


Genome-Wide CRISPR Screen Identifies KEAP1 Perturbation as a Vulnerability of ARID1A-Deficient Cells

 ,
,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
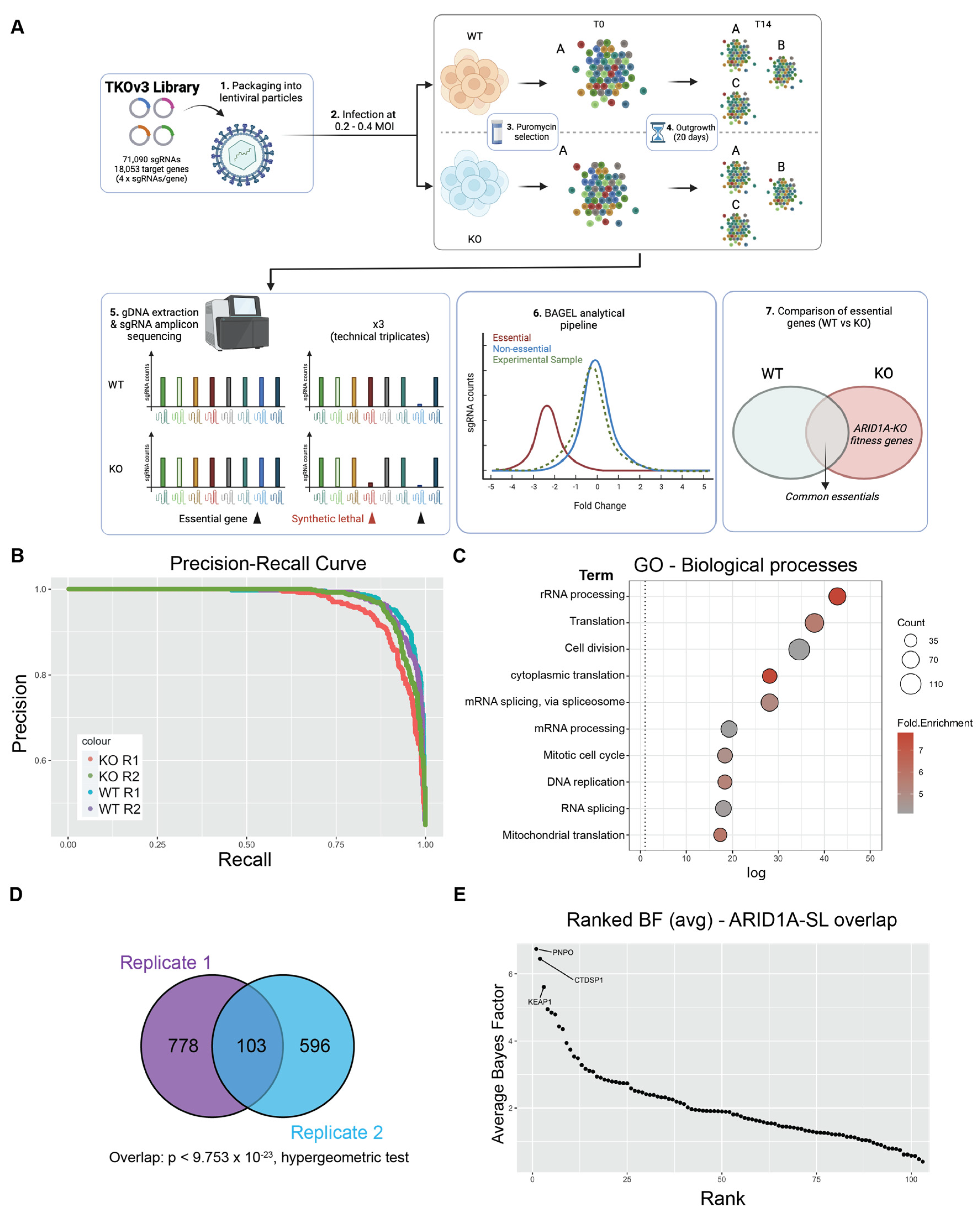
2.1. Genome-Wide CRISPR Screen Identifies Genes Important for Fitness in ARID1A-KO CCOC Cells
2.2. Validation of KEAP1 as a Fitness Gene in ARID1A-Deficient Cells
2.3. Gene Expression Analysis Reveals a Dysregulation of NRF2 Target Genes in ARID1A-KO Cells
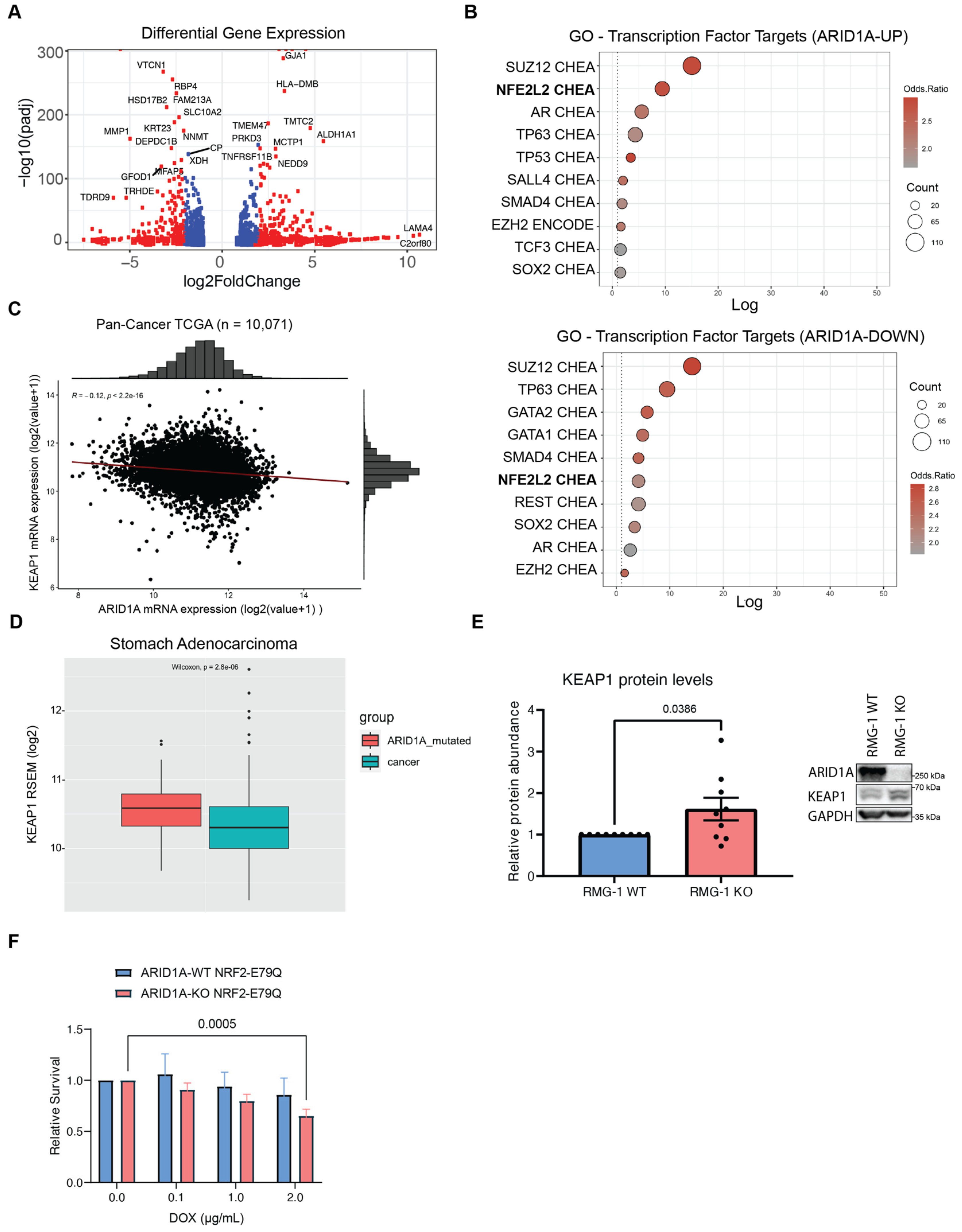
2.4. Aberrant NRF2 Signalling Does Not Significantly Impair the Growth of ARID1A-KO Cells
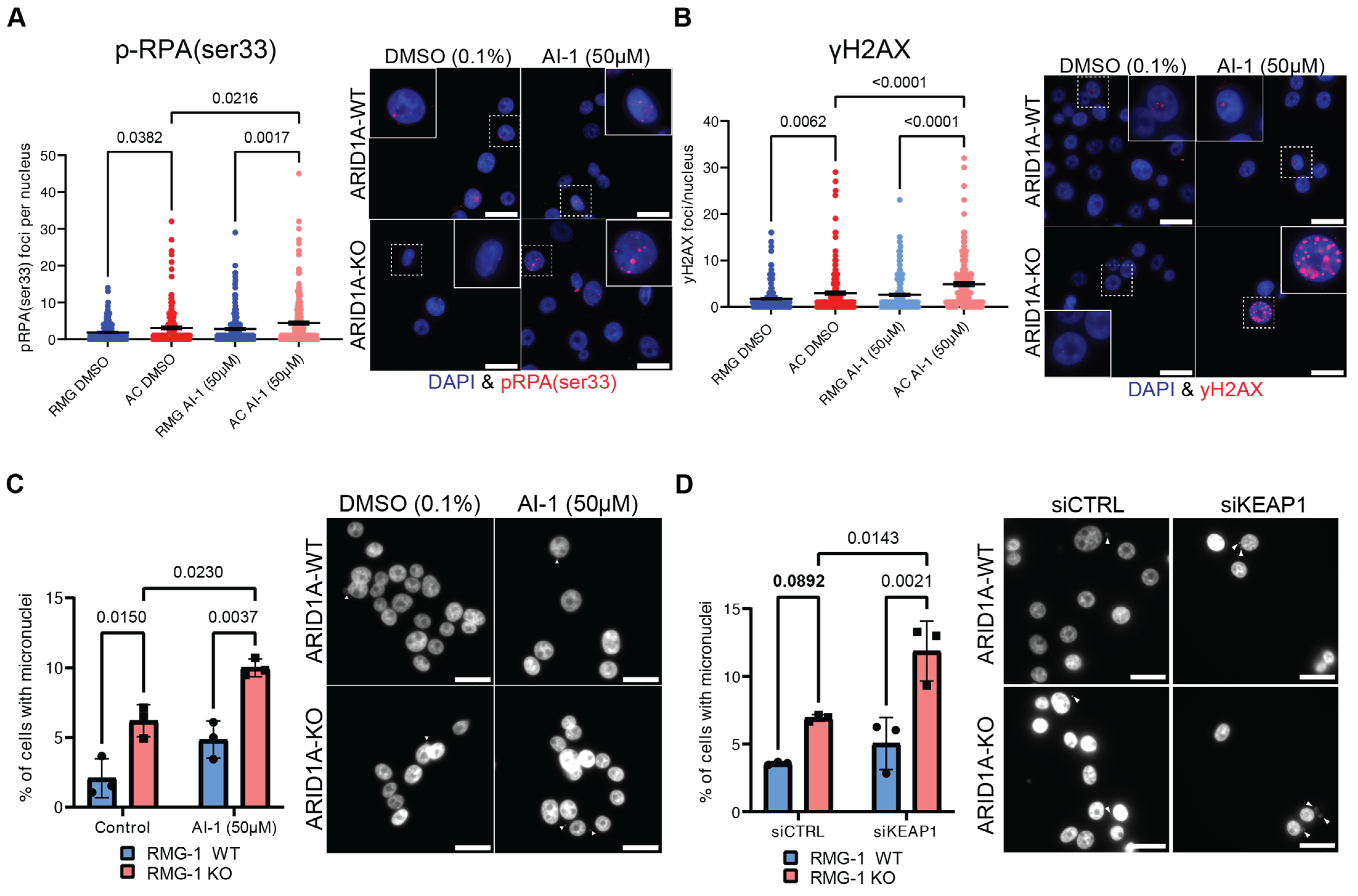
2.5. KEAP1 Perturbation Exacerbates Genome Instability in ARID1A-Deficient Cells
2.6. Dual Inhibition of KEAP1 and ATR Potentiates Killing of ARID1A-KO Cells
3. Discussion
4. Methods
4.1. Cell Culture Conditions
4.2. Generation of NRF2E79Q Dox-Inducible Cell Lines
4.3. TKOv3 Library Cloning and Viral Preparation
4.4. TKOv3 Library Viral Transduction and Titration
4.5. CRISPR Knockout Pool Generation
4.6. Genomic DNA Extraction and Precipitation
4.7. Library Preparation and Sequencing
4.8. Amplicon Scoring, Guide-RNA Enrichment and Hit Identification
4.9. Gene Ontology (GO) Analysis
4.10. Incucyte Growth Assay
4.11. RNA Interference
4.12. Crystal Violet Proliferation Assay
4.13. Immunofluorescence
4.14. Western Blotting
4.15. TCGA Dataset Analysis
4.16. Differential Gene Expression Analysis
4.17. Culture of Patient-Derived Endometrial Progenitor Cells
4.18. Statistical Analysis, Data Availability, and Reproducibility
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wiegand, K.C.; Shah, S.P.; Al-Agha, O.M.; Zhao, Y.; Tse, K.; Zeng, T.; Senz, J.; McConechy, M.K.; Anglesio, M.S.; Kalloger, S.E.; et al. ARID1A Mutations in Endometriosis-Associated Ovarian Carcinomas. N. Engl. J. Med. 2010, 363, 1532–1543. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Wang, T.-L.; Shih, I.-M.; Mao, T.-L.; Nakayama, K.; Roden, R.; Glas, R.; Slamon, D.; Diaz, L.A.; Vogelstein, B.; et al. Frequent Mutations of Chromatin Remodeling Gene ARID1A in Ovarian Clear Cell Carcinoma. Science 2010, 330, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Ashizawa, M.; Saito, M.; Min, A.K.T.; Ujiie, D.; Saito, K.; Sato, T.; Kikuchi, T.; Okayama, H.; Fujita, S.; Endo, H.; et al. Prognostic Role of ARID1A Negative Expression in Gastric Cancer. Sci. Rep. 2019, 9, 6769. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Chen, Y.; Pan, K.; Wang, W.; Chen, S.; Chen, J.; Zhao, J.; Lv, L.; Pan, Q.; Li, Y.; et al. Decreased Expression of the ARID1A Gene Is Associated with Poor Prognosis in Primary Gastric Cancer. PLoS ONE 2012, 7, e40364. [Google Scholar] [CrossRef]
- Zhu, Y.P.; Sheng, L.L.; Wu, J.; Yang, M.; Cheng, X.F.; Wu, N.N.; Ye, X.B.; Cai, J.; Wang, L.; Shen, Q.; et al. Loss of ARID1A Expression Is Associated with Poor Prognosis in Patients with Gastric Cancer. Hum. Pathol. 2018, 78, 28–35. [Google Scholar] [CrossRef]
- Wang, T.; Guo, J.; Liu, W.; Guo, Q.; Cheng, L.; Zheng, R.; Hu, X. Downregulation of ARID1A Is Correlated with Poor Prognosis in Non-Small Cell Lung Cancer. Transl. Cancer Res. 2020, 9, 4896–4905. [Google Scholar] [CrossRef]
- Yang, H.; Huo, J.; Li, X. Identification and Validation of a Five-Gene Prognostic Signature for Hepatocellular Carcinoma. World J. Surg. Oncol. 2021, 19, 90. [Google Scholar] [CrossRef]
- Yim, S.Y.; Kang, S.H.; Shin, J.-H.; Jeong, Y.S.; Sohn, B.H.; Um, S.H.; Lee, J.-S. Low ARID1A Expression Is Associated with Poor Prognosis in Hepatocellular Carcinoma. Cells 2020, 9, 2002. [Google Scholar] [CrossRef]
- He, F.; Li, J.; Xu, J.; Zhang, S.; Xu, Y.; Zhao, W.; Yin, Z.; Wang, X. Decreased Expression of ARID1A Associates with Poor Prognosis and Promotes Metastases of Hepatocellular Carcinoma. J. Exp. Clin. Cancer Res. 2015, 34, 47. [Google Scholar] [CrossRef]
- Cho, H.D.; Lee, J.E.; Jung, H.Y.; Oh, M.-H.; Lee, J.-H.; Jang, S.-H.; Kim, K.-J.; Han, S.W.; Kim, S.Y.; Kim, H.J.; et al. Loss of Tumor Suppressor ARID1A Protein Expression Correlates with Poor Prognosis in Patients with Primary Breast Cancer. J. Breast Cancer 2015, 18, 339–346. [Google Scholar] [CrossRef]
- Ayhan, A.; Mao, T.-L.; Seckin, T.; Wu, C.-H.; Guan, B.; Ogawa, H.; Futagami, M.; Mizukami, H.; Yokoyama, Y.; Kurman, R.J.; et al. Loss of ARID1A Expression Is an Early Molecular Event in Tumor Progression from Ovarian Endometriotic Cyst to Clear Cell and Endometrioid Carcinoma. Int. J. Gynecol. Cancer 2012, 22, 1310–1315. [Google Scholar] [CrossRef] [PubMed]
- Lowery, W.J.; Schildkraut, J.M.; Akushevich, L.; Bentley, R.; Marks, J.R.; Huntsman, D.; Berchuck, A. Loss of ARID1A-Associated Protein Expression Is a Frequent Event in Clear Cell and Endometrioid Ovarian Cancers. Int. J. Gynecol. Cancer 2012, 22, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Maeda, D.; Mao, T.-L.; Fukayama, M.; Nakagawa, S.; Yano, T.; Taketani, Y.; Shih, I.-M. Clinicopathological Significance of Loss of ARID1A Immunoreactivity in Ovarian Clear Cell Carcinoma. Int. J. Mol. Sci. 2010, 11, 5120–5128. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Tsuda, H.; Takano, M.; Tamai, S.; Matsubara, O. PIK3CA Mutations and Loss of ARID1A Protein Expression Are Early Events in the Development of Cystic Ovarian Clear Cell Adenocarcinoma. Virchows Arch. Int. J. Pathol. 2012, 460, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Han, H.H.; Kim, Y.T.; Lee, J.H.; Kim, B.G.; Kang, S.; Cho, N.H. Ovarian Clear Cell Carcinoma Sub-Typing by ARID1A Expression. Yonsei Med. J. 2017, 58, 59–66. [Google Scholar] [CrossRef]
- Katagiri, A.; Nakayama, K.; Rahman, M.T.; Rahman, M.; Katagiri, H.; Ishikawa, M.; Ishibashi, T.; Iida, K.; Otsuki, Y.; Nakayama, S.; et al. Frequent Loss of Tumor Suppressor ARID1A Protein Expression in Adenocarcinomas/Adenosquamous Carcinomas of the Uterine Cervix. Int. J. Gynecol. Cancer 2012, 22, 208–212. [Google Scholar] [CrossRef]
- Heinze, K.; Nazeran, T.M.; Lee, S.; Krämer, P.; Cairns, E.S.; Chiu, D.S.; Leung, S.C.; Kang, E.Y.; Meagher, N.S.; Kennedy, C.J.; et al. Validated Biomarker Assays Confirm That ARID1A Loss Is Confounded with MMR Deficiency, CD8+ TIL Infiltration, and Provides No Independent Prognostic Value in Endometriosis-Associated Ovarian Carcinomas. J. Pathol. 2022, 256, 388–401. [Google Scholar] [CrossRef]
- Mandal, J.; Mandal, P.; Wang, T.-L.; Shih, I.-M. Treating ARID1A Mutated Cancers by Harnessing Synthetic Lethality and DNA Damage Response. J. Biomed. Sci. 2022, 29, 71. [Google Scholar] [CrossRef]
- Tsai, S.; Fournier, L.-A.; Chang, E.Y.-C.; Wells, J.P.; Minaker, S.W.; Zhu, Y.D.; Wang, A.Y.-H.; Wang, Y.; Huntsman, D.G.; Stirling, P.C. ARID1A Regulates R-Loop Associated DNA Replication Stress. PLoS Genet. 2021, 17, e1009238. [Google Scholar] [CrossRef]
- Zhao, B.; Lin, J.; Rong, L.; Wu, S.; Deng, Z.; Fatkhutdinov, N.; Zundell, J.; Fukumoto, T.; Liu, Q.; Kossenkov, A.; et al. ARID1A Promotes Genomic Stability through Protecting Telomere Cohesion. Nat. Commun. 2019, 10, 4067. [Google Scholar] [CrossRef]
- Trizzino, M.; Barbieri, E.; Petracovici, A.; Wu, S.; Welsh, S.A.; Owens, T.A.; Licciulli, S.; Zhang, R.; Gardini, A. The Tumor Suppressor ARID1A Controls Global Transcription via Pausing of RNA Polymerase II. Cell Rep. 2018, 23, 3933–3945. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Fatkhutdinov, N.; Rosin, L.; Luppino, J.M.; Iwasaki, O.; Tanizawa, H.; Tang, H.-Y.; Kossenkov, A.V.; Gardini, A.; Noma, K.-I.; et al. ARID1A Spatially Partitions Interphase Chromosomes. Sci. Adv. 2019, 5, eaaw5294. [Google Scholar] [CrossRef] [PubMed]
- Goldman, A.R.; Bitler, B.G.; Schug, Z.; Conejo-Garcia, J.R.; Zhang, R.; Speicher, D.W. The Primary Effect on the Proteome of ARID1A-Mutated Ovarian Clear Cell Carcinoma Is Downregulation of the Mevalonate Pathway at the Post-Transcriptional Level. Mol. Cell. Proteom. MCP 2016, 15, 3348–3360. [Google Scholar] [CrossRef] [PubMed]
- Ogiwara, H.; Takahashi, K.; Sasaki, M.; Kuroda, T.; Yoshida, H.; Watanabe, R.; Maruyama, A.; Makinoshima, H.; Chiwaki, F.; Sasaki, H.; et al. Targeting the Vulnerability of Glutathione Metabolism in ARID1A-Deficient Cancers. Cancer Cell 2019, 35, 177–190.e8. [Google Scholar] [CrossRef]
- Zhang, X.; Shetty, M.; Clemente, V.; Linder, S.; Bazzaro, M. Targeting Mitochondrial Metabolism in Clear Cell Carcinoma of the Ovaries. Int. J. Mol. Sci. 2021, 22, 4750. [Google Scholar] [CrossRef]
- Zhang, W.; Chronis, C.; Chen, X.; Zhang, H.; Spalinskas, R.; Pardo, M.; Chen, L.; Wu, G.; Zhu, Z.; Yu, Y.; et al. The BAF and PRC2 Complex Subunits Dpf2 and Eed Antagonistically Converge on Tbx3 to Control ESC Differentiation. Cell Stem Cell 2019, 24, 138–152.e8. [Google Scholar] [CrossRef]
- Kadoch, C.; Copeland, R.A.; Keilhack, H. PRC2 and SWI/SNF Chromatin Remodeling Complexes in Health and Disease. Biochemistry 2016, 55, 1600–1614. [Google Scholar] [CrossRef]
- Wilson, M.R.; Reske, J.J.; Koeman, J.; Adams, M.; Joshi, N.R.; Fazleabas, A.T.; Chandler, R.L. SWI/SNF Antagonism of PRC2 Mediates Estrogen-Induced Progesterone Receptor Expression. Cells 2022, 11, 1000. [Google Scholar] [CrossRef]
- Bitler, B.G.; Aird, K.M.; Garipov, A.; Li, H.; Amatangelo, M.; Kossenkov, A.V.; Schultz, D.C.; Liu, Q.; Shih, I.-M.; Conejo-Garcia, J.R.; et al. Synthetic Lethality by Targeting EZH2 Methyltransferase Activity in ARID1A-Mutated Cancers. Nat. Med. 2015, 21, 231–238. [Google Scholar] [CrossRef]
- Yamada, L.; Saito, M.; Thar Min, A.K.; Saito, K.; Ashizawa, M.; Kase, K.; Nakajima, S.; Onozawa, H.; Okayama, H.; Endo, H.; et al. Selective Sensitivity of EZH2 Inhibitors Based on Synthetic Lethality in ARID1A-Deficient Gastric Cancer. Gastric Cancer 2021, 24, 60–71. [Google Scholar] [CrossRef]
- Sen, M.; Wang, X.; Hamdan, F.H.; Rapp, J.; Eggert, J.; Kosinsky, R.L.; Wegwitz, F.; Kutschat, A.P.; Younesi, F.S.; Gaedcke, J.; et al. ARID1A Facilitates KRAS Signaling-Regulated Enhancer Activity in an AP1-Dependent Manner in Colorectal Cancer Cells. Clin. Epigenet. 2019, 11, 92. [Google Scholar] [CrossRef] [PubMed]
- Berns, K.; Caumanns, J.J.; Hijmans, E.M.; Gennissen, A.M.C.; Severson, T.M.; Evers, B.; Wisman, G.B.A.; Jan Meersma, G.; Lieftink, C.; Beijersbergen, R.L.; et al. ARID1A Mutation Sensitizes Most Ovarian Clear Cell Carcinomas to BET Inhibitors. Oncogene 2018, 37, 4611–4625. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Lyu, J.; Yang, E.J.; Liu, Y.; Zhang, B.; Shim, J.S. Targeting AURKA-CDC25C Axis to Induce Synthetic Lethality in ARID1A-Deficient Colorectal Cancer Cells. Nat. Commun. 2018, 9, 3212. [Google Scholar] [CrossRef]
- Hart, T.; Tong, A.H.Y.; Chan, K.; Van Leeuwen, J.; Seetharaman, A.; Aregger, M.; Chandrashekhar, M.; Hustedt, N.; Seth, S.; Noonan, A.; et al. Evaluation and Design of Genome-Wide CRISPR/SpCas9 Knockout Screens. G3 Genes Genomes Genet. 2017, 7, 2719–2727. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Kim, E.; Hart, T. Improved Analysis of CRISPR Fitness Screens and Reduced Off-Target Effects with the BAGEL2 Gene Essentiality Classifier. Genome Med. 2021, 13, 2. [Google Scholar] [CrossRef]
- Mittal, P.; Roberts, C.W.M. The SWI/SNF Complex in Cancer—Biology, Biomarkers and Therapy. Nat. Rev. Clin. Oncol. 2020, 17, 435–448. [Google Scholar] [CrossRef]
- Mathur, R. ARID1A Loss in Cancer: Towards a Mechanistic Understanding. Pharmacol. Ther. 2018, 190, 15–23. [Google Scholar] [CrossRef]
- Hodges, C.; Kirkland, J.G.; Crabtree, G.R. The Many Roles of BAF (mSWI/SNF) and PBAF Complexes in Cancer. Cold Spring Harb. Perspect. Med. 2016, 6, a026930. [Google Scholar] [CrossRef]
- Lebedev, T.; Kousar, R.; Patrick, B.; Usama, M.; Lee, M.-K.; Tan, M.; Li, X.-G. Targeting ARID1A-Deficient Cancers: An Immune-Metabolic Perspective. Cells 2023, 12, 952. [Google Scholar] [CrossRef]
- Feng, X.; Tang, M.; Dede, M.; Su, D.; Pei, G.; Jiang, D.; Wang, C.; Chen, Z.; Li, M.; Nie, L.; et al. Genome-Wide CRISPR Screens Using Isogenic Cells Reveal Vulnerabilities Conferred by Loss of Tumor Suppressors. Sci. Adv. 2022, 8, eabm6638. [Google Scholar] [CrossRef] [PubMed]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576.e16. [Google Scholar] [CrossRef] [PubMed]
- Helming, K.C.; Wang, X.; Wilson, B.G.; Vazquez, F.; Haswell, J.R.; Manchester, H.E.; Kim, Y.; Kryukov, G.V.; Ghandi, M.; Aguirre, A.J.; et al. ARID1B Is a Specific Vulnerability in ARID1A-Mutant Cancers. Nat. Med. 2014, 20, 251–254. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef] [PubMed]
- Hur, W.; Sun, Z.; Jiang, T.; Mason, D.E.; Peters, E.C.; Zhang, D.D.; Luesch, H.; Schultz, P.G.; Gray, N.S. A Small-Molecule Inducer of the Antioxidant Response Element. Chem. Biol. 2010, 17, 537–547. [Google Scholar] [CrossRef]
- Hu, C.; Eggler, A.L.; Mesecar, A.D.; van Breemen, R.B. Modification of Keap1 Cysteine Residues by Sulforaphane. Chem. Res. Toxicol. 2011, 24, 515–521. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Taguchi, K.; Yamamoto, M. The KEAP1-NRF2 System in Cancer. Front. Oncol. 2017, 7, 85. [Google Scholar] [CrossRef]
- Kerins, M.J.; Ooi, A. A Catalogue of Somatic NRF2 Gain-of-Function Mutations in Cancer. Sci. Rep. 2018, 8, 12846. [Google Scholar] [CrossRef]
- ENCODE Project Consortium An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 2012, 489, 57–74. [CrossRef]
- Lachmann, A.; Xu, H.; Krishnan, J.; Berger, S.I.; Mazloom, A.R.; Ma’ayan, A. ChEA: Transcription Factor Regulation Inferred from Integrating Genome-Wide ChIP-X Experiments. Bioinformatics 2010, 26, 2438–2444. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and Collaborative HTML5 Gene List Enrichment Analysis Tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Krupina, K.; Goginashvili, A.; Cleveland, D.W. Causes and Consequences of Micronuclei. Curr. Opin. Cell Biol. 2021, 70, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Williamson, C.T.; Miller, R.; Pemberton, H.N.; Jones, S.E.; Campbell, J.; Konde, A.; Badham, N.; Rafiq, R.; Brough, R.; Gulati, A.; et al. ATR Inhibitors as a Synthetic Lethal Therapy for Tumours Deficient in ARID1A. Nat. Commun. 2016, 7, 13837. [Google Scholar] [CrossRef]
- Shen, J.; Peng, Y.; Wei, L.; Zhang, W.; Yang, L.; Lan, L.; Kapoor, P.; Ju, Z.; Mo, Q.; Shih, I.-M.; et al. ARID1A Deficiency Impairs the DNA Damage Checkpoint and Sensitizes Cells to PARP Inhibitors. Cancer Discov. 2015, 5, 752–767. [Google Scholar] [CrossRef]
- Kadoch, C.; Williams, R.T.; Calarco, J.P.; Miller, E.L.; Weber, C.M.; Braun, S.M.G.; Pulice, J.L.; Chory, E.J.; Crabtree, G.R. Dynamics of BAF-Polycomb Complex Opposition on Heterochromatin in Normal and Oncogenic States. Nat. Genet. 2017, 49, 213–222. [Google Scholar] [CrossRef]
- Alldredge, J.K.; Eskander, R.N. EZH2 Inhibition in ARID1A Mutated Clear Cell and Endometrioid Ovarian and Endometrioid Endometrial Cancers. Gynecol. Oncol. Res. Pract. 2017, 4, 17. [Google Scholar] [CrossRef]
- Kim, K.H.; Kim, W.; Howard, T.P.; Vazquez, F.; Tsherniak, A.; Wu, J.N.; Wang, W.; Haswell, J.R.; Walensky, L.D.; Hahn, W.C.; et al. SWI/SNF-Mutant Cancers Depend on Catalytic and Non-Catalytic Activity of EZH2. Nat. Med. 2015, 21, 1491–1496. [Google Scholar] [CrossRef]
- Kopacz, A.; Kloska, D.; Forman, H.J.; Jozkowicz, A.; Grochot-Przeczek, A. Beyond Repression of Nrf2: An Update on Keap1. Free Radic. Biol. Med. 2020, 157, 63–74. [Google Scholar] [CrossRef]
- Chen, F.; Xiao, M.; Feng, J.; Wufur, R.; Liu, K.; Hu, S.; Zhang, Y. Different Inhibition of Nrf2 by Two Keap1 Isoforms α and β to Shape Malignant Behaviour of Human Hepatocellular Carcinoma. Int. J. Mol. Sci. 2022, 23, 10342. [Google Scholar] [CrossRef]
- Shah, S.Z.A.; Zhao, D.; Hussain, T.; Sabir, N.; Mangi, M.H.; Yang, L. P62-Keap1-NRF2-ARE Pathway: A Contentious Player for Selective Targeting of Autophagy, Oxidative Stress and Mitochondrial Dysfunction in Prion Diseases. Front. Mol. Neurosci. 2018, 11, 310. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.-S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The Selective Autophagy Substrate P62 Activates the Stress Responsive Transcription Factor Nrf2 through Inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Tamberg, N.; Tahk, S.; Koit, S.; Kristjuhan, K.; Kasvandik, S.; Kristjuhan, A.; Ilves, I. Keap1-MCM3 Interaction Is a Potential Coordinator of Molecular Machineries of Antioxidant Response and Genomic DNA Replication in Metazoa. Sci. Rep. 2018, 8, 12136. [Google Scholar] [CrossRef] [PubMed]
- Mulvaney, K.M.; Matson, J.P.; Siesser, P.F.; Tamir, T.Y.; Goldfarb, D.; Jacobs, T.M.; Cloer, E.W.; Harrison, J.S.; Vaziri, C.; Cook, J.G.; et al. Identification and Characterization of MCM3 as a Kelch-like ECH-Associated Protein 1 (KEAP1) Substrate. J. Biol. Chem. 2016, 291, 23719–23733. [Google Scholar] [CrossRef]
- Kopacz, A.; Rojo, A.I.; Patibandla, C.; Lastra-Martínez, D.; Piechota-Polanczyk, A.; Kloska, D.; Jozkowicz, A.; Sutherland, C.; Cuadrado, A.; Grochot-Przeczek, A. Overlooked and Valuable Facts to Know in the NRF2/KEAP1 Field. Free Radic. Biol. Med. 2022, 192, 37–49. [Google Scholar] [CrossRef]
- Lv, Y.; Lv, X.; Zhang, J.; Cao, G.; Xu, C.; Zhang, B.; Lin, W. BRD4 Targets the KEAP1-Nrf2-G6PD Axis and Suppresses Redox Metabolism in Small Cell Lung Cancer. Antioxidants 2022, 11, 661. [Google Scholar] [CrossRef]
- Hussong, M.; Börno, S.T.; Kerick, M.; Wunderlich, A.; Franz, A.; Sültmann, H.; Timmermann, B.; Lehrach, H.; Hirsch-Kauffmann, M.; Schweiger, M.R. The Bromodomain Protein BRD4 Regulates the KEAP1/NRF2-Dependent Oxidative Stress Response. Cell Death Dis. 2014, 5, e1195. [Google Scholar] [CrossRef]
- Huang, M.; Zhu, L.; Garcia, J.S.; Li, M.X.; Gentles, A.J.; Mitchell, B.S. Brd4 Regulates the Expression of Essential Autophagy Genes and Keap1 in AML Cells. Oncotarget 2018, 9, 11665–11676. [Google Scholar] [CrossRef]
- Song, S.; Nguyen, V.; Schrank, T.; Mulvaney, K.; Walter, V.; Wei, D.; Orvis, T.; Desai, N.; Zhang, J.; Hayes, D.N.; et al. Loss of SWI/SNF Chromatin Remodeling Alters NRF2 Signaling in Non-Small Cell Lung Carcinoma. Mol. Cancer Res. MCR 2020, 18, 1777–1788. [Google Scholar] [CrossRef]
- Hart, T.; Chandrashekhar, M.; Aregger, M.; Steinhart, Z.; Brown, K.R.; MacLeod, G.; Mis, M.; Zimmermann, M.; Fradet-Turcotte, A.; Sun, S.; et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 2015, 163, 1515–1526. [Google Scholar] [CrossRef]
- Mair, B.; Tomic, J.; Masud, S.N.; Tonge, P.; Weiss, A.; Usaj, M.; Tong, A.H.Y.; Kwan, J.J.; Brown, K.R.; Titus, E.; et al. Essential Gene Profiles for Human Pluripotent Stem Cells Identify Uncharacterized Genes and Substrate Dependencies. Cell Rep. 2019, 27, 599–615. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioinformatics Resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Cochrane, D.R.; Campbell, K.R.; Greening, K.; Ho, G.C.; Hopkins, J.; Bui, M.; Douglas, J.M.; Sharlandjieva, V.; Munzur, A.D.; Lai, D.; et al. Single Cell Transcriptomes of Normal Endometrial Derived Organoids Uncover Novel Cell Type Markers and Cryptic Differentiation of Primary Tumours. J. Pathol. 2020, 252, 201–214. [Google Scholar] [CrossRef]
- Hart, T.; Moffat, J. BAGEL: A Computational Framework for Identifying Essential Genes from Pooled Library Screens. BMC Bioinform. 2016, 17, 164. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fournier, L.-A.; Kalantari, F.; Wells, J.P.; Lee, J.S.; Trigo-Gonzalez, G.; Moksa, M.M.; Smith, T.; White, J.; Shanks, A.; Wang, S.L.; et al. Genome-Wide CRISPR Screen Identifies KEAP1 Perturbation as a Vulnerability of ARID1A-Deficient Cells. Cancers 2024, 16, 2949. https://doi.org/10.3390/cancers16172949
Fournier L-A, Kalantari F, Wells JP, Lee JS, Trigo-Gonzalez G, Moksa MM, Smith T, White J, Shanks A, Wang SL, et al. Genome-Wide CRISPR Screen Identifies KEAP1 Perturbation as a Vulnerability of ARID1A-Deficient Cells. Cancers. 2024; 16(17):2949. https://doi.org/10.3390/cancers16172949
Chicago/Turabian StyleFournier, Louis-Alexandre, Forouh Kalantari, James P. Wells, Joon Seon Lee, Genny Trigo-Gonzalez, Michelle M. Moksa, Theodore Smith, Justin White, Alynn Shanks, Siyun L. Wang, and et al. 2024. "Genome-Wide CRISPR Screen Identifies KEAP1 Perturbation as a Vulnerability of ARID1A-Deficient Cells" Cancers 16, no. 17: 2949. https://doi.org/10.3390/cancers16172949
APA StyleFournier, L.-A., Kalantari, F., Wells, J. P., Lee, J. S., Trigo-Gonzalez, G., Moksa, M. M., Smith, T., White, J., Shanks, A., Wang, S. L., Su, E., Wang, Y., Huntsman, D. G., Hirst, M., & Stirling, P. C. (2024). Genome-Wide CRISPR Screen Identifies KEAP1 Perturbation as a Vulnerability of ARID1A-Deficient Cells. Cancers, 16(17), 2949. https://doi.org/10.3390/cancers16172949