; Spinette, A.; Larsimont, D.; Andry, G.; Lundeberg, J.; Maenhaut, C.; Detours, V. Transcriptional Output, Cell-Type Densities, and Normalization in Spatial Transcriptomics. J. Mol. Cell Biol. 2020, 12, 906–908. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Ni, Z.; Mohanty, C.; Bacher, R.; Kendziorski, C. Normalization by Distributional Resampling of High Throughput Single-Cell RNA-Sequencing Data. Bioinformatics 2021, 37, 4123–4128. [Google Scholar] [CrossRef]
- Zou, L.S.; Zhao, T.; Cable, D.M.; Murray, E.; Aryee, M.J.; Chen, F.; Irizarry, R.A. Detection of Allele-Specific Expression in Spatial Transcriptomics with SpASE. Genome Biol. 2024, 25, 180. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Guo, Q.; Liu, Y.; Hou, Q.; Liao, M.; Guo, Y.; Zang, Y.; Wang, F.; Liu, H.; Luan, X.; et al. Single-Cell and Spatial Transcriptomics Reveal POSTN+ Cancer-Associated Fibroblasts Correlated with Immune Suppression and Tumour Progression in Non-Small Cell Lung Cancer. Clin. Transl. Med. 2023, 13, e1515. [Google Scholar] [CrossRef]
- Wolf, F.A.; Angerer, P.; Theis, F.J. SCANPY: Large-Scale Single-Cell Gene Expression Data Analysis. Genome Biol. 2018, 19, 15. [Google Scholar] [CrossRef]
- Palla, G.; Spitzer, H.; Klein, M.; Fischer, D.; Schaar, A.C.; Kuemmerle, L.B.; Rybakov, S.; Ibarra, I.L.; Holmberg, O.; Virshup, I.; et al. Squidpy: A Scalable Framework for Spatial Omics Analysis. Nat. Methods 2022, 19, 171–178. [Google Scholar] [CrossRef]
- Odhiambo, J.; Onsongo, W.; Osman, S. An Analytical Comparison Between Python Vs R Programming Languages Which One Is the Best for Machine Learning and Deep Learning? 2020. Available online: https://www.researchgate.net/publication/341419997_An_Analytical_Comparison_Between_Python_Vs_R_Programming_Languages_Which_one_is_the_best_for_Machine_Learning_and_Deep_Learning (accessed on 3 May 2024).
- Ahlmann-Eltze, C.; Patil, I. Ggsignif: R Package for Displaying Significance Brackets for “Ggplot2” 2021. Available online: https://const-ae.github.io/ggsignif/#citation (accessed on 3 May 2024).
- Kolde, R. Pheatmap: Pretty Heatmaps; 2019. Available online: https://cran.r-project.org/web/packages/pheatmap/pheatmap.pdf (accessed on 3 May 2024).
- Patil, I. Visualizations with Statistical Details: The “ggstatsplot” Approach. JOSS 2021, 6, 3167. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex Heatmaps Reveal Patterns and Correlations in Multidimensional Genomic Data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Waskom, M. Seaborn: Statistical Data Visualization. JOSS 2021, 6, 3021. [Google Scholar] [CrossRef]
- Zhao, E.; Stone, M.R.; Ren, X.; Guenthoer, J.; Smythe, K.S.; Pulliam, T.; Williams, S.R.; Uytingco, C.R.; Taylor, S.E.B.; Nghiem, P.; et al. Spatial Transcriptomics at Subspot Resolution with BayesSpace. Nat. Biotechnol. 2021, 39, 1375–1384. [Google Scholar] [CrossRef]
- Liu, Y.; Xun, Z.; Ma, K.; Liang, S.; Li, X.; Zhou, S.; Sun, L.; Liu, Y.; Du, Y.; Guo, X.; et al. Identification of a Tumour Immune Barrier in the HCC Microenvironment That Determines the Efficacy of Immunotherapy. J. Hepatol. 2023, 78, 770–782. [Google Scholar] [CrossRef] [PubMed]
- Kleshchevnikov, V.; Shmatko, A.; Dann, E.; Aivazidis, A.; King, H.W.; Li, T.; Elmentaite, R.; Lomakin, A.; Kedlian, V.; Gayoso, A.; et al. Cell2location Maps Fine-Grained Cell Types in Spatial Transcriptomics. Nat. Biotechnol. 2022, 40, 661–671. [Google Scholar] [CrossRef]
- Du, J.; Qiu, C.; Li, W.-S.; Wang, B.; Han, X.-L.; Lin, S.-W.; Fu, X.-H.; Hou, J.; Huang, Z.-F. Spatial Transcriptomics Analysis Reveals That CCL17 and CCL22 Are Robust Indicators of a Suppressive Immune Environment in Angioimmunoblastic T Cell Lymphoma (AITL). Front. Biosci. 2022, 27, 270. [Google Scholar] [CrossRef]
- Jin, S.; Guerrero-Juarez, C.F.; Zhang, L.; Chang, I.; Ramos, R.; Kuan, C.-H.; Myung, P.; Plikus, M.V.; Nie, Q. Inference and Analysis of Cell-Cell Communication Using CellChat. Nat. Commun. 2021, 12, 1088. [Google Scholar] [CrossRef]
- Guo, W.; Zhou, B.; Yang, Z.; Liu, X.; Huai, Q.; Guo, L.; Xue, X.; Tan, F.; Li, Y.; Xue, Q.; et al. Integrating Microarray-Based Spatial Transcriptomics and Single-Cell RNA-Sequencing Reveals Tissue Architecture in Esophageal Squamous Cell Carcinoma. EBioMedicine 2022, 84, 104281. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, Z.; Feng, Y.; Gao, J.; Wang, B.; Lian, C.; Diao, B. Integration Analysis of Single-Cell and Spatial Transcriptomics Reveal the Cellular Heterogeneity Landscape in Glioblastoma and Establish a Polygenic Risk Model. Front. Oncol. 2023, 13, 1109037. [Google Scholar] [CrossRef]
- Wei, R.; He, S.; Bai, S.; Sei, E.; Hu, M.; Thompson, A.; Chen, K.; Krishnamurthy, S.; Navin, N.E. Spatial Charting of Single-Cell Transcriptomes in Tissues. Nat. Biotechnol. 2022, 40, 1190–1199. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. ClusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integrative Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Al-Holou, W.N.; Wang, H.; Ravikumar, V.; Shankar, S.; Oneka, M.; Fehmi, Z.; Verhaak, R.G.; Kim, H.; Pratt, D.; Camelo-Piragua, S.; et al. Subclonal Evolution and Expansion of Spatially Distinct THY1-Positive Cells Is Associated with Recurrence in Glioblastoma. Neoplasia 2023, 36, 100872. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Pan, B.; Wang, F.; Lyu, S.; Zhai, J.; Hu, X.; Liu, Z.; Li, L.; Lang, R.; He, Q.; et al. Spatial Transcriptomics Reveals the Heterogeneity and FGG+CRP+ Inflammatory Cancer-Associated Fibroblasts Replace Islets in Pancreatic Ductal Adenocarcinoma. Front. Oncol. 2023, 13, 1112576. [Google Scholar] [CrossRef] [PubMed]
- Cherry, C.; Maestas, D.R.; Han, J.; Andorko, J.I.; Cahan, P.; Fertig, E.J.; Garmire, L.X.; Elisseeff, J.H. Computational Reconstruction of the Signalling Networks Surrounding Implanted Biomaterials from Single-Cell Transcriptomics. Nat. Biomed. Eng. 2021, 5, 1228–1238. [Google Scholar] [CrossRef]
- Zhang, S.; Yuan, L.; Danilova, L.; Mo, G.; Zhu, Q.; Deshpande, A.; Bell, A.T.F.; Elisseeff, J.; Popel, A.S.; Anders, R.A.; et al. Spatial Transcriptomics Analysis of Neoadjuvant Cabozantinib and Nivolumab in Advanced Hepatocellular Carcinoma Identifies Independent Mechanisms of Resistance and Recurrence. Genome Med. 2023, 15, 72. [Google Scholar] [CrossRef]
- Cao, E.Y.; Ouyang, J.F.; Rackham, O.J.L. GeneSwitches: Ordering Gene Expression and Functional Events in Single-Cell Experiments. Bioinformatics 2020, 36, 3273–3275. [Google Scholar] [CrossRef]
- Dries, R.; Zhu, Q.; Dong, R.; Eng, C.-H.L.; Li, H.; Liu, K.; Fu, Y.; Zhao, T.; Sarkar, A.; Bao, F.; et al. Giotto: A Toolbox for Integrative Analysis and Visualization of Spatial Expression Data. Genome Biol. 2021, 22, 78. [Google Scholar] [CrossRef]
- Vo, T.; Balderson, B.; Jones, K.; Ni, G.; Crawford, J.; Millar, A.; Tolson, E.; Singleton, M.; Kojic, M.; Robertson, T.; et al. Spatial Transcriptomic Analysis of Sonic Hedgehog Medulloblastoma Identifies That the Loss of Heterogeneity and Promotion of Differentiation Underlies the Response to CDK4/6 Inhibition. Genome Med. 2023, 15, 29. [Google Scholar] [CrossRef]
- Shi, Z.-D.; Sun, Z.; Zhu, Z.-B.; Liu, X.; Chen, J.-Z.; Hao, L.; Zhu, J.-F.; Pang, K.; Wu, D.; Dong, Y.; et al. Integrated Single-Cell and Spatial Transcriptomic Profiling Reveals Higher Intratumour Heterogeneity and Epithelial-Fibroblast Interactions in Recurrent Bladder Cancer. Clin. Transl. Med. 2023, 13, e1338. [Google Scholar] [CrossRef]
- Korsunsky, I.; Millard, N.; Fan, J.; Slowikowski, K.; Zhang, F.; Wei, K.; Baglaenko, Y.; Brenner, M.; Loh, P.; Raychaudhuri, S. Fast, Sensitive and Accurate Integration of Single-Cell Data with Harmony. Nat. Methods 2019, 16, 1289–1296. [Google Scholar] [CrossRef]
- Fu, R.; Norris, G.A.; Willard, N.; Griesinger, A.M.; Riemondy, K.A.; Amani, V.; Grimaldo, E.; Harris, F.; Hankinson, T.C.; Mitra, S.; et al. Spatial Transcriptomic Analysis Delineates Epithelial and Mesenchymal Subpopulations and Transition Stages in Childhood Ependymoma. Neuro Oncol. 2023, 25, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Yousuf, S.; Qiu, M.; Voith von Voithenberg, L.; Hulkkonen, J.; Macinkovic, I.; Schulz, A.R.; Hartmann, D.; Mueller, F.; Mijatovic, M.; Ibberson, D.; et al. Spatially Resolved Multi-Omics Single-Cell Analyses Inform Mechanisms of Immune Dysfunction in Pancreatic Cancer. Gastroenterology 2023, 165, 891–908.e14. [Google Scholar] [CrossRef]
- Qiu, X.; Hill, A.; Packer, J.; Lin, D.; Ma, Y.-A.; Trapnell, C. Single-Cell MRNA Quantification and Differential Analysis with Census. Nat. Methods 2017, 14, 309–315. [Google Scholar] [CrossRef]
- Liu, H.-T.; Chen, S.-Y.; Peng, L.-L.; Zhong, L.; Zhou, L.; Liao, S.-Q.; Chen, Z.-J.; Wang, Q.-L.; He, S.; Zhou, Z.-H. Spatially Resolved Transcriptomics Revealed Local Invasion-Related Genes in Colorectal Cancer. Front. Oncol. 2023, 13, 1089090. [Google Scholar] [CrossRef]
- Raredon, M.S.B.; Yang, J.; Kothapalli, N.; Lewis, W.; Kaminski, N.; Niklason, L.E.; Kluger, Y. Comprehensive Visualization of Cell-Cell Interactions in Single-Cell and Spatial Transcriptomics with NICHES. Bioinformatics 2023, 39, btac775. [Google Scholar] [CrossRef] [PubMed]
- Tashireva, L.; Grigoryeva, E.; Alifanov, V.; Iamshchikov, P.; Zavyalova, M.; Perelmuter, V. Spatial Heterogeneity of Integrins and Their Ligands in Primary Breast Tumors. Discov. Med. 2023, 35, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Browaeys, R.; Saelens, W.; Saeys, Y. NicheNet: Modeling Intercellular Communication by Linking Ligands to Target Genes. Nat. Methods 2020, 17, 159–162. [Google Scholar] [CrossRef]
- Griss, J.; Viteri, G.; Sidiropoulos, K.; Nguyen, V.; Fabregat, A.; Hermjakob, H. ReactomeGSA-Efficient Multi-Omics Comparative Pathway Analysis. Mol. Cell Proteom. 2020, 19, 2115–2125. [Google Scholar] [CrossRef]
- Aibar, S.; González-Blas, C.B.; Moerman, T.; Huynh-Thu, V.A.; Imrichova, H.; Hulselmans, G.; Rambow, F.; Marine, J.-C.; Geurts, P.; Aerts, J.; et al. SCENIC: Single-Cell Regulatory Network Inference and Clustering. Nat. Methods 2017, 14, 1083–1086. [Google Scholar] [CrossRef]
- Wu, Y.; Yang, S.; Ma, J.; Chen, Z.; Song, G.; Rao, D.; Cheng, Y.; Huang, S.; Liu, Y.; Jiang, S.; et al. Spatiotemporal Immune Landscape of Colorectal Cancer Liver Metastasis at Single-Cell Level. Cancer Discov. 2022, 12, 134–153. [Google Scholar] [CrossRef]
- Yang, T.; Liu, J.; Liu, F.; Lei, J.; Chen, S.; Ma, Z.; Ke, P.; Yang, Q.; Wen, J.; He, Y.; et al. Integrative Analysis of Disulfidptosis and Immune Microenvironment in Hepatocellular Carcinoma: A Putative Model and Immunotherapeutic Strategies. Front. Immunol. 2023, 14, 1294677. [Google Scholar] [CrossRef]
- Kueckelhaus, J.; Frerich, S.; Kada-Benotmane, J.; Koupourtidou, C.; Ninkovic, J.; Dichgans, M.; Beck, J.; Schnell, O.; Heiland, D. Inferring histology-associated gene expression gradients in spatial transcriptomic studies. Nat. Commun. 2024, 15, 7280. [Google Scholar] [CrossRef]
- Andrieux, G.; Das, T.; Griffin, M.; Straehle, J.; Paine, S.M.L.; Beck, J.; Boerries, M.; Heiland, D.H.; Smith, S.J.; Rahman, R.; et al. Spatially Resolved Transcriptomic Profiles Reveal Unique Defining Molecular Features of Infiltrative 5ALA-Metabolizing Cells Associated with Glioblastoma Recurrence. Genome Med. 2023, 15, 48. [Google Scholar] [CrossRef] [PubMed]
- Elosua-Bayes, M.; Nieto, P.; Mereu, E.; Gut, I.; Heyn, H. SPOTlight: Seeded NMF Regression to Deconvolute Spatial Transcriptomics Spots with Single-Cell Transcriptomes. Nucleic Acids Res. 2021, 49, e50. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Looney, A.P.; Liu, L.; Wu, E.; Fong, V.; Hsu, A.; Chak, S.; Naikawadi, R.P.; Wolters, P.J.; Abate, A.R.; et al. Reference-Based Analysis of Lung Single-Cell Sequencing Reveals a Transitional Profibrotic Macrophage. Nat. Immunol. 2019, 20, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Street, K.; Risso, D.; Fletcher, R.B.; Das, D.; Ngai, J.; Yosef, N.; Purdom, E.; Dudoit, S. Slingshot: Cell Lineage and Pseudotime Inference for Single-Cell Transcriptomics. BMC Genom. 2018, 19, 477. [Google Scholar] [CrossRef] [PubMed]
- Ni, Z.; Prasad, A.; Chen, S.; Halberg, R.B.; Arkin, L.M.; Drolet, B.A.; Newton, M.A.; Kendziorski, C. SpotClean Adjusts for Spot Swapping in Spatial Transcriptomics Data. Nat. Commun. 2022, 13, 2971. [Google Scholar] [CrossRef]
- Hao, Y.; Stuart, T.; Kowalski, M.; Choudhary, S.; Hoffman, P.; Hartman, A.; Srivastava, A.; Molla, G.; Madad, S.; Fernandez-Granda, C.; et al. Dictionary Learning for Integrative, Multimodal, and Massively Scalable Single-Cell Analysis. Nat. Biotechnol. 2024, 42, 293–304. [Google Scholar] [CrossRef]
- Mihaylov, I.; Kańduła, M.; Krachunov, M.; Vassilev, D. A Novel Framework for Horizontal and Vertical Data Integration in Cancer Studies with Application to Survival Time Prediction Models. Biol. Direct 2019, 14, 22. [Google Scholar] [CrossRef]
- Moncada, R.; Barkley, D.; Wagner, F.; Chiodin, M.; Devlin, J.C.; Baron, M.; Hajdu, C.H.; Simeone, D.M.; Yanai, I. Integrating Microarray-Based Spatial Transcriptomics and Single-Cell RNA-Seq Reveals Tissue Architecture in Pancreatic Ductal Adenocarcinomas. Nat. Biotechnol. 2020, 38, 333–342. [Google Scholar] [CrossRef]
- Zhu, L.; Jiang, M.; Wang, H.; Sun, H.; Zhu, J.; Zhao, W.; Fang, Q.; Yu, J.; Chen, P.; Wu, S.; et al. A narrative review of tumor heterogeneity and challenges to tumor drug therapy. Ann. Transl. Med. 2021, 9, 1351. [Google Scholar] [CrossRef]
- Hass, R.; von der Ohe, J.; Ungefroren, H. Impact of the Tumor Microenvironment on Tumor Heterogeneity and Consequences for Cancer Cell Plasticity and Stemness. Cancers 2020, 12, 3716. [Google Scholar] [CrossRef] [PubMed]
- Moffet, J.J.D.; Fatunla, O.E.; Freytag, L.; Kriel, J.; Jones, J.J.; Roberts-Thomson, S.J.; Pavenko, A.; Scoville, D.K.; Zhang, L.; Liang, Y.; et al. Spatial Architecture of High-Grade Glioma Reveals Tumor Heterogeneity within Distinct Domains. Neurooncol Adv. 2023, 5, vdad142. [Google Scholar] [CrossRef] [PubMed]
- Powell, N.R.; Silvola, R.M.; Howard, J.S.; Badve, S.; Skaar, T.C.; Ipe, J. Quantification of Spatial Pharmacogene Expression Heterogeneity in Breast Tumors. Cancer Rep. 2023, 6, e1686. [Google Scholar] [CrossRef]
- Levy-Jurgenson, A.; Tekpli, X.; Yakhini, Z. Assessing Heterogeneity in Spatial Data Using the HTA Index with Applications to Spatial Transcriptomics and Imaging. Bioinformatics 2021, 37, 3796–3804. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology Consortium. The Gene Ontology (GO) Database and Informatics Resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar] [CrossRef]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautès-Fridman, C.; Fridman, W.H.; et al. Estimating the Population Abundance of Tissue-Infiltrating Immune and Stromal Cell Populations Using Gene Expression. Genome Biol. 2016, 17, 218. [Google Scholar] [CrossRef]
- Hong, J.H.; Yong, C.H.; Heng, H.L.; Chan, J.Y.; Lau, M.C.; Chen, J.; Lee, J.Y.; Lim, A.H.; Li, Z.; Guan, P.; et al. Integrative Multiomics Enhancer Activity Profiling Identifies Therapeutic Vulnerabilities in Cholangiocarcinoma of Different Etiologies. Gut 2023, 73, 966–984. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Heming, M.; Haessner, S.; Wolbert, J.; Lu, I.-N.; Li, X.; Brokinkel, B.; Müther, M.; Holling, M.; Stummer, W.; Thomas, C.; et al. Intratumor Heterogeneity and T Cell Exhaustion in Primary CNS Lymphoma. Genome Med. 2022, 14, 109. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Zhang, Y.; Cheng, R.; Zhang, D.; Li, F.; Guo, Y.; Qiu, Z.; Dong, X.; Ban, X.; Sun, B.; et al. Spatial Maps of Hepatocellular Carcinoma Transcriptomes Highlight an Unexplored Landscape of Heterogeneity and a Novel Gene Signature for Survival. Cancer Cell Int. 2022, 22, 57. [Google Scholar] [CrossRef] [PubMed]
- Pham, D.; Tan, X.; Balderson, B.; Xu, J.; Grice, L.F.; Yoon, S.; Willis, E.F.; Tran, M.; Lam, P.Y.; Raghubar, A.; et al. Robust Mapping of Spatiotemporal Trajectories and Cell-Cell Interactions in Healthy and Diseased Tissues. Nat. Commun. 2023, 14, 7739. [Google Scholar] [CrossRef]
- Zheng, T.; Liu, Q.; Xing, F.; Zeng, C.; Wang, W. Disulfidptosis: A New Form of Programmed Cell Death. J. Exp. Clin. Cancer Res. 2023, 42, 137. [Google Scholar] [CrossRef]
- Zhou, R.; Yang, G.; Zhang, Y.; Wang, Y. Spatial Transcriptomics in Development and Disease. Mol. Biomed. 2023, 4, 32. [Google Scholar] [CrossRef]
- Jung, N.; Kim, T.-K. Spatial Transcriptomics in Neuroscience. Exp. Mol. Med. 2023, 55, 2105–2115. [Google Scholar] [CrossRef]
- Baker, M. 1,500 Scientists Lift the Lid on Reproducibility. Nature 2016, 533, 452–454. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.; Hardin, J.; Stoebel, D.M. Selecting Between-Sample RNA-Seq Normalization Methods from the Perspective of Their Assumptions. Brief. Bioinform. 2017, 19, 776–792. [Google Scholar] [CrossRef]
- Wang, M.; Song, W.-M.; Ming, C.; Wang, Q.; Zhou, X.; Xu, P.; Krek, A.; Yoon, Y.; Ho, L.; Orr, M.E.; et al. Guidelines for Bioinformatics of Single-Cell Sequencing Data Analysis in Alzheimer’s Disease: Review, Recommendation, Implementation and Application. Mol. Neurodegener. 2022, 17, 17. [Google Scholar] [CrossRef]
- Bhuva, D.D.; Tan, C.W.; Marceaux, C.; Chen, J.; Kharbanda, M.; Jin, X.; Liu, N.; Feher, K.; Putri, G.; Asselin-Labat, M.-L.; et al. Library Size Confounds Biology in Spatial Transcriptomics Data. Genome Biol. 2024, 25, 99. [Google Scholar] [CrossRef]
- Atta, L.; Clifton, K.; Anant, M.; Aihara, G.; Fan, J. Gene Count Normalization in Single-Cell Imaging-Based Spatially Resolved Transcriptomics. Genome Biol. 2024, 25, 153. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Dong, W.; Socher, R.; Li, L.-J.; Li, K.; Fei-Fei, L. ImageNet: A Large-Scale Hierarchical Image Database. In Proceedings of the 2009 IEEE Conference on Computer Vision and Pattern Recognition, Miami, FL, USA, 20–25 June 2009; pp. 248–255. [Google Scholar]
- Riasatian, A.; Babaie, M.; Maleki, D.; Kalra, S.; Valipour, M.; Hemati, S.; Zaveri, M.; Safarpoor, A.; Shafiei, S.; Afshari, M.; et al. Fine-Tuning and Training of Densenet for Histopathology Image Representation Using TCGA Diagnostic Slides. Med. Image Anal. 2021, 70, 102032. [Google Scholar] [CrossRef]
- Sikaroudi, M.; Hosseini, M.; Gonzalez, R.; Rahnamayan, S.; Tizhoosh, H.R. Generalization of Vision Pre-Trained Models for Histopathology. Sci. Rep. 2023, 13, 6065. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, S.; Zhang, J.; Wang, M.; Zhang, J.; Yang, W.; Huang, J.; Han, X. Transformer-Based Unsupervised Contrastive Learning for Histopathological Image Classification. Med. Image Anal. 2022, 81, 102559. [Google Scholar] [CrossRef] [PubMed]
- Singh, Y.; Farrelly, C.M.; Hathaway, Q.A.; Leiner, T.; Jagtap, J.; Carlsson, G.E.; Erickson, B.J. Topological Data Analysis in Medical Imaging: Current State of the Art. Insights Imaging 2023, 14, 58. [Google Scholar] [CrossRef] [PubMed]
- Chazal, F.; Michel, B. An Introduction to Topological Data Analysis: Fundamental and Practical Aspects for Data Scientists. Front. Artif. Intell. 2021, 4, 667963. [Google Scholar] [CrossRef]
- Vandaele, R.; Nervo, G.A.; Gevaert, O. Topological Image Modification for Object Detection and Topological Image Processing of Skin Lesions. Sci. Rep. 2020, 10, 21061. [Google Scholar] [CrossRef]
- Sprang, M.; Andrade-Navarro, M.A.; Fontaine, J.-F. Batch Effect Detection and Correction in RNA-Seq Data Using Machine-Learning-Based Automated Assessment of Quality. BMC Bioinform. 2022, 23, 279. [Google Scholar] [CrossRef]
- Altman, N. Batches and Blocks, Sample Pools and Subsamples in the Design and Analysis of Gene Expression Studies. In Batch Effects and Noise in Microarray Experiments; Wiley-Blackwell: Hoboken, NJ, USA, 2009; pp. 33–50. ISBN 978-0-470-74138-2. [Google Scholar]
- Liu, N.; Bhuva, D.D.; Mohamed, A.; Bokelund, M.; Kulasinghe, A.; Tan, C.W.; Davis, M.J. StandR: Spatial Transcriptomic Analysis for GeoMx DSP Data. Nucleic Acids Res. 2024, 52, e2. [Google Scholar] [CrossRef]
- Yu, X.; Xie, L.; Ge, J.; Li, H.; Zhong, S.; Liu, X. Integrating Single-Cell RNA-Seq and Spatial Transcriptomics Reveals MDK-NCL Dependent Immunosuppressive Environment in Endometrial Carcinoma. Front. Immunol. 2023, 14, 1145300. [Google Scholar] [CrossRef]
- Peng, Z.; Ye, M.; Ding, H.; Feng, Z.; Hu, K. Spatial Transcriptomics Atlas Reveals the Crosstalk between Cancer-Associated Fibroblasts and Tumor Microenvironment Components in Colorectal Cancer. J. Transl. Med. 2022, 20, 302. [Google Scholar] [CrossRef]
- Nyamundanda, G.; Poudel, P.; Patil, Y.; Sadanandam, A. A Novel Statistical Method to Diagnose, Quantify and Correct Batch Effects in Genomic Studies. Sci. Rep. 2017, 7, 10849. [Google Scholar] [CrossRef]
- Shinn, M. Phantom Oscillations in Principal Component Analysis. Proc. Natl. Acad. Sci. USA 2023, 120, e2311420120. [Google Scholar] [CrossRef] [PubMed]
- Elhaik, E. Principal Component Analyses (PCA)-Based Findings in Population Genetic Studies Are Highly Biased and Must Be Reevaluated. Sci. Rep. 2022, 12, 14683. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Papkovich, A.; Anderson-Trocmé, L.; Gravel, S. A Review of UMAP in Population Genetics. J. Hum. Genet. 2021, 66, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Kobak, D.; Berens, P. The Art of Using T-SNE for Single-Cell Transcriptomics. Nat. Commun. 2019, 10, 5416. [Google Scholar] [CrossRef]
- Sun, S.; Zhu, J.; Ma, Y.; Zhou, X. Accuracy, Robustness and Scalability of Dimensionality Reduction Methods for Single-Cell RNA-Seq Analysis. Genome Biol. 2019, 20, 269. [Google Scholar] [CrossRef]
- Rahaman, M.M.; Millar, E.K.A.; Meijering, E. Breast Cancer Histopathology Image-Based Gene Expression Prediction Using Spatial Transcriptomics Data and Deep Learning. Sci. Rep. 2023, 13, 13604. [Google Scholar] [CrossRef]
- Van Herck, Y.; Antoranz, A.; Andhari, M.D.; Milli, G.; Bechter, O.; De Smet, F.; Bosisio, F.M. Multiplexed Immunohistochemistry and Digital Pathology as the Foundation for Next-Generation Pathology in Melanoma: Methodological Comparison and Future Clinical Applications. Front. Oncol. 2021, 11, 636681. [Google Scholar] [CrossRef]
- Gao, Z.-J.; Fang, Z.; Yuan, J.-P.; Sun, S.-R.; Li, B. Integrative Multi-Omics Analyses Unravel the Immunological Implication and Prognostic Significance of CXCL12 in Breast Cancer. Front. Immunol. 2023, 14, 1188351. [Google Scholar] [CrossRef]
- Sun, H.; Li, Y.; Zhang, Y.; Zhao, X.; Dong, X.; Guo, Y.; Mo, J.; Che, N.; Ban, X.; Li, F.; et al. The Relevance between Hypoxia-Dependent Spatial Transcriptomics and the Prognosis and Efficacy of Immunotherapy in Claudin-Low Breast Cancer. Front. Immunol. 2022, 13, 1042835. [Google Scholar] [CrossRef]
- Tomiyama, T.; Itoh, S.; Iseda, N.; Toshida, K.; Kosai-Fujimoto, Y.; Tomino, T.; Kurihara, T.; Nagao, Y.; Morita, K.; Harada, N.; et al. Clinical Significance of Signal Regulatory Protein Alpha (SIRPα) Expression in Hepatocellular Carcinoma. Ann. Surg. Oncol. 2023, 30, 3378–3389. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Cheng, R.; Zhang, D.; Guo, Y.; Li, F.; Li, Y.; Li, Y.; Bai, X.; Mo, J.; Huang, C. MIF Promotes Cell Invasion by the LRP1-UPAR Interaction in Pancreatic Cancer Cells. Front. Oncol. 2022, 12, 1028070. [Google Scholar] [CrossRef] [PubMed]
- Agostini, A.; Guerriero, I.; Piro, G.; Quero, G.; Roberto, L.; Esposito, A.; Caggiano, A.; Priori, L.; Scaglione, G.; De Sanctis, F.; et al. Talniflumate Abrogates Mucin Immune Suppressive Barrier Improving Efficacy of Gemcitabine and Nab-Paclitaxel Treatment in Pancreatic Cancer. J. Transl. Med. 2023, 21, 843. [Google Scholar] [CrossRef]
- Peng, Z.; Ren, Z.; Tong, Z.; Zhu, Y.; Zhu, Y.; Hu, K. Interactions between MFAP5 + Fibroblasts and Tumor-Infiltrating Myeloid Cells Shape the Malignant Microenvironment of Colorectal Cancer. J. Transl. Med. 2023, 21, 405. [Google Scholar] [CrossRef]
- Larroquette, M.; Guegan, J.-P.; Besse, B.; Cousin, S.; Brunet, M.; Le Moulec, S.; Le Loarer, F.; Rey, C.; Soria, J.-C.; Barlesi, F.; et al. Spatial Transcriptomics of Macrophage Infiltration in Non-Small Cell Lung Cancer Reveals Determinants of Sensitivity and Resistance to Anti-PD1/PD-L1 Antibodies. J. Immunother. Cancer 2022, 10, e003890. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, Y.; Ouyang, J.; Yi, H.; Wang, S.; Liu, D.; Dai, Y.; Song, K.; Pei, W.; Hong, Z.; et al. TRPV1 Inhibition Suppresses Non-Small Cell Lung Cancer Progression by Inhibiting Tumour Growth and Enhancing the Immune Response. Cell. Oncol. 2023, 47, 779–791. [Google Scholar] [CrossRef]
- Alsaleh, L.; Li, C.; Couetil, J.L.; Ye, Z.; Huang, K.; Zhang, J.; Chen, C.; Johnson, T.S. Spatial Transcriptomic Analysis Reveals Associations between Genes and Cellular Topology in Breast and Prostate Cancers. Cancers 2022, 14, 4856. [Google Scholar] [CrossRef]
- Gong, S.; Sun, N.; Meyer, L.S.; Tetti, M.; Koupourtidou, C.; Krebs, S.; Masserdotti, G.; Blum, H.; Rainey, W.E.; Reincke, M.; et al. Primary Aldosteronism: Spatial Multiomics Mapping of Genotype-Dependent Heterogeneity and Tumor Expansion of Aldosterone-Producing Adenomas. Hypertension 2023, 80, 1555–1567. [Google Scholar] [CrossRef]
- Lindskrog, S.V.; Schmøkel, S.S.; Nordentoft, I.; Lamy, P.; Knudsen, M.; Prip, F.; Strandgaard, T.; Jensen, J.B.; Dyrskjøt, L. Single-Nucleus and Spatially Resolved Intratumor Subtype Heterogeneity in Bladder Cancer. Eur. Urol. Open Sci. 2023, 51, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Curry, J.; Alnemri, A.; Philips, R.; Fiorella, M.; Sussman, S.; Stapp, R.; Solomides, C.; Harshyne, L.; South, A.; Luginbuhl, A.; et al. CD8+ and FoxP3+ T-Cell Cellular Density and Spatial Distribution After Programmed Death-Ligand 1 Check Point Inhibition. Laryngoscope 2023, 133, 1875–1884. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhang, D.; Huang, C.; Guo, Y.; Yang, Z.; Yao, N.; Dong, X.; Cheng, R.; Zhao, N.; Meng, J.; et al. Hypoxic Microenvironment Induced Spatial Transcriptome Changes in Pancreatic Cancer. Cancer Biol. Med. 2021, 18, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Eckhoff, A.M.; Fletcher, A.A.; Landa, K.; Iyer, M.; Nussbaum, D.P.; Shi, C.; Nair, S.K.; Allen, P.J. Multidimensional Immunophenotyping of Intraductal Papillary Mucinous Neoplasms Reveals Novel T Cell and Macrophage Signature. Ann. Surg. Oncol. 2022, 29, 7781–7788. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Software | Language | Examples of Applications |
|---|---|---|
| BayesSpace [48] | R | Utilized by Liu et al. to better exhibit spatial expression of features. The spots were enhanced using the spatialEnhance function, and the expression features were enhanced with the enhanceFeatures function [49]. |
| Cell2location [50] | Python | Utilized by Du et al. to run the deconvolution of spatial tissue locations. To identify the spatial co-occurrence patterns of different cell types, they performed non-negative matrix factorization (NMF) of cell type abundance estimates [51]. |
| CellChat [52] | R | In general, this tool is used to map cellular interactions. It was applied by Liu et al. to evaluate the interaction weights of identified spatial clusters [49]. Guo et al. performed cell communication analysis. To create cell communication networks, they applied CellChat’s functions such as createCellChat function, computeCommunProb, computeCommunProbPathway, and aggregateNet [53]. Liu et al. inferred the cell–cell interactome by assessing the gene expression of ligand–receptor pairs across cell types in bulk-seq and ST [54]. |
| CellTrek [55] | R | Employed by Liu et al. to investigate spatial transcriptomics at a single-cell resolution. CellTrek directly mapped cells back to their spatial location by co-embedding the same-tissue single-cell transcriptomics and ST datasets. This approach allowed them to identify malignant cell clusters directly in ST data [54]. |
| clusterProfiler [56] | R | Utilized by Al-Holou et al. to perform gene ontology enrichment analysis, aiming to identify enriched processes amongst the previously identified differentially up- and down-regulated genes [57]. A similar approach was taken by Ren et al. [58]. |
| Domino [59] | R | Used by Zhang et al. to analyze the signaling networks based on the gene regulatory module activities, quantified for each spot by the AUCell/SCENIC tool [60]. |
| GeneSwitches [61] | R | Applied by Liu et al. to identify the order in which functional events are acquired or lost during the transition of malignant cells by processing single-cell data together with pseudotime trajectories to order pathways along the pseudotime. They filtered genes for pathway analysis using the filter switchgenes functionality and used the find switch pathways module to find significantly changed pathways with pseudotime trajectories [54]. |
| Giotto [62] | R, Python | Vo et al. applied the PAGE function, which calculates an enrichment score based on the fold change of cell type marker genes for each spot [62,63]. Cell–cell spatial communication and interactions are available through cellProximityEnrichment and spatCellCellcom functions and were utilized by Shi et al. [64]. |
| Harmony [65] | R | Employed by, among many, Fu et al. and Yousuf et al. to, e.g., correct batch effects among samples through dataset merging for samples-integrated analysis [66,67]. |
| Monocle2 [68] | R | Liu et al. utilized this tool to simulate the dynamics of temporal development for cell trajectory analysis (aka pseudotime trajectory analysis) by using the spot-resolved expression patterns of previously identified key genes [69]. |
| NICHES [70] | R | NICHES (Niche Interactions and Communication Heterogeneity in Extracellular Signaling) library was applied by Tashireva et al. to assess ligand–receptor interaction between neighboring tissue spots [71]. |
| NicheNet [72] | R | Used by Zhi et al. to access cell–cell interactions between specific cell populations [64]. |
| ReactomeGSA [73] | R, web | Used to score the Hallmark gene set in spatial spots after immune cells scoring through the CIBERSORT algorithm for each spot in the Guo et al. study [53]. |
| SCENIC [74] | R, Python | Employed by Yousuf et al. to perform inference of regulatory modules analysis between transcription factors and downstream regulated genes [67]. |
| scMetabolism [75] | R | Utilized by Yang et al. to evaluate the metabolic activity of each spot [76]. |
| spacerx [37] | R | Used to conduct spatial deconvolution in the Yang et al. study [76]. |
| SPATA2 [77] | R | Used by Andrieux et al. to infer copy number variation (CNV), using the InferCNV function. As a result, gains or loss of chromosomes were identified [78]. |
| SPOTLight [79] | R | Utilized by Yang et al. and Liu et al. to identify cell types at spatial spots of interest after their deconvolution [76]. To infer the cell composition of each spot, SPOTLight combined single-cell data and cell-type marker gene information with ST spots and performed a deconvolution based on a NMF decomposition [69]. |
| SingleR [80] | R | Utilized by Vo et al. to map a dominant reference cell type to each spot based on Spearman’s correlation coefficient and nearest label classification [63]. |
| Slingshot [81] | R | Used to infer pseudotime trajectories in the Vo et al. study [63]. |
| SpotClean [82] | R | Applied by Yousuf et al. to remove ambient RNA present in their data [67]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maciejewski, K.; Czerwinska, P. Scoping Review: Methods and Applications of Spatial Transcriptomics in Tumor Research. Cancers 2024, 16, 3100. https://doi.org/10.3390/cancers16173100
Maciejewski K, Czerwinska P. Scoping Review: Methods and Applications of Spatial Transcriptomics in Tumor Research. Cancers. 2024; 16(17):3100. https://doi.org/10.3390/cancers16173100
Chicago/Turabian StyleMaciejewski, Kacper, and Patrycja Czerwinska. 2024. "Scoping Review: Methods and Applications of Spatial Transcriptomics in Tumor Research" Cancers 16, no. 17: 3100. https://doi.org/10.3390/cancers16173100
APA StyleMaciejewski, K., & Czerwinska, P. (2024). Scoping Review: Methods and Applications of Spatial Transcriptomics in Tumor Research. Cancers, 16(17), 3100. https://doi.org/10.3390/cancers16173100