
Mutational Signatures in Colorectal Cancer: Translational Insights, Clinical Applications, and Limitations


{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
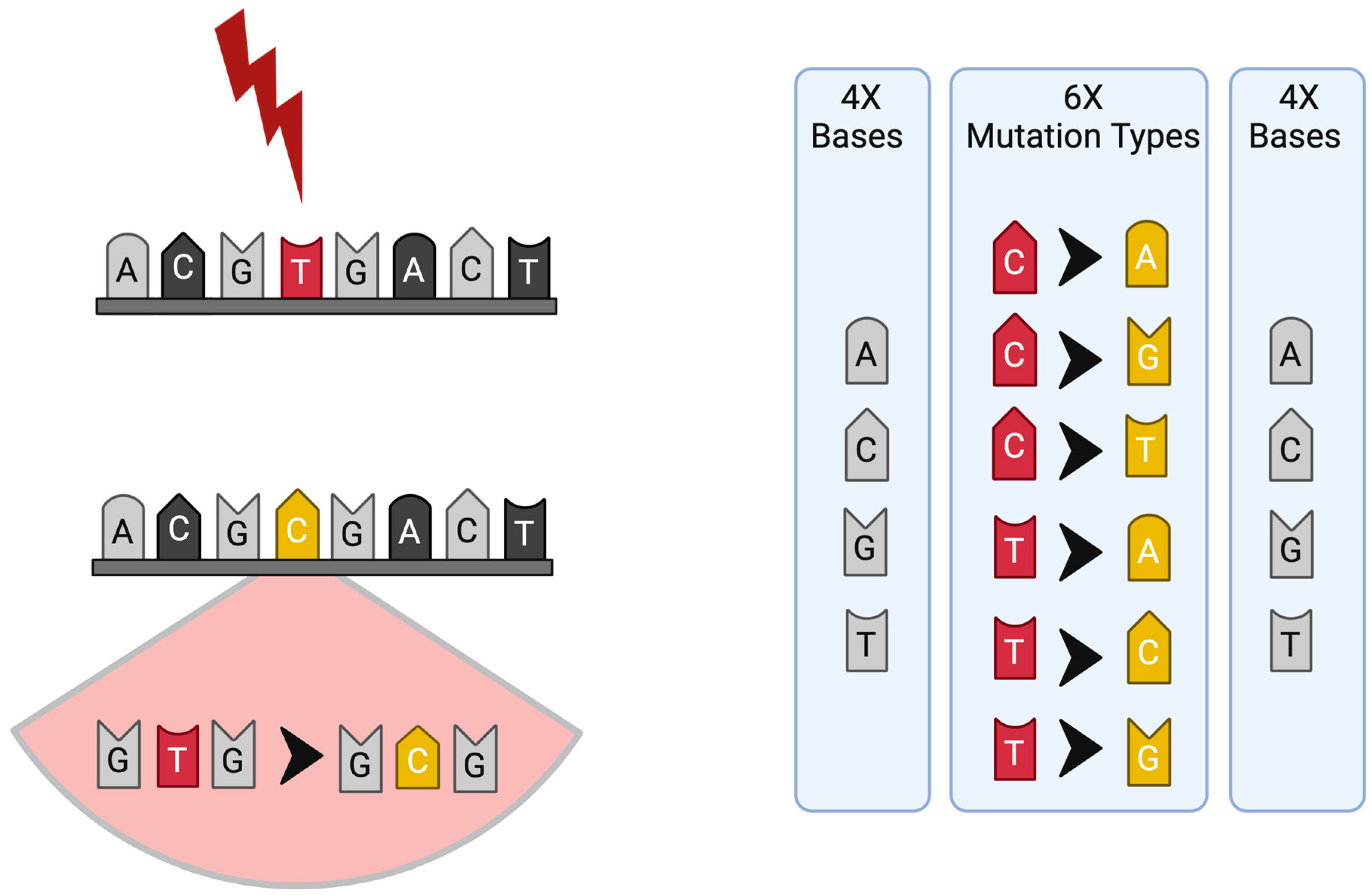
1. Introduction: Historical Perspective on Mutational Signature
2. Mutational Signature Characterizing Colorectal Cancer Tumors
2.1. Specific Signatures of MSI/MMRd CRC
- SBS6 is associated with MMRd cells and is commonly found in cancers with MMR deficiency, including those associated with Lynch syndrome. It demonstrates a markedly elevated incidence of C>T transitions at NpCpG trinucleotides.
- SBS14 is linked to MMR deficiency and is often associated with tumors exhibiting high microsatellite instability.
- SBS15 is associated with MMR deficiency and UV light exposure and is characterized by a high number of C>T transitions at dipyrimidines.
- SBS20 has been linked to defective DNA mismatch repair, though it is less prevalent and is often found in MSI-high cancers.
- SBS21 is also linked to MMR deficiency, though its relationship with this process is less well understood than that of other MMR-related signatures.
- SBS26 is specifically associated with defective mismatch repair, which is characterized by a high level of C>A and C>T mutations. It could be related to the high consumption of alcohol.
- SBS44 is associated with defective DNA mismatch repair and often found in combination with other MMR-related signatures or with a co-occurrence of colibactin-related SBS88.
2.2. Specific Signature of MSS/MMRp POLE-Mutant CRC
- SBS10A: This variant is characterized by a high number of T>G mutations and is linked to mutations in the exonuclease domain of POLE.
- SBS10B is analogous to SBS10A, yet it may exhibit disparate mutation contexts and slightly variant mutation spectra. Nevertheless, it remains associated with POLE mutations.
2.3. Specific Signature of MSS/MMRp POLE wt CRC
- SBS1 is frequently linked to the spontaneous deamination of 5-methylcytosine, which results in C>T transitions. It has been identified as exhibiting a clock-like pattern, whereby the number of mutations in most cancers and normal cells can be correlated with the age of the individual or the tumor. Furthermore, SBS1 has been observed in colon adenomas.
- SBS5 is a similar clock-like signature with an unknown etiology that has been observed in numerous cancer types.
- SBS17A has been linked to oxidative damage and has been associated with gastric cancers. However, it has also been identified in CRC.
- SBS17B: This is similar to SBS17A and may be linked to oxidative damage, and some studies associated SBS17b with fluorouracil (5FU) chemotherapy.
- SBS18 is distinguished by C>A transversions, which may be associated with reactive oxygen species.
- SBS23 is a rare and poorly understood phenomenon that has been observed on occasion in CRC.
- SBS28 is a rare phenomenon with an unclear etiology. In some cases, it was found related to the emergence of SBS10a, SBS10b, and SBS17b signatures.
- SBS37 is less prevalent and the mechanisms underlying its occurrence remain unclear. It has been observed in a range of cancers, albeit infrequently.
- SBS40 is a ubiquitous signature with an unknown etiology, present in a multitude of cancer types (until COSMIC version 3.3) and was superseded by SBS40a, SBS40b, and SBS40c in COSMIC version 3.4.
- SBS44 has been previously associated with defective mismatch repair. Additionally, it can manifest in microsatellite-stable (MSS) tumors, albeit to a lesser extent. It can co-occur with SBS88.
- SBS93 is frequently found in MSS primary tumors but almost absent in MSI; it has been associated with esophageal and gastric cancers and could be related to diet or smoking but these data should be confirmed in further studies.
2.4. Additional Mutational Signatures Found in All CRCs
3. Perspective on Clinical Applications and Benefits of Mutational Signature Analysis in Colorectal Cancer
3.1. Improving Minimal Residual Disease Tests in Stage II (High-Risk) and Stage III CRC after Surgery
3.2. CRC Stratification Based on the Effect of Genetic and Epigenetic Changes
3.3. Identification of Potential Vulnerabilities by Mutational Signatures Analysis
3.4. Evaluation of Drug Effects and Guiding the Therapy
4. Limitations of Mutational Signature Analysis
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; Van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef] [PubMed]
- Petljak, M.; Alexandrov, L.B.; Brammeld, J.S.; Price, S.; Wedge, D.C.; Grossmann, S.; Dawson, K.J.; Ju, Y.S.; Iorio, F.; Tubio, J.M.C.; et al. Characterizing Mutational Signatures in Human Cancer Cell Lines Reveals Episodic APOBEC Mutagenesis. Cell 2019, 176, 1282–1294.e20. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Stratton, M.R. Mutational signatures: The patterns of somatic mutations hidden in cancer genomes. Curr. Opin. Genet. Dev. 2014, 24, 52–60. [Google Scholar] [CrossRef]
- Bergstrom, E.N.; Huang, M.N.; Mahto, U.; Barnes, M.; Stratton, M.R.; Rozen, S.G.; Alexandrov, L.B. SigProfilerMatrixGenerator: A tool for visualizing and exploring patterns of small mutational events. BMC Genom. 2019, 20, 685. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Battuello, P.; Corti, G.; Bartolini, A.; Lorenzato, A.; Sogari, A.; Russo, M.; Di Nicolantonio, F.; Bardelli, A.; Crisafulli, G. Mutational signatures of colorectal cancers according to distinct computational workflows. Brief. Bioinform. 2024, 25, bbae249. [Google Scholar] [CrossRef]
- Setlow, R.B.; Carrier, W.L. The disappearance of thymine dimers from DNA: An error-correcting mechanism. Proc. Natl. Acad. Sci. USA 1964, 51, 226–231. [Google Scholar] [CrossRef]
- Rupert, C.S. Photoreactivation of transforming DNA by an enzyme from bakers’ yeast. J. Gen. Physiol. 1960, 43, 573–595. [Google Scholar] [CrossRef]
- Kelner, A. Photoreactivation of ultraviolet-irradiated escherichia coli, with special reference to the dose-reduction principle and to ultraviolet-induced mutation. J. Bacteriol. 1949, 58, 511–522. [Google Scholar] [CrossRef]
- Bergstrom, E.N.; Luebeck, J.; Petljak, M.; Khandekar, A.; Barnes, M.; Zhang, T.; Steele, C.D.; Pillay, N.; Landi, M.T.; Bafna, V.; et al. Mapping clustered mutations in cancer reveals APOBEC3 mutagenesis of ecDNA. Nature 2022, 602, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Mas-Ponte, D.; Supek, F. DNA mismatch repair promotes APOBEC3-mediated diffuse hypermutation in human cancers. Nat. Genet. 2020, 52, 958–968. [Google Scholar] [CrossRef] [PubMed]
- Otlu, B.; Alexandrov, L.B. Evaluating topography of mutational signatures with SigProfilerTopography. bioRxiv 2024. [Google Scholar] [CrossRef]
- Pleguezuelos-Manzano, C.; Puschhof, J.; Rosendahl Huber, A.; van Hoeck, A.; Wood, H.M.; Nomburg, J.; Gurjao, C.; Manders, F.; Dalmasso, G.; Stege, P.B.; et al. Mutational signature in colorectal cancer caused by genotoxic pks. Nature 2020, 580, 269–273. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Campbell, P.J.; Stratton, M.R. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013, 3, 246–259. [Google Scholar] [CrossRef] [PubMed]
- Steele, C.D.; Bbasi, A.; Islam, S.M.A.; Bowes, A.L.; Khandekar., A.; Haase, K.; Hames-Fathi, S.; Ajayi, D.; Verfaillie, A.; Dhami, P.; et al. Signatures of copy number alterations in human cancer. Nature 2022, 606, 984–991. [Google Scholar] [CrossRef]
- Martínez-Ruiz, C.; Black, J.R.M.; Puttick, C.; Hill, M.S.; Demeulemeester, J.; Larose Cadieux, E.; Thol, K.; Jones, T.P.; Veeriah, S.; Naceur-Lombardelli, C.; et al. Genomic-transcriptomic evolution in lung cancer and metastasis. Nature 2023, 616, 543–552. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer, J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer, J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef]
- Ganesh, K.; Stadler, Z.K.; Cercek, A.; Mendelsohn, R.B.; Shia, J.; Segal, N.H.; Diaz, L.A. Immunotherapy in colorectal cancer: Rationale, challenges and potential. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 361–375. [Google Scholar] [CrossRef]
- Koopman, M.; Kortman, G.A.; Mekenkamp, L.; Ligtenberg, M.J.; Hoogerbrugge, N.; Antonini, N.F.; Punt, C.J.; van Krieken, J.H. Deficient mismatch repair system in patients with sporadic advanced colorectal cancer. Br. J. Cancer 2009, 100, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Sinicrope, F.A.; Sargent, D.J. Molecular pathways: Microsatellite instability in colorectal cancer: Prognostic, predictive, and therapeutic implications. Clin. Cancer Res. 2012, 18, 1506–1512. [Google Scholar] [CrossRef]
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006, 7, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Cohen, R.; Rousseau, B.; Vidal, J.; Colle, R.; Diaz, L.A.; André, T. Immune Checkpoint Inhibition in Colorectal Cancer: Microsatellite Instability and Beyond. Target. Oncol. 2020, 15, 11–24. [Google Scholar] [CrossRef]
- Network, C.G.A. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S.; et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015, 5, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Rospo, G.; Lorenzato, A.; Amirouchene-Angelozzi, N.; Magrì, A.; Cancelliere, C.; Corti, G.; Negrino, C.; Amodio, V.; Montone, M.; Bartolini, A.; et al. Evolving neoantigen profiles in colorectal cancers with DNA repair defects. Genome Med. 2019, 11, 42. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, B.; Foote, M.B.; Maron, S.B.; Diplas, B.H.; Lu, S.; Argilés, G.; Cercek, A.; Diaz, L.A. The Spectrum of Benefit from Checkpoint Blockade in Hypermutated Tumors. N. Engl. J. Med. 2021, 384, 1168–1170. [Google Scholar] [CrossRef]
- Castellsagué, E.; Li, R.; Aligue, R.; González, S.; Sanz, J.; Martin, E.; Velasco, À.; Capellá, G.; Stewart, C.J.R.; Vidal, A.; et al. Novel POLE pathogenic germline variant in a family with multiple primary tumors results in distinct mutational signatures. Hum. Mutat. 2019, 40, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Mur, P.; García-Mulero, S.; Del Valle, J.; Magraner-Pardo, L.; Vidal, A.; Pineda, M.; Cinnirella, G.; Martín-Ramos, E.; Pons, T.; López-Doriga, A.; et al. Role of POLE and POLD1 in familial cancer. Genet. Med. 2020, 22, 2089–2100. [Google Scholar] [CrossRef]
- Lu, C.; Guan, J.; Lu, S.; Jin, Q.; Rousseau, B.; Lu, T.; Stephens, D.; Zhang, H.; Zhu, J.; Yang, M.; et al. DNA Sensing in Mismatch Repair-Deficient Tumor Cells Is Essential for Anti-tumor Immunity. Cancer Cell 2021, 39, 96–108.e6. [Google Scholar] [CrossRef]
- Németh, E.; Lovrics, A.; Gervai, J.Z.; Seki, M.; Rospo, G.; Bardelli, A.; Szüts, D. Two main mutational processes operate in the absence of DNA mismatch repair. DNA Repair 2020, 89, 102827. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Zhu, X.; Yang, H.; Oh, J.; Barbour, J.A.; Wong, J.W.H. Deficiency of replication-independent DNA mismatch repair drives a 5-methylcytosine deamination mutational signature in cancer. Sci. Adv. 2021, 7, eabg4398. [Google Scholar] [CrossRef]
- Georgeson, P.; Pope, B.J.; Rosty, C.; Clendenning, M.; Mahmood, K.; Joo, J.E.; Walker, R.; Hutchinson, R.A.; Preston, S.; Como, J.; et al. Evaluating the utility of tumour mutational signatures for identifying hereditary colorectal cancer and polyposis syndrome carriers. Gut 2021, 70, 2138–2149. [Google Scholar] [CrossRef]
- Walker, R.; Mahmood, K.; Joo, J.E.; Clendenning, M.; Georgeson, P.; Como, J.; Joseland, S.; Preston, S.G.; Antill, Y.; Austin, R.; et al. A tumor focused approach to resolving the etiology of DNA mismatch repair deficient tumors classified as suspected Lynch syndrome. J. Transl. Med. 2023, 21, 282. [Google Scholar] [CrossRef] [PubMed]
- Hodel, K.P.; Sun, M.J.S.; Ungerleider, N.; Park, V.S.; Williams, L.G.; Bauer, D.L.; Immethun, V.E.; Wang, J.; Suo, Z.; Lu, H.; et al. POLE Mutation Spectra Are Shaped by the Mutant Allele Identity, Its Abundance, and Mismatch Repair Status. Mol. Cell 2020, 78, 1166–1177.e6. [Google Scholar] [CrossRef] [PubMed]
- Kucab, J.E.; Zou, X.; Morganella, S.; Joel, M.; Nanda, A.S.; Nagy, E.; Gomez, C.; Degasperi, A.; Harris, R.; Jackson, S.P.; et al. A Compendium of Mutational Signatures of Environmental Agents. Cell 2019, 177, 821–836.e16. [Google Scholar] [CrossRef] [PubMed]
- Secrier, M.; Li, X.; de Silva, N.; Eldridge, M.D.; Contino, G.; Bornschein, J.; MacRae, S.; Grehan, N.; O’Donovan, M.; Miremadi, A.; et al. Mutational signatures in esophageal adenocarcinoma define etiologically distinct subgroups with therapeutic relevance. Nat. Genet. 2016, 48, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Drost, J.; van Boxtel, R.; Blokzijl, F.; Mizutani, T.; Sasaki, N.; Sasselli, V.; de Ligt, J.; Behjati, S.; Grolleman, J.E.; van Wezel, T.; et al. Use of CRISPR-modified human stem cell organoids to study the origin of mutational signatures in cancer. Science 2017, 358, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Cornish, A.J.; Gruber, A.J.; Kinnersley, B.; Chubb, D.; Frangou, A.; Caravagna, G.; Noyvert, B.; Lakatos, E.; Wood, H.M.; Thorn, S.; et al. The genomic landscape of 2023 colorectal cancers. Nature 2024. [Google Scholar] [CrossRef]
- Degasperi, A.; Zou, X.; Amarante, T.D.; Martinez-Martinez, A.; Koh, G.C.C.; Dias, J.M.L.; Heskin, L.; Chmelova, L.; Rinaldi, G.; Wang, V.Y.W.; et al. Substitution mutational signatures in whole-genome-sequenced cancers in the UK population. Science 2022, 376, abl9283. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, S.I.; Sotiriou, S.K.; Pateras, I.S.; Santoni, F.; Sougioultzis, S.; Edgren, H.; Almusa, H.; Robyr, D.; Guipponi, M.; Saarela, J.; et al. A single-nucleotide substitution mutator phenotype revealed by exome sequencing of human colon adenomas. Cancer Res. 2012, 72, 6279–6289. [Google Scholar] [CrossRef]
- Dionellis, V.S.; Norkin, M.; Karamichali, A.; Rossetti, G.G.; Huelsken, J.; Ordonez-Moran, P.; Halazonetis, T.D. Genomic Instability Profiles at the Single Cell Level in Mouse Colorectal Cancers of Defined Genotypes. Cancers 2021, 13, 1267. [Google Scholar] [CrossRef]
- Dziubańska-Kusibab, P.J.; Berger, H.; Battistini, F.; Bouwman, B.A.M.; Iftekhar, A.; Katainen, R.; Cajuso, T.; Crosetto, N.; Orozco, M.; Aaltonen, L.A.; et al. Colibactin DNA-damage signature indicates mutational impact in colorectal cancer. Nat. Med. 2020, 26, 1063–1069. [Google Scholar] [CrossRef]
- Georgeson, P.; Steinfelder, R.S.; Harrison, T.A.; Pope, B.J.; Zaidi, S.H.; Qu, C.; Lin, Y.; Joo, J.E.; Mahmood, K.; Clendenning, M.; et al. Genotoxic colibactin mutational signature in colorectal cancer is associated with clinicopathological features, specific genomic alterations and better survival. medRxiv 2024. [Google Scholar] [CrossRef]
- Dougherty, M.W.; Valdés-Mas, R.; Wernke, K.M.; Gharaibeh, R.Z.; Yang, Y.; Brant, J.O.; Riva, A.; Muehlbauer, M.; Elinav, E.; Puschhof, J.; et al. The microbial genotoxin colibactin exacerbates mismatch repair mutations in colorectal tumors. Neoplasia 2023, 43, 100918. [Google Scholar] [CrossRef]
- Boot, A.; Huang, M.N.; Ng, A.W.T.; Ho, S.C.; Lim, J.Q.; Kawakami, Y.; Chayama, K.; The, B.T.; Nakagawa, H.; Rozen, S.G. In-depth characterization of the cisplatin mutational signature in human cell lines and in esophageal and liver tumors. Genome Res. 2018, 28, 654–665. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Jones, P.H.; Wedge, D.C.; Sale, J.E.; Campbell, P.J.; Nik-Zainal, S.; Stratton, M.R. Clock-like mutational processes in human somatic cells. Nat. Genet. 2015, 47, 1402–1407. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Arena, S.; Corti, G.; Durinikova, E.; Montone, M.; Reilly, N.M.; Russo, M.; Lorenzato, A.; Arcella, P.; Lazzari, L.; Rospo, G.; et al. A Subset of Colorectal Cancers with Cross-Sensitivity to Olaparib and Oxaliplatin. Clin. Cancer Res. 2020, 26, 1372–1384. [Google Scholar] [CrossRef]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 2017, 23, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Pignochino, Y.; Crisafulli, G.; Giordano, G.; Merlini, A.; Berrino, E.; Centomo, M.L.; Chiabotto, G.; Brusco, S.; Basiricò, M.; Maldi, E.; et al. PARP1 Inhibitor and Trabectedin Combination Does Not Increase Tumor Mutational Burden in Advanced Sarcomas-A Preclinical and Translational Study. Cancers 2021, 13, 6295. [Google Scholar] [CrossRef] [PubMed]
- Crisafulli, G.; Sartore-Bianchi, A.; Lazzari, L.; Pietrantonio, F.; Amatu, A.; Macagno, M.; Barault, L.; Cassingena, A.; Bartolini, A.; Luraghi, P.; et al. Temozolomide Treatment Alters Mismatch Repair and Boosts Mutational Burden in Tumor and Blood of Colorectal Cancer Patients. Cancer Discov. 2022, 12, 1656–1675. [Google Scholar] [CrossRef] [PubMed]
- Fang, A.; Zhu, X.; Yang, H.; Oh, J.; Barbour, J.A.; Wong, J.W.H. Alcohol and colorectal cancer risk, subclassified by mutational signatures of DNA mismatch repair deficiency. J. Natl. Cancer Inst. 2024, 116, 1255–1263. [Google Scholar] [CrossRef]
- Taieb, J.; André, T.; Auclin, E. Refining adjuvant therapy for non-metastatic colon cancer, new standards and perspectives. Cancer Treat. Rev. 2019, 75, 1–11. [Google Scholar] [CrossRef]
- Collienne, M.; Arnold, D. The Optimal Duration of Adjuvant Chemotherapy in Colon Cancer. Cancers 2020, 12, 2509. [Google Scholar] [CrossRef]
- Lonardi, S.; Pietrantonio, F.; Tarazona Llavero, N.; Montagut Viladot, C.; Sartore Bianchi, A.; Zampino, M.G.; Elez Fernandez, M.E.; Santos Vivas, C.; Mandalà, M.; Tamberi, S.; et al. LBA28-The PEGASUS trial: Post-surgical liquid biopsy-guided treatment of stage III and high-risk stage II colon cancer patients, A.o.O.s. S1254–S1335. Editor. 23 October 2023. Available online: https://oncologypro.esmo.org/meeting-resources/esmo-congress-2023/the-pegasus-trial-post-surgical-liquid-biopsy-guided-treatment-of-stage-iii-and-high-risk-stage-ii-colon-cancer-patients (accessed on 21 August 2024).
- Tie, J.; Cohen, J.D.; Lahouel, K.; Lo, S.N.; Wang, Y.; Kosmider, S.; Wong, R.; Shapiro, J.; Lee, M.; Harris, S.; et al. Circulating Tumor DNA Analysis Guiding Adjuvant Therapy in Stage II Colon Cancer. N. Engl. J. Med. 2022, 386, 2261–2272. [Google Scholar] [CrossRef]
- Woolston, A.; Barber, L.J.; Griffiths, B.; Pich, O.; Lopez-Bigas, N.; Matthews, N.; Rao, S.; Watkins, D.; Chau, I.; Starling, N.; et al. Mutational signatures impact the evolution of anti-EGFR antibody resistance in colorectal cancer. Nat. Ecol. Evol. 2021, 5, 1024–1032. [Google Scholar] [CrossRef]
- Blokzijl, F.; Janssen, R.; van Boxtel, R.; Cuppen, E. MutationalPatterns: Comprehensive genome-wide analysis of mutational processes. Genome Med. 2018, 10, 33. [Google Scholar] [CrossRef]
- Lahtz, C.; Pfeifer, G. Epigenetic changes of DNA repair genes in cancer. J. Mol. Cell Biol. 2011, 3, 51–58. [Google Scholar] [CrossRef]
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 2018, 23, 239–254.e6. [Google Scholar] [CrossRef] [PubMed]
- Diouf, B.; Cheng, Q.; Krynetskaia, N.F.; Yang, W.; Cheok, M.; Pei, D.; Fan, Y.; Cheng, C.; Krynetskiy, E.Y.; Geng, H.; et al. Somatic deletions of genes regulating MSH2 protein stability cause DNA mismatch repair deficiency and drug resistance in human leukemia cells. Nat. Med. 2011, 17, 1298–1303. [Google Scholar] [CrossRef]
- Levatić, J.; Salvadores, M.; Fuster-Tormo, F.; Supek, F. 2 Mutational signatures are markers of drug sensitivity of cancer cells. Nat. Commun. 2022, 13, 2926. [Google Scholar] [CrossRef] [PubMed]
- Morano, F.; Raimondi, A.; Pagani, F.; Lonardi, S.; Salvatore, L.; Cremolini, C.; Murgioni, S.; Randon, G.; Palermo, F.; Antonuzzo, L. Temozolomide Followed by Combination With Low-Dose Ipilimumab and Nivolumab in Patients With Microsatellite-Stable, O6-Methylguanine-DNA Methyltransferase-Silenced Metastatic Colorectal Cancer: The MAYA Trial. J. Clin. Oncol. 2022, 40, 1562–1573. [Google Scholar] [CrossRef] [PubMed]
- Germano, G.; Lamba, S.; Rospo, G.; Barault, L.; Magrì, A.; Maione, F.; Russo, M.; Crisafulli, G.; Bartolini, A.; Lerda, G.; et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 2017, 552, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.M.A.; Alexandrov, L.B. Bioinformatic Methods to Identify Mutational Signatures in Cancer. Methods Mol. Biol. 2021, 2185, 447–473. [Google Scholar]
- Maura, F.; Degasperi, A.; Nadeu, F.; Leongamornlert, D.; Davies, H.; Moore, L.; Royo, R.; Ziccheddu, B.; Puente, X.S.; Avet-Loiseau, H.; et al. A practical guide for mutational signature analysis in hematological malignancies. Nat. Commun. 2019, 10, 2969. [Google Scholar] [CrossRef]
- Crisafulli, G.; Mussolin, B.; Cassingena, A.; Montone, M.; Bartolini, A.; Barault, L.; Martinetti, A.; Morano, F.; Pietrantonio, F.; Sartore-Bianchi, A.; et al. Whole exome sequencing analysis of urine trans-renal tumour DNA in metastatic colorectal cancer patients. ESMO Open 2019, 4, e000572. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crisafulli, G. Mutational Signatures in Colorectal Cancer: Translational Insights, Clinical Applications, and Limitations. Cancers 2024, 16, 2956. https://doi.org/10.3390/cancers16172956
Crisafulli G. Mutational Signatures in Colorectal Cancer: Translational Insights, Clinical Applications, and Limitations. Cancers. 2024; 16(17):2956. https://doi.org/10.3390/cancers16172956
Chicago/Turabian StyleCrisafulli, Giovanni. 2024. "Mutational Signatures in Colorectal Cancer: Translational Insights, Clinical Applications, and Limitations" Cancers 16, no. 17: 2956. https://doi.org/10.3390/cancers16172956
APA StyleCrisafulli, G. (2024). Mutational Signatures in Colorectal Cancer: Translational Insights, Clinical Applications, and Limitations. Cancers, 16(17), 2956. https://doi.org/10.3390/cancers16172956