
Towards Understanding Tumour Colonisation by Probiotic Bacterium E. coli Nissle 1917

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Fit for Purpose: Inherent Properties of Probiotic E. coli Nissle 1917
3. Anti-Cancer Activity of Genetically Engineered EcN in Pre-Clinical Tumour Models
4. First in Human Testing of Engineered EcN in Cancer Patients: Points for Consideration
5. How Does EcN Colonise Tumour Tissue?
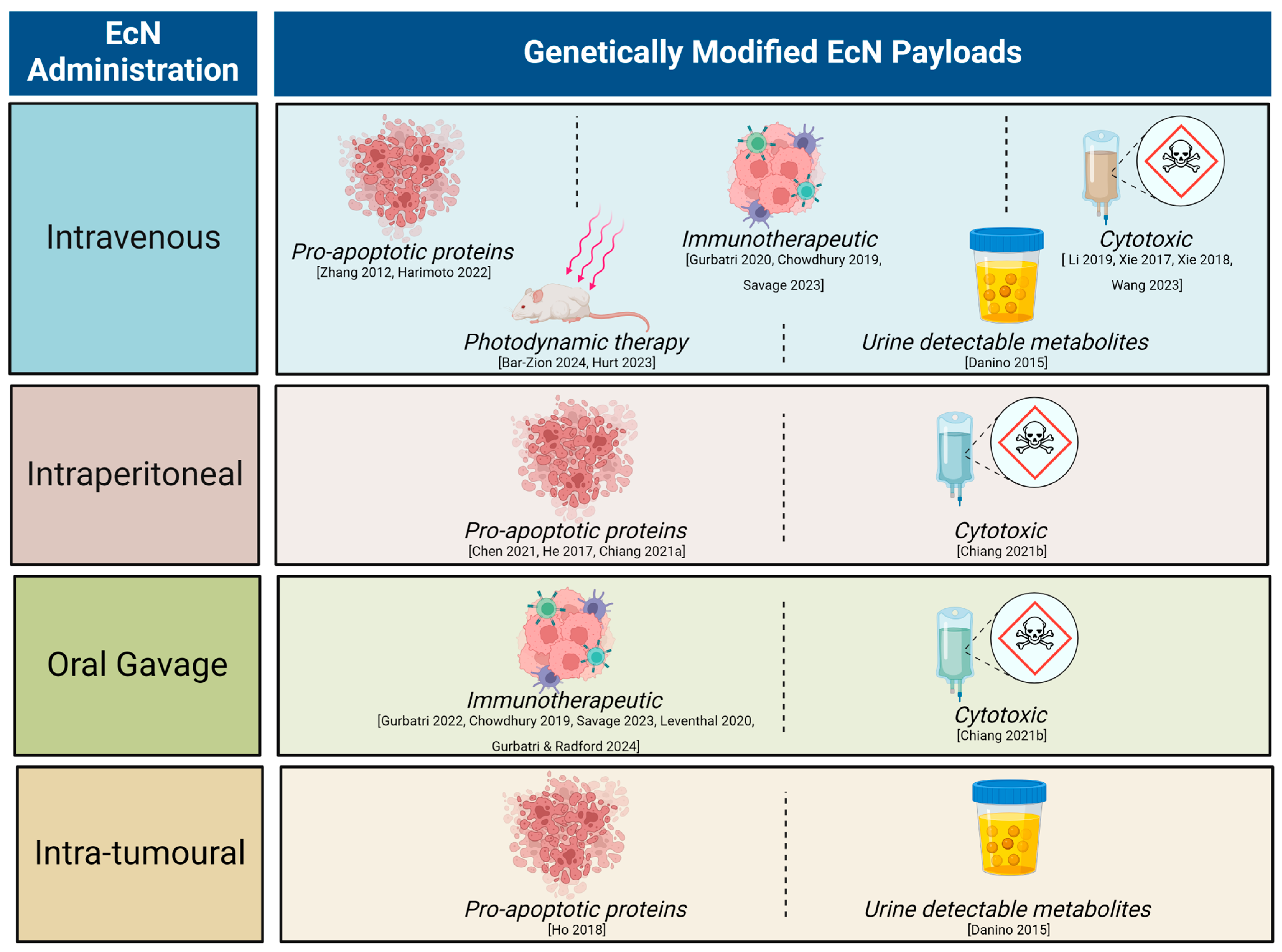
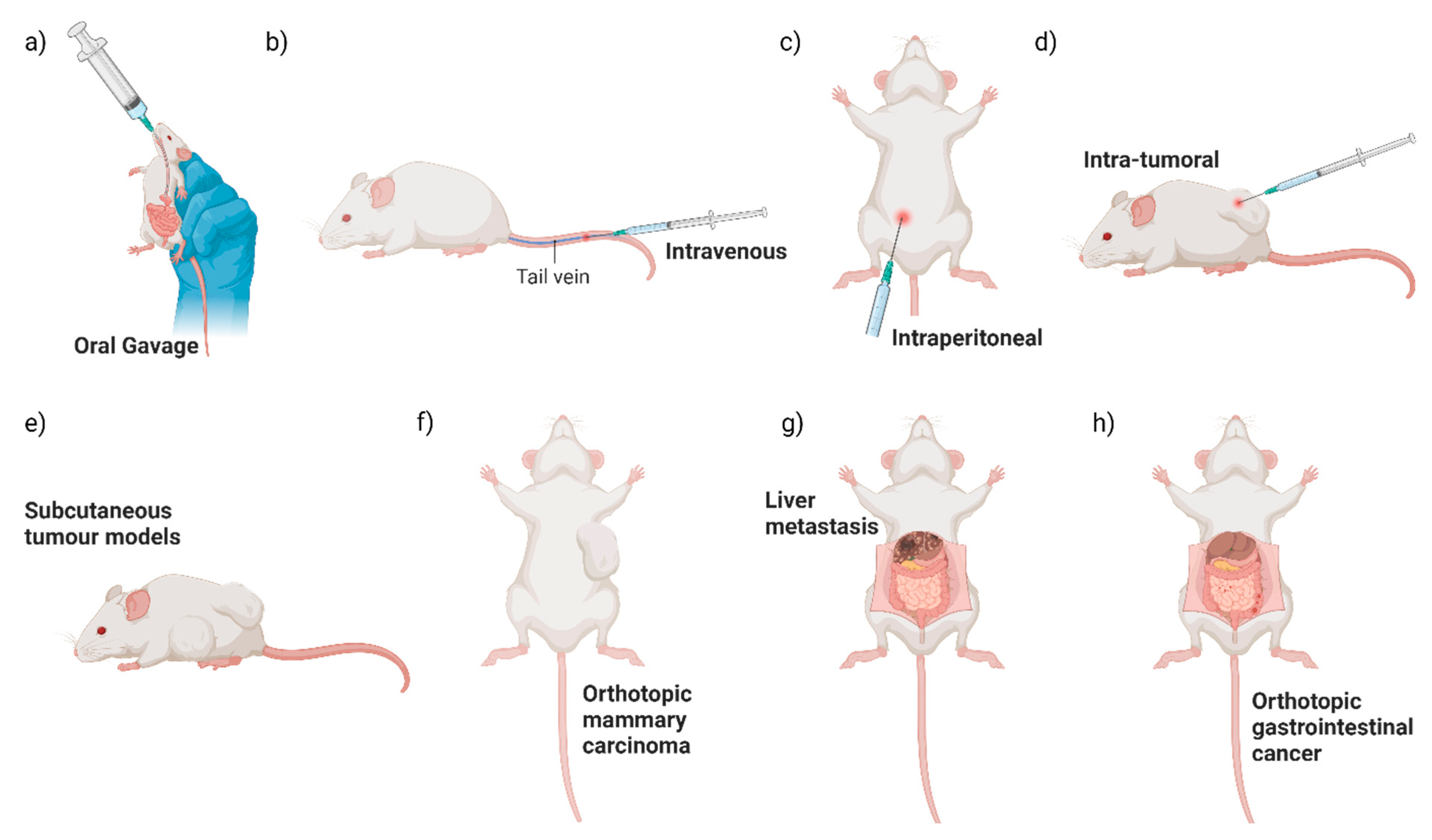
5.1. Influence of Route of Bacterial Administration on Tumour Colonisation: Evidence from Pre-Clinical Studies
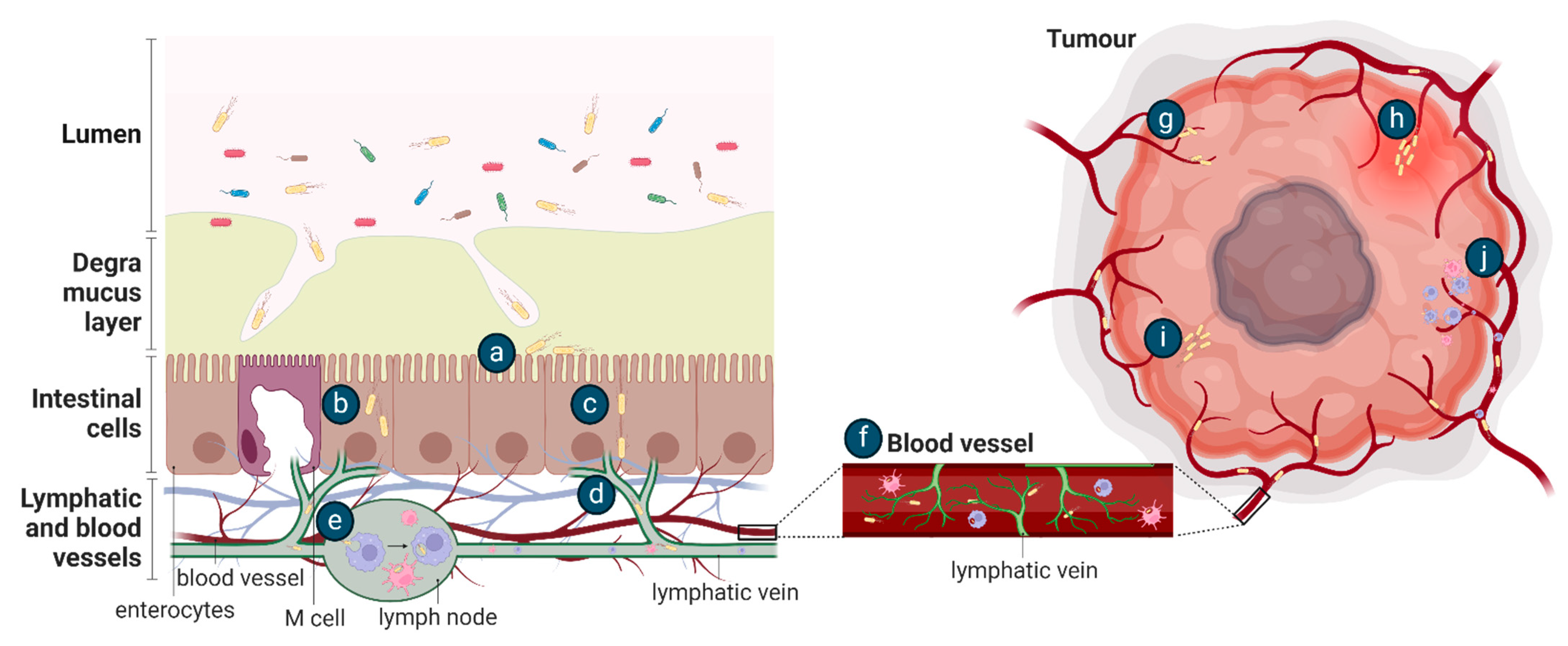
5.2. Learning from Host Infection by Enteric Pathogens
6. Inherent Tumour Properties Underlying Colonisation by Bacteria
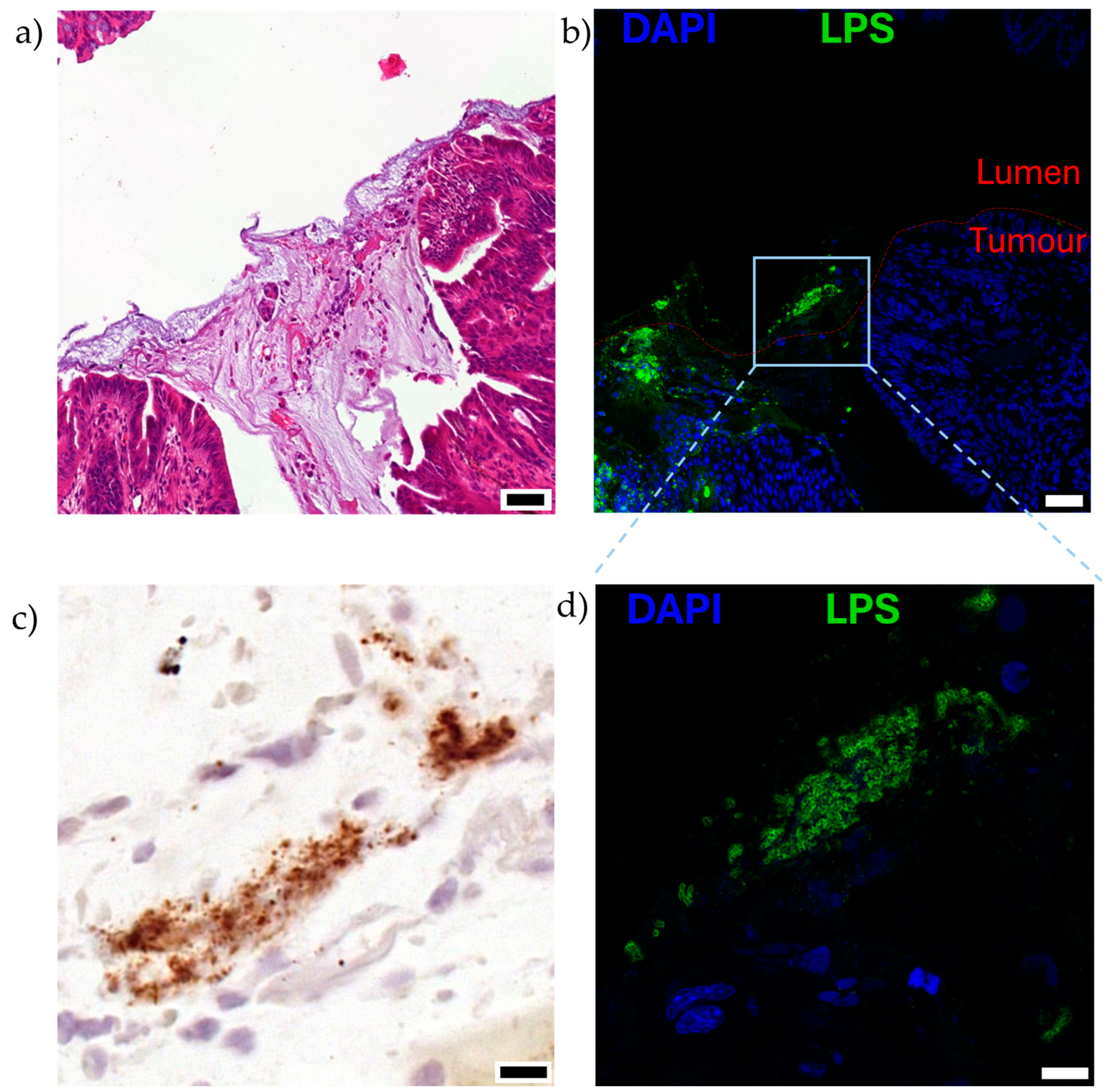
6.1. Aberrant Tumour Vasculature and Inflammation Permit Bacterial Entry
6.2. Variable Oxygen Supply in Tumour Tissue
6.3. Tumour Necrosis—The Ultimate Metabolic Niche for Bacteria?
6.4. Aberrant Immune Surveillance in the Tumour Microenvironment
7. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Coley, W.B. The classic: The treatment of malignant tumors by repeated inoculations of erysipelas: With a report of ten original cases. Clin. Orthop. Relat. Res. 1991, 262, 3–11. [Google Scholar] [CrossRef]
- Coley, W.B. Contribution to the knowledge of sarcoma. Ann. Surg. 1891, 14, 199–220. [Google Scholar] [CrossRef]
- Nauts, H.C.; Swift, W.E.; Coley, B.L. The treatment of malignant tumors by bacterial toxins as developed by the late william b. Coley, m.D., reviewed in the light of modern research. Cancer Res. 1946, 6, 205–216. [Google Scholar]
- Kamat, A.M.; Flaig, T.W.; Grossman, H.B.; Konety, B.; Lamm, D.; O’Donnell, M.A.; Uchio, E.; Efstathiou, J.A.; Taylor, J.A. Consensus statement on best practice management regarding the use of intravesical immunotherapy with bcg for bladder cancer. Nat. Rev. Urol. 2015, 12, 225–235. [Google Scholar] [CrossRef]
- Silverstein, M.J. Malignant melanoma metastatic to the bladder. JAMA 1974, 229, 688. [Google Scholar] [CrossRef]
- Brader, P.; Stritzker, J.; Riedl, C.C.; Zanzonico, P.; Cai, S.; Burnazi, E.M.; Ghani, E.R.; Hricak, H.; Szalay, A.A.; Fong, Y.; et al. Escherichia coli nissle 1917 facilitates tumor detection by positron emission tomography and optical imaging. Clin. Cancer Res. 2008, 14, 2295–2302. [Google Scholar] [CrossRef]
- Chen, J.; Li, X.; Liu, Y.; Su, T.; Lin, C.; Shao, L.; Li, L.; Li, W.; Niu, G.; Yu, J.; et al. Engineering a probiotic strain of escherichia coli to induce the regression of colorectal cancer through production of 5-aminolevulinic acid. Microb. Biotechnol. 2021, 14, 2130–2139. [Google Scholar] [CrossRef]
- Gurbatri, C.R.; Radford, G.A.; Vrbanac, L.; Im, J.; Thomas, E.M.; Coker, C.; Taylor, S.R.; Jang, Y.; Sivan, A.; Rhee, K.; et al. Engineering tumor-colonizing e. Coli nissle 1917 for detection and treatment of colorectal neoplasia. Nat. Commun. 2024, 15, 646. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Helbig, L.; Fu, J.; Bian, X.; Herrmann, J.; Baumann, M.; Stewart, A.F.; Muller, R.; Li, A.; Zips, D.; et al. Expressing cytotoxic compounds in escherichia coli nissle 1917 for tumor-targeting therapy. Res. Microbiol. 2019, 170, 74–79. [Google Scholar] [CrossRef]
- Stritzker, J.; Weibel, S.; Hill, P.J.; Oelschlaeger, T.A.; Goebel, W.; Szalay, A.A. Tumor-specific colonization, tissue distribution, and gene induction by probiotic escherichia coli nissle 1917 in live mice. Int. J. Med. Microbiol. 2007, 297, 151–162. [Google Scholar] [CrossRef]
- Xie, S.; Zhao, L.; Song, X.; Tang, M.; Mo, C.; Li, X. Doxorubicin-conjugated escherichia coli nissle 1917 swimmers to achieve tumor targeting and responsive drug release. J. Control. Release 2017, 268, 390–399. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.; Xia, L.; Zhang, X.; Ding, X.; Yan, F.; Wu, F. Escherichia coli nissle 1917 targets and restrains mouse b16 melanoma and 4t1 breast tumors through expression of azurin protein. Appl. Environ. Microbiol. 2012, 78, 7603–7610. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.; Van Zijl, P.; Fox, M.; Mauchline, M.; Giaccia, A.; Minton, N.; Brown, J. Anaerobic bacteria as a gene delivery system that is controlled by the tumor microenvironment. Gene Ther. 1997, 4, 791–796. [Google Scholar] [CrossRef]
- Kim, J.-E.; Phan, T.X.; Nguyen, V.H.; Dinh-Vu, H.-V.; Zheng, J.H.; Yun, M.; Park, S.-G.; Hong, Y.; Choy, H.E.; Szardenings, M.; et al. Salmonella typhimurium suppresses tumor growth via the pro-inflammatory cytokine interleukin-1β. Theranostics 2015, 5, 1328–1342. [Google Scholar] [CrossRef] [PubMed]
- Leschner, S.; Westphal, K.; Dietrich, N.; Viegas, N.; Jablonska, J.; Lyszkiewicz, M.; Lienenklaus, S.; Falk, W.; Gekara, N.; Loessner, H.; et al. Tumor invasion of salmonella enterica serovar typhimurium is accompanied by strong hemorrhage promoted by tnf-alpha. PLoS ONE 2009, 4, e6692. [Google Scholar] [CrossRef]
- Zhang, M.; Forbes, N.S. Trg-deficient salmonella colonize quiescent tumor regions by exclusively penetrating or proliferating. J. Control. Release 2015, 199, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Yang, M.; Li, X.; Jiang, P.; Baranov, E.; Li, S.; Xu, M.; Penman, S.; Hoffman, R.M. Tumor-targeting bacterial therapy with amino acid auxotrophs of gfp-expressing salmonella typhimurium. Proc. Natl. Acad. Sci. USA 2005, 102, 755–760. [Google Scholar] [CrossRef]
- Ganai, S.; Arenas, R.B.; Sauer, J.P.; Bentley, B.; Forbes, N.S. In tumors salmonella migrate away from vasculature toward the transition zone and induce apoptosis. Cancer Gene Ther. 2011, 18, 457–466. [Google Scholar] [CrossRef]
- Cronin, M.; Morrissey, D.; Rajendran, S.; El Mashad, S.M.; van Sinderen, D.; O’Sullivan, G.C.; Tangney, M. Orally administered bifidobacteria as vehicles for delivery of agents to systemic tumors. Mol. Ther. 2010, 18, 1397–1407. [Google Scholar] [CrossRef]
- Sender, R.; Fuchs, S.; Milo, R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef]
- Sonnenborn, U.; Schulze, J. The non-pathogenicescherichia colistrain nissle 1917—Features of a versatile probiotic. Microb. Ecol. Health Dis. 2009, 21, 122–158. [Google Scholar]
- Russell, B.J.; Brown, S.D.; Siguenza, N.; Mai, I.; Saran, A.R.; Lingaraju, A.; Maissy, E.S.; Machado, A.C.D.; Pinto, A.F.; Sanchez, C. Intestinal transgene delivery with native e. Coli chassis allows persistent physiological changes. Cell 2022, 185, 3263–3277.e3215. [Google Scholar] [CrossRef]
- Joeres-Nguyen-Xuan, T.H.; Boehm, S.K.; Joeres, L.; Schulze, J.; Kruis, W. Survival of the probiotic escherichia coli nissle 1917 (ecn) in the gastrointestinal tract given in combination with oral mesalamine to healthy volunteers. Inflamm. Bowel Dis. 2010, 16, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Schultz, M.; Watzl, S.; Oelschlaeger, T.A.; Rath, H.C.; Göttl, C.; Lehn, N.; Schölmerich, J.; Linde, H.-J. Green fluorescent protein for detection of the probiotic microorganism escherichia coli strain nissle 1917 (ecn) in vivo. J. Microbiol. Methods 2005, 61, 389–398. [Google Scholar] [CrossRef]
- Deriu, E.; Liu, J.Z.; Pezeshki, M.; Edwards, R.A.; Ochoa, R.J.; Contreras, H.; Libby, S.J.; Fang, F.C.; Raffatellu, M. Probiotic bacteria reduce salmonella typhimurium intestinal colonization by competing for iron. Cell Host Microbe 2013, 14, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Sassone-Corsi, M.; Nuccio, S.-P.; Liu, H.; Hernandez, D.; Vu, C.T.; Takahashi, A.A.; Edwards, R.A.; Raffatellu, M. Microcins mediate competition among enterobacteriaceae in the inflamed gut. Nature 2016, 540, 280–283. [Google Scholar] [CrossRef]
- Alvarez, C.-S.; Giménez, R.; Cañas, M.-A.; Vera, R.; Díaz-Garrido, N.; Badia, J.; Baldomà, L. Extracellular vesicles and soluble factors secreted by escherichia coli nissle 1917 and ecor63 protect against enteropathogenic e. Coli-induced intestinal epithelial barrier dysfunction. BMC Microbiol. 2019, 19, 166. [Google Scholar] [CrossRef]
- Zhang, Y.; Ji, W.; He, L.; Chen, Y.; Ding, X.; Sun, Y.; Hu, S.; Yang, H.; Huang, W.; Zhang, Y.; et al. E. Coli Nissle 1917-derived minicells for targeted delivery of chemotherapeutic drug to hypoxic regions for cancer therapy. Theranostics 2018, 8, 1690–1705. [Google Scholar] [CrossRef]
- Stritzker, J.; Weibel, S.; Seubert, C.; Götz, A.; Tresch, A.; Van Rooijen, N.; Oelschlaeger, T.A.; Hill, P.J.; Gentschev, I.; Szalay, A.A. Enterobacterial tumor colonization in mice depends on bacterial metabolism and macrophages but is independent of chemotaxis and motility. Int. J. Med. Microbiol. 2010, 300, 449–456. [Google Scholar] [CrossRef]
- Leventhal, D.S.; Sokolovska, A.; Li, N.; Plescia, C.; Kolodziej, S.A.; Gallant, C.W.; Christmas, R.; Gao, J.-R.; James, M.J.; Abin-Fuentes, A.; et al. Immunotherapy with engineered bacteria by targeting the sting pathway for anti-tumor immunity. Nat. Commun. 2020, 11, 2739. [Google Scholar] [CrossRef]
- He, L.; Yang, H.; Liu, F.; Chen, Y.; Tang, S.; Ji, W.; Tang, J.; Liu, Z.; Sun, Y.; Hu, S.; et al. Escherichia coli nissle 1917 engineered to express tum-5 can restrain murine melanoma growth. Oncotarget 2017, 8, 85772–85782. [Google Scholar] [CrossRef]
- He, L.; Yang, H.; Tang, J.; Liu, Z.; Chen, Y.; Lu, B.; He, H.; Tang, S.; Sun, Y.; Liu, F.; et al. Intestinal probiotics e. Coli nissle 1917 as a targeted vehicle for delivery of p53 and tum-5 to solid tumors for cancer therapy. J. Biol. Eng. 2019, 13, 58. [Google Scholar] [CrossRef] [PubMed]
- Harimoto, T.; Hahn, J.; Chen, Y.Y.; Im, J.; Zhang, J.; Hou, N.; Li, F.; Coker, C.; Gray, K.; Harr, N.; et al. A programmable encapsulation system improves delivery of therapeutic bacteria in mice. Nat. Biotechnol. 2022, 40, 1259–1269. [Google Scholar] [CrossRef]
- Gurbatri, C.R.; Lia, I.; Vincent, R.; Coker, C.; Castro, S.; Treuting, P.M.; Hinchliffe, T.E.; Arpaia, N.; Danino, T. Engineered probiotics for local tumor delivery of checkpoint blockade nanobodies. Sci. Transl. Med. 2020, 12, 1–12. [Google Scholar] [CrossRef]
- Danino, T.; Prindle, A.; Kwong, G.A.; Skalak, M.; Li, H.; Allen, K.; Hasty, J.; Bhatia, S.N. Programmable probiotics for detection of cancer in urine. Sci. Transl. Med. 2015, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.-J.; Huang, P.-H. Metabolic engineering of probiotic escherichia coli for cytolytic therapy of tumors. Sci. Rep. 2021, 11, 5853. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.-J.; Hong, Y.-H. In situ delivery of biobutyrate by probiotic escherichia coli for cancer therapy. Sci. Rep. 2021, 11, 18172. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Cheng, S.; Wang, X.; Pang, Y.; Liu, J. Camouflaging bacteria by wrapping with cell membranes. Nat. Commun. 2019, 10, 3452. [Google Scholar] [CrossRef] [PubMed]
- Canale, F.P.; Basso, C.; Antonini, G.; Perotti, M.; Li, N.; Sokolovska, A.; Neumann, J.; James, M.J.; Geiger, S.; Jin, W.; et al. Metabolic modulation of tumours with engineered bacteria for immunotherapy. Nature 2021, 598, 662–666. [Google Scholar] [CrossRef]
- Chowdhury, S.; Castro, S.; Coker, C.; Hinchliffe, T.E.; Arpaia, N.; Danino, T. Programmable bacteria induce durable tumor regression and systemic antitumor immunity. Nat. Med. 2019, 25, 1057–1063. [Google Scholar] [CrossRef]
- Yu, Y.A.; Shabahang, S.; Timiryasova, T.M.; Zhang, Q.; Beltz, R.; Gentschev, I.; Goebel, W.; Szalay, A.A. Visualization of tumors and metastases in live animals with bacteria and vaccinia virus encoding light-emitting proteins. Nat. Biotechnol. 2004, 22, 313–320. [Google Scholar] [CrossRef]
- Ho, C.L.; Tan, H.Q.; Chua, K.J.; Kang, A.; Lim, K.H.; Ling, K.L.; Yew, W.S.; Lee, Y.S.; Thiery, J.P.; Chang, M.W. Engineered commensal microbes for diet-mediated colorectal-cancer chemoprevention. Nat. Biomed. Eng. 2018, 2, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Grozdanov, L.; ZäHringer, U.; Blum-Oehler, G.; Brade, L.; Henne, A.; Knirel, Y.A.; Schombel, U.; Schulze, J.R.; Sonnenborn, U.; Gottschalk, G.; et al. A single nucleotide exchange in the wzy gene is responsible for the semirough o6 lipopolysaccharide phenotype and serum sensitivity of escherichia coli strain nissle 1917. J. Bacteriol. 2002, 184, 5912–5925. [Google Scholar] [CrossRef]
- Lasaro, M.A.; Salinger, N.; Zhang, J.; Wang, Y.; Zhong, Z.; Goulian, M.; Zhu, J. F1c fimbriae play an important role in biofilm formation and intestinal colonization by the escherichia coli commensal strain nissle 1917. Appl. Environ. Microbiol. 2009, 75, 246–251. [Google Scholar] [CrossRef]
- Altenhoefer, A.; Oswald, S.; Sonnenborn, U.; Enders, C.; Schulze, J.; Hacker, J.; Oelschlaeger, T.A. The probiotic escherichia coli strain nissle 1917 interferes with invasion of human intestinal epithelial cells by different enteroinvasive bacterial pathogens. FEMS Immunol. Med. Microbiol. 2004, 40, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Boudeau, J.; Glasser, A.L.; Julien, S.; Colombel, J.F.; Darfeuille-Michaud, A. Inhibitory effect of probiotic escherichia coli strain nissle 1917 on adhesion to and invasion of intestinal epithelial cells by adherent-invasive e. Coli strains isolated from patients with crohn’s disease. Aliment. Pharmacol. Ther. 2003, 18, 45–56. [Google Scholar] [CrossRef]
- Sonnenborn, U. Escherichia colistrain nissle 1917—From bench to bedside and back: History of a specialescherichia colistrain with probiotic properties. FEMS Microbiol. Lett. 2016, 363, fnw212. [Google Scholar] [CrossRef]
- Gurbatri, C.R.; Arpaia, N.; Danino, T. Engineering bacteria as interactive cancer therapies. Science 2022, 378, 858–864. [Google Scholar] [CrossRef]
- Raman, V.; Deshpande, C.P.; Khanduja, S.; Howell, L.M.; Van Dessel, N.; Forbes, N.S. Build-a-bug workshop: Using microbial-host interactions and synthetic biology tools to create cancer therapies. Cell Host Microbe. 2023, 31, 1574–1592. [Google Scholar] [CrossRef]
- Yu, X.; Lin, C.; Yu, J.; Qi, Q.; Wang, Q. Bioengineered escherichia coli nissle 1917 for tumour-targeting therapy. Microb. Biotechnol. 2020, 13, 629–636. [Google Scholar] [CrossRef]
- Xie, S.; Chen, M.; Song, X.; Zhang, Z.; Zhang, Z.; Chen, Z.; Li, X. Bacterial microbots for acid-labile release of hybrid micelles to promote the synergistic antitumor efficacy. Acta Biomater. 2018, 78, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Yin, Q.; Huang, H.y.; Wang, Z.; Song, H.; Luo, X. Probiotic escherichia coli nissle 1917 propelled micro-robot with ph sensitivity for hypoxia targeted intestinal tumor therapy. Colloids Surf. B Biointerfaces 2023, 225, 113277. [Google Scholar] [CrossRef] [PubMed]
- Savage, T.M.; Vincent, R.L.; Rae, S.S.; Huang, L.H.; Ahn, A.; Pu, K.; Li, F.; de los Santos-Alexis, K.; Coker, C.; Danino, T.; et al. Chemokines expressed by engineered bacteria recruit and orchestrate antitumor immunity. Sci. Adv. 2023, 9, eadc9436. [Google Scholar] [CrossRef]
- Bar-Zion, A.; Nourmahnad, A.; Mittelstein, D.R.; Shivaei, S.; Yoo, S.; Buss, M.T.; Hurt, R.C.; Malounda, D.; Abedi, M.H.; Lee-Gosselin, A.; et al. Acoustically triggered mechanotherapy using genetically encoded gas vesicles. Nat. Nanotechnol. 2021, 16, 1403–1412. [Google Scholar] [CrossRef]
- Hurt, R.C.; Buss, M.T.; Duan, M.; Wong, K.; You, M.Y.; Sawyer, D.P.; Swift, M.B.; Dutka, P.; Barturen-Larrea, P.; Mittelstein, D.R.; et al. Genomically mined acoustic reporter genes for real-time in vivo monitoring of tumors and tumor-homing bacteria. Nat. Biotechnol. 2023, 41, 919–931. [Google Scholar] [CrossRef]
- Luke, J.J.; Piha-Paul, S.A.; Medina, T.; Verschraegen, C.F.; Varterasian, M.; Brennan, A.M.; Riese, R.J.; Sokolovska, A.; Strauss, J.; Hava, D.L.; et al. Phase i study of synb1891, an engineered e. Coli nissle strain expressing sting agonist, with and without atezolizumab in advanced malignancies. Clin. Cancer Res. 2023, 29, 2435–2444. [Google Scholar] [CrossRef]
- Chien, T.; Harimoto, T.; Kepecs, B.; Gray, K.; Coker, C.; Hou, N.; Pu, K.; Azad, T.; Nolasco, A.; Pavlicova, M.; et al. Enhancing the tropism of bacteria via genetically programmed biosensors. Nat. Biomed. Eng. 2021, 6, 94–104. [Google Scholar] [CrossRef]
- Din, M.O.; Danino, T.; Prindle, A.; Skalak, M.; Selimkhanov, J.; Allen, K.; Julio, E.; Atolia, E.; Tsimring, L.S.; Bhatia, S.N.; et al. Synchronized cycles of bacterial lysis for in vivo delivery. Nature 2016, 536, 81–85. [Google Scholar] [CrossRef]
- Danino, T.; Mondragon-Palomino, O.; Tsimring, L.; Hasty, J. A synchronized quorum of genetic clocks. Nature 2010, 463, 326–330. [Google Scholar] [CrossRef]
- Rosendahl Huber, A.; Pleguezuelos-Manzano, C.; Puschhof, J.; Ubels, J.; Boot, C.; Saftien, A.; Verheul, M.; Trabut, L.T.; Groenen, N.; van Roosmalen, M.; et al. Improved detection of colibactin-induced mutations by genotoxic e. Coli in organoids and colorectal cancer. Cancer Cell 2024, 42, 487–496.e486. [Google Scholar] [CrossRef]
- Iftekhar, A.; Berger, H.; Bouznad, N.; Heuberger, J.; Boccellato, F.; Dobrindt, U.; Hermeking, H.; Sigal, M.; Meyer, T.F. Genomic aberrations after short-term exposure to colibactin-producing e. Coli transform primary colon epithelial cells. Nat. Commun. 2021, 12, 1003. [Google Scholar] [CrossRef] [PubMed]
- Nougayrède, J.P.; Chagneau, C.V.; Motta, J.P.; Bossuet-Greif, N.; Belloy, M.; Taieb, F.; Gratadoux, J.J.; Thomas, M.; Langella, P.; Oswald, E. A toxic friend: Genotoxic and mutagenic activity of the probiotic strain escherichia coli nissle 1917. mSphere 2021, 6, e0062421. [Google Scholar] [CrossRef] [PubMed]
- Gaab, M.E.; Lozano, P.O.; Ibañez, D.; Manese, K.D.; Riego, F.M.; Tiongco, R.E.; Albano, P.M. A meta-analysis on the association of colibactin-producing pks+ escherichia coli with the development of colorectal cancer. Lab. Med. 2023, 54, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Pleguezuelos-Manzano, C.; Puschhof, J.; Rosendahl Huber, A.; Van Hoeck, A.; Wood, H.M.; Nomburg, J.; Gurjao, C.; Manders, F.; Dalmasso, G.; Stege, P.B.; et al. Mutational signature in colorectal cancer caused by genotoxic pks+ e. Coli. Nature 2020, 580, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Kalantari, A.; James, M.J.; Renaud, L.A.; Perreault, M.; Monahan, C.E.; McDonald, M.N.; Hava, D.L.; Isabella, V.M. Robust performance of a live bacterial therapeutic chassis lacking the colibactin gene cluster. PLoS ONE 2023, 18, e0280499. [Google Scholar] [CrossRef] [PubMed]
- Massip, C.; Branchu, P.; Bossuet-Greif, N.; Chagneau, C.V.; Gaillard, D.; Martin, P.; Boury, M.; Sécher, T.; Dubois, D.; Nougayrède, J.-P.; et al. Deciphering the interplay between the genotoxic and probiotic activities of escherichia coli nissle 1917. PLoS Pathog. 2019, 15, e1008029. [Google Scholar] [CrossRef]
- Tang-Fichaux, M.; Chagneau, C.V.; Bossuet-Greif, N.; Nougayrède, J.-P.; Oswald, É.; Branchu, P.; D’Orazio, S.E.F. The polyphosphate kinase of Escherichia coli is required for full production of the genotoxin colibactin. mSphere 2020, 5, e01195-20. [Google Scholar] [CrossRef] [PubMed]
- Isabella, V.M.; Ha, B.N.; Castillo, M.J.; Lubkowicz, D.J.; Rowe, S.E.; Millet, Y.A.; Anderson, C.L.; Li, N.; Fisher, A.B.; West, K.A.; et al. Development of a synthetic live bacterial therapeutic for the human metabolic disease phenylketonuria. Nat. Biotechnol. 2018, 36, 857–864. [Google Scholar] [CrossRef]
- Kurtz, C.B.; Millet, Y.A.; Puurunen, M.K.; Perreault, M.; Charbonneau, M.R.; Isabella, V.M.; Kotula, J.W.; Antipov, E.; Dagon, Y.; Denney, W.S.; et al. An engineered e. Coli nissle improves hyperammonemia and survival in mice and shows dose-dependent exposure in healthy humans. Sci. Transl. Med. 2019, 11, 1–14. [Google Scholar] [CrossRef]
- Lynch, J.P.; González-Prieto, C.; Reeves, A.Z.; Bae, S.; Powale, U.; Godbole, N.P.; Tremblay, J.M.; Schmidt, F.I.; Ploegh, H.L.; Kansra, V.; et al. Engineered escherichia coli for the in situ secretion of therapeutic nanobodies in the gut. Cell Host Microbe 2023, 31, 634–649. [Google Scholar] [CrossRef]
- Abed, J.; Maalouf, N.; Manson, A.L.; Earl, A.M.; Parhi, L.; Emgård, J.E.M.; Klutstein, M.; Tayeb, S.; Almogy, G.; Atlan, K.A.; et al. Colon cancer-associated fusobacterium nucleatum may originate from the oral cavity and reach colon tumors via the circulatory system. Front. Cell. Infect. Microbiol. 2020, 10, 400. [Google Scholar] [CrossRef]
- Crull, K.; Bumann, D.; Weiss, S. Influence of infection route and virulence factors on colonization of solid tumors bysalmonella entericaserovar typhimurium. FEMS Immunol. Med. Microbiol. 2011, 62, 75–83. [Google Scholar] [CrossRef]
- Zhou, J.; Li, M.; Chen, Q.; Li, X.; Chen, L.; Dong, Z.; Zhu, W.; Yang, Y.; Liu, Z.; Chen, Q. Programmable probiotics modulate inflammation and gut microbiota for inflammatory bowel disease treatment after effective oral delivery. Nat. Commun. 2022, 13, 3432. [Google Scholar] [CrossRef] [PubMed]
- Van ‘t Hof, M.; Mohite, O.S.; Monk, J.M.; Weber, T.; Palsson, B.O.; Sommer, M.O.A. High-quality genome-scale metabolic network reconstruction of probiotic bacterium escherichia coli nissle 1917. BMC Bioinform. 2022, 23, 566. [Google Scholar] [CrossRef] [PubMed]
- Grozdanov, L.; Raasch, C.; Schulze, J.R.; Sonnenborn, U.; Gottschalk, G.; Hacker, J.R.; Dobrindt, U. Analysis of the genome structure of the nonpathogenic probiotic escherichia coli strain nissle 1917. J. Bacteriol. 2004, 186, 5432–5441. [Google Scholar] [CrossRef] [PubMed]
- Wiles, T.J.; Kulesus, R.R.; Mulvey, M.A. Origins and virulence mechanisms of uropathogenic escherichia coli. Exp. Mol. Pathol. 2008, 85, 11–19. [Google Scholar] [CrossRef]
- Martinez, J.J. Type 1 pilus-mediated bacterial invasion of bladder epithelial cells. EMBO J. 2000, 19, 2803–2812. [Google Scholar] [CrossRef]
- Wright, K.J.; Seed, P.C.; Hultgren, S.J. Development of intracellular bacterial communities of uropathogenic escherichia coli depends on type 1 pili. Cell. Microbiol. 2007, 9, 2230–2241. [Google Scholar] [CrossRef]
- Blum, G.; Marre, R.; Hacker, J. Properties of escherichia coli strains of serotype o6. Infection 1995, 23, 234–236. [Google Scholar] [CrossRef]
- Palmela, C.; Chevarin, C.; Xu, Z.; Torres, J.; Sevrin, G.; Hirten, R.; Barnich, N.; Ng, S.C.; Colombel, J.F. Adherent-invasive escherichia coli in inflammatory bowel disease. Gut 2018, 67, 574–587. [Google Scholar] [CrossRef]
- Gibold, L.; Garenaux, E.; Dalmasso, G.; Gallucci, C.; Cia, D.; Mottet-Auselo, B.; Faïs, T.; Darfeuille-Michaud, A.; Nguyen, H.T.T.; Barnich, N.; et al. The vat-aiec protease promotes crossing of the intestinal mucus layer by crohn’s disease-associated escherichia coli. Cell. Microbiol. 2016, 18, 617–631. [Google Scholar] [CrossRef] [PubMed]
- Barnich, N.; Carvalho, F.A.; Glasser, A.L.; Darcha, C.; Jantscheff, P.; Allez, M.; Peeters, H.; Bommelaer, G.; Desreumaux, P.; Colombel, J.F.; et al. Ceacam6 acts as a receptor for adherent-invasive e. Coli, supporting ileal mucosa colonization in crohn disease. J. Clin. Investig. 2007, 117, 1566–1574. [Google Scholar] [CrossRef]
- Denizot, J.; Sivignon, A.; Barreau, F.; Darcha, C.; Chan, C.H.F.; Stanners, C.P.; Hofman, P.; Darfeuille-Michaud, A.; Barnich, N. Adherent-invasive escherichia coli induce claudin-2 expression and barrier defect in ceabac10 mice and crohn’s disease patients§. Inflamm. Bowel Dis. 2012, 18, 294–304. [Google Scholar] [CrossRef]
- Wine, E.; Ossa, J.C.; Gray-Owen, S.D.; Sherman, P.M. Adherent-invasive escherichia coli, strain lf82 disrupts apical junctional complexes in polarized epithelia. BMC Microbiol. 2009, 9, 180. [Google Scholar] [CrossRef]
- Baranov, V.; Hammarstrom, S. Carcinoembryonic antigen (cea) and cea-related cell adhesion molecule 1 (ceacam1), apically expressed on human colonic m cells, are potential receptors for microbial adhesion. Histochem. Cell Biol. 2004, 121, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Glasser, A.-L.; Boudeau, J.; Barnich, N.; Perruchot, M.-H.; Colombel, J.-F.; Darfeuille-Michaud, A. Adherent invasive escherichia coli strains from patients with crohn’s disease survive and replicate within macrophages without inducing host cell death. Infect. Immun. 2001, 69, 5529–5537. [Google Scholar] [CrossRef]
- Voedisch, S.; Koenecke, C.; David, S.; Herbrand, H.; FöRster, R.; Rhen, M.; Pabst, O. Mesenteric lymph nodes confine dendritic cell-mediated dissemination of salmonella enterica serovar typhimurium and limit systemic disease in mice. Infect. Immun. 2009, 77, 3170–3180. [Google Scholar] [CrossRef]
- Abed, J.; Emgård, J.E.M.; Zamir, G.; Faroja, M.; Almogy, G.; Grenov, A.; Sol, A.; Naor, R.; Pikarsky, E.; Atlan, K.A.; et al. Fap2 mediates fusobacterium nucleatum colorectal adenocarcinoma enrichment by binding to tumor-expressed gal-galnac. Cell Host Microbe 2016, 20, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Cummins, J.; Tangney, M. Bacteria and tumours: Causative agents or opportunistic inhabitants? Infect. Agents Cancer 2013, 8, 11. [Google Scholar] [CrossRef]
- Liu, X.; Cheng, Y.; Shao, L.; Ling, Z. Alterations of the predominant fecal microbiota and disruption of the gut mucosal barrier in patients with early-stage colorectal cancer. Biomed. Res. Int. 2020, 2020, 2948282. [Google Scholar] [CrossRef]
- Nzakizwanayo, J.; Kumar, S.; Ogilvie, L.A.; Patel, B.A.; Dedi, C.; Macfarlane, W.M.; Jones, B.V. Disruption of escherichia coli nissle 1917 k5 capsule biosynthesis, through loss of distinct kfi genes, modulates interaction with intestinal epithelial cells and impact on cell health. PLoS ONE 2015, 10, e0120430. [Google Scholar] [CrossRef] [PubMed]
- Gautreaux, M.D.; Deitch, E.A.; Berg, R.D. Bacterial translocation from the gastrointestinal tract to various segments of the mesenteric lymph node complex. Infect. Immun. 1994, 62, 2132–2134. [Google Scholar] [CrossRef]
- Forbes, N.S.; Munn, L.L.; Fukumura, D.; Jain, R.K. Sparse initial entrapment of systemically injected salmonella typhimurium leads to heterogeneous accumulation within tumors. Cancer Res. 2003, 63, 5188–5193. [Google Scholar] [PubMed]
- Nejman, D.; Livyatan, I.; Fuks, G.; Gavert, N.; Zwang, Y.; Geller, L.T.; Rotter-Maskowitz, A.; Weiser, R.; Mallel, G.; Gigi, E.; et al. The human tumor microbiome is composed of tumor type–specific intracellular bacteria. Science 2020, 368, 973–980. [Google Scholar] [CrossRef]
- Yu, Y.A.; Zhang, Q.; Szalay, A.A. Establishment and characterization of conditions required for tumor colonization by intravenously delivered bacteria. Biotechnol. Bioeng. 2008, 100, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Kasinskas, R.W.; Forbes, N.S. Salmonella typhimurium specifically chemotax and proliferate in heterogeneous tumor tissue in vitro. Biotechnol. Bioeng. 2006, 94, 710–721. [Google Scholar] [CrossRef]
- Westphal, K.; Leschner, S.; Jablonska, J.; Loessner, H.; Weiss, S. Containment of tumor-colonizing bacteria by host neutrophils. Cancer Res. 2008, 68, 2952–2960. [Google Scholar] [CrossRef]
- Weibel, S.; Stritzker, J.; Eck, M.; Goebel, W.; Szalay, A.A. Colonization of experimental murine breast tumours by escherichia coli k-12 significantly alters the tumour microenvironment. Cell Microbiol. 2008, 10, 1235–1248. [Google Scholar] [CrossRef]
- Wang, C.; Dang, T.; Baste, J.; Joshi, A.A.; Bhushan, A. A novel standalone microfluidic device for local control of oxygen tension for intestinal-bacteria interactions. FASEB J. 2021, 35, e21291. [Google Scholar] [CrossRef]
- Drees, J.J.; Mertensotto, M.J.; Augustin, L.B.; Schottel, J.L.; Saltzman, D.A. Vasculature disruption enhances bacterial targeting of autochthonous tumors. J. Cancer 2015, 6, 843–848. [Google Scholar] [CrossRef]
- Autieri, S.M.; Lins, J.J.; Leatham, M.P.; Laux, D.C.; Conway, T.; Cohen, P.S. L-fucose stimulates utilization of d-ribose by escherichia coli mg1655 fucao and e. Coli nissle 1917 fucao mutants in the mouse intestine and in m9 minimal medium. Infect. Immun. 2007, 75, 5465–5475. [Google Scholar] [CrossRef] [PubMed]
- Kasinskas, R.W.; Forbes, N.S. Salmonella typhimurium lacking ribose chemoreceptors localize in tumor quiescence and induce apoptosis. Cancer Res. 2007, 67, 3201–3209. [Google Scholar] [CrossRef]
- Schären, O.P.; Hapfelmeier, S. Robust microbe immune recognition in the intestinal mucosa. Genes. Immun. 2021, 22, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Galeano Niño, J.L.; Wu, H.; Lacourse, K.D.; Kempchinsky, A.G.; Baryiames, A.; Barber, B.; Futran, N.; Houlton, J.; Sather, C.; Sicinska, E.; et al. Effect of the intratumoral microbiota on spatial and cellular heterogeneity in cancer. Nature 2022, 611, 810–817. [Google Scholar] [CrossRef]
- Hahn, J.; Ding, S.; Im, J.; Harimoto, T.; Leong, K.W.; Danino, T. Bacterial therapies at the interface of synthetic biology and nanomedicine. Nat. Rev. Bioeng. 2024, 2, 120–135. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.P.; Goers, L.; Lesser, C.F. Emerging strategies for engineering escherichia coli nissle 1917-based therapeutics. Trends Pharmacol. Sci. 2022, 43, 772–786. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Radford, G.A.; Vrbanac, L.; de Nys, R.T.; Worthley, D.L.; Wright, J.A.; Hasty, J.; Woods, S.L. Towards Understanding Tumour Colonisation by Probiotic Bacterium E. coli Nissle 1917. Cancers 2024, 16, 2971. https://doi.org/10.3390/cancers16172971
Radford GA, Vrbanac L, de Nys RT, Worthley DL, Wright JA, Hasty J, Woods SL. Towards Understanding Tumour Colonisation by Probiotic Bacterium E. coli Nissle 1917. Cancers. 2024; 16(17):2971. https://doi.org/10.3390/cancers16172971
Chicago/Turabian StyleRadford, Georgette A., Laura Vrbanac, Rebekah T. de Nys, Daniel L. Worthley, Josephine A. Wright, Jeff Hasty, and Susan L. Woods. 2024. "Towards Understanding Tumour Colonisation by Probiotic Bacterium E. coli Nissle 1917" Cancers 16, no. 17: 2971. https://doi.org/10.3390/cancers16172971
APA StyleRadford, G. A., Vrbanac, L., de Nys, R. T., Worthley, D. L., Wright, J. A., Hasty, J., & Woods, S. L. (2024). Towards Understanding Tumour Colonisation by Probiotic Bacterium E. coli Nissle 1917. Cancers, 16(17), 2971. https://doi.org/10.3390/cancers16172971