Optimizing Timing of Intraperitoneal Chemotherapy to Enhance Intravenous Carboplatin Concentration

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
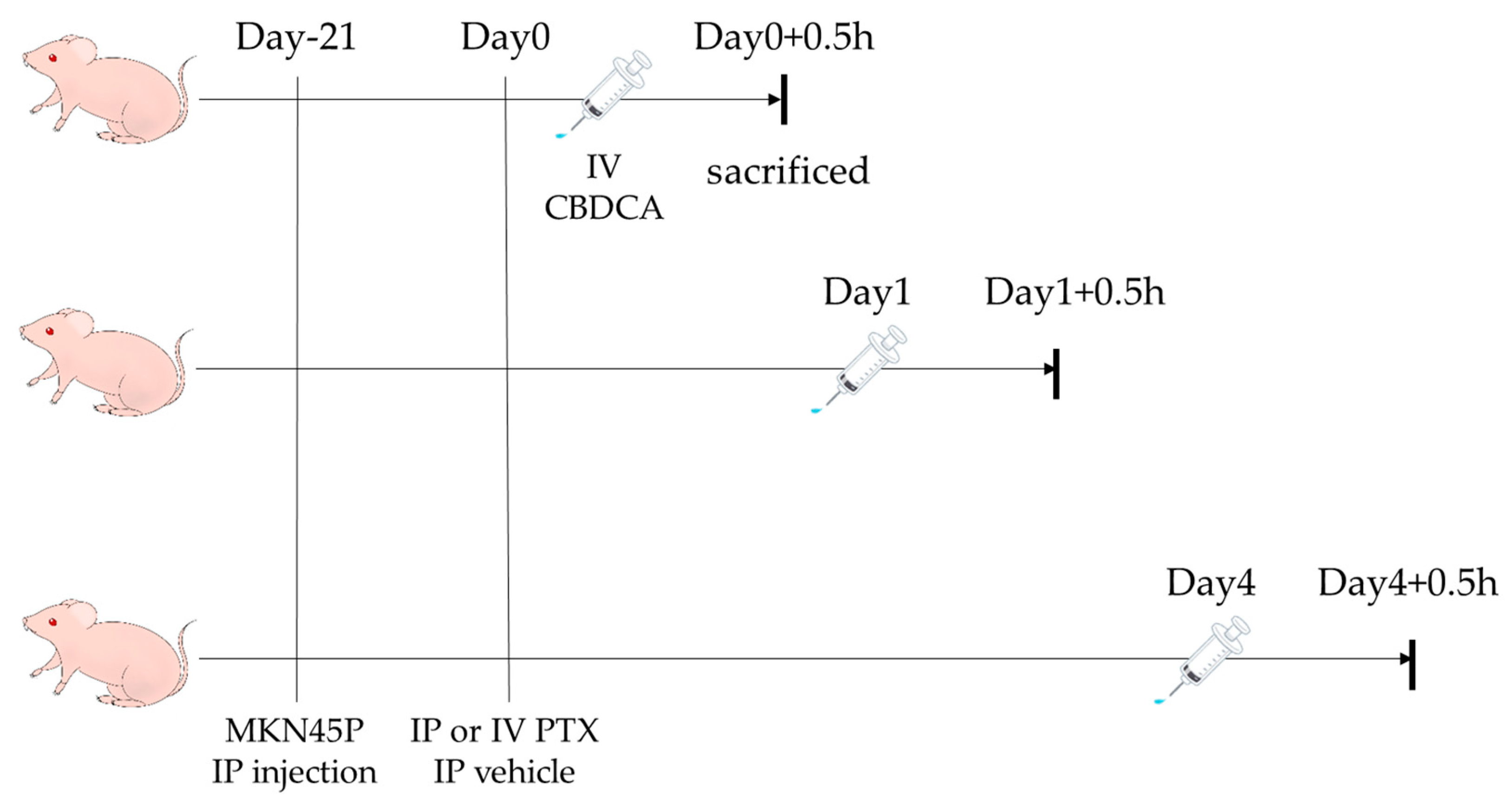
2. Materials and Methods
2.1. Cell Culture
2.2. Drugs and Animals
2.3. Measurement of PTX and CBDCA Concentrations in Tumors with LC-MS/MS
2.4. Preparation of Tumor Samples
2.5. Preparation of Calibration Standards and QC Samples
2.6. Analysis of PTX
2.7. Analysis of CBDCA
2.8. Ethics
2.9. Statistical Analysis
3. Results
3.1. Concentrations of PTX in Peritoneal Metastases of Gastric Cancer
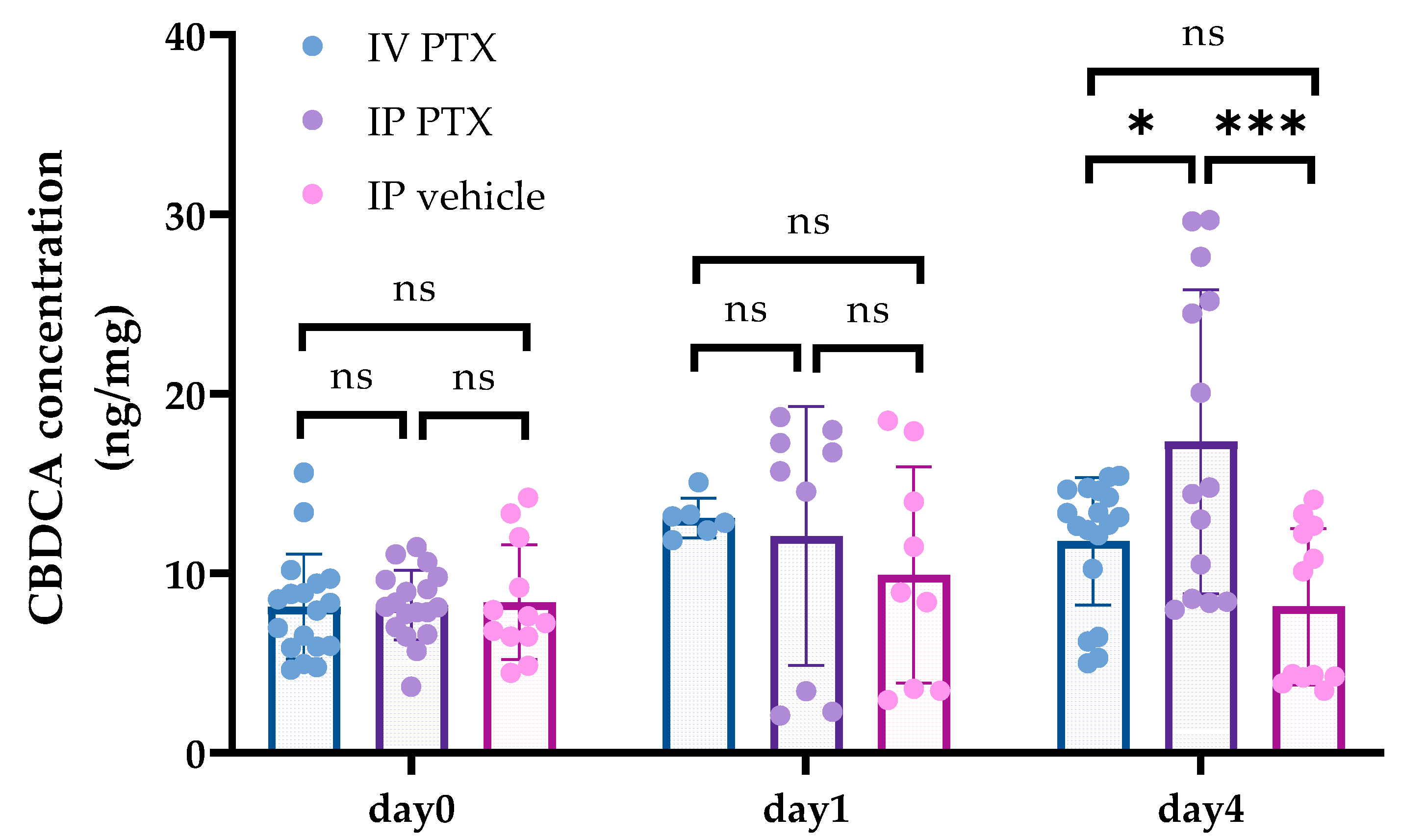
3.2. Concentrations of CBDCA in Tumors after PTX Administration
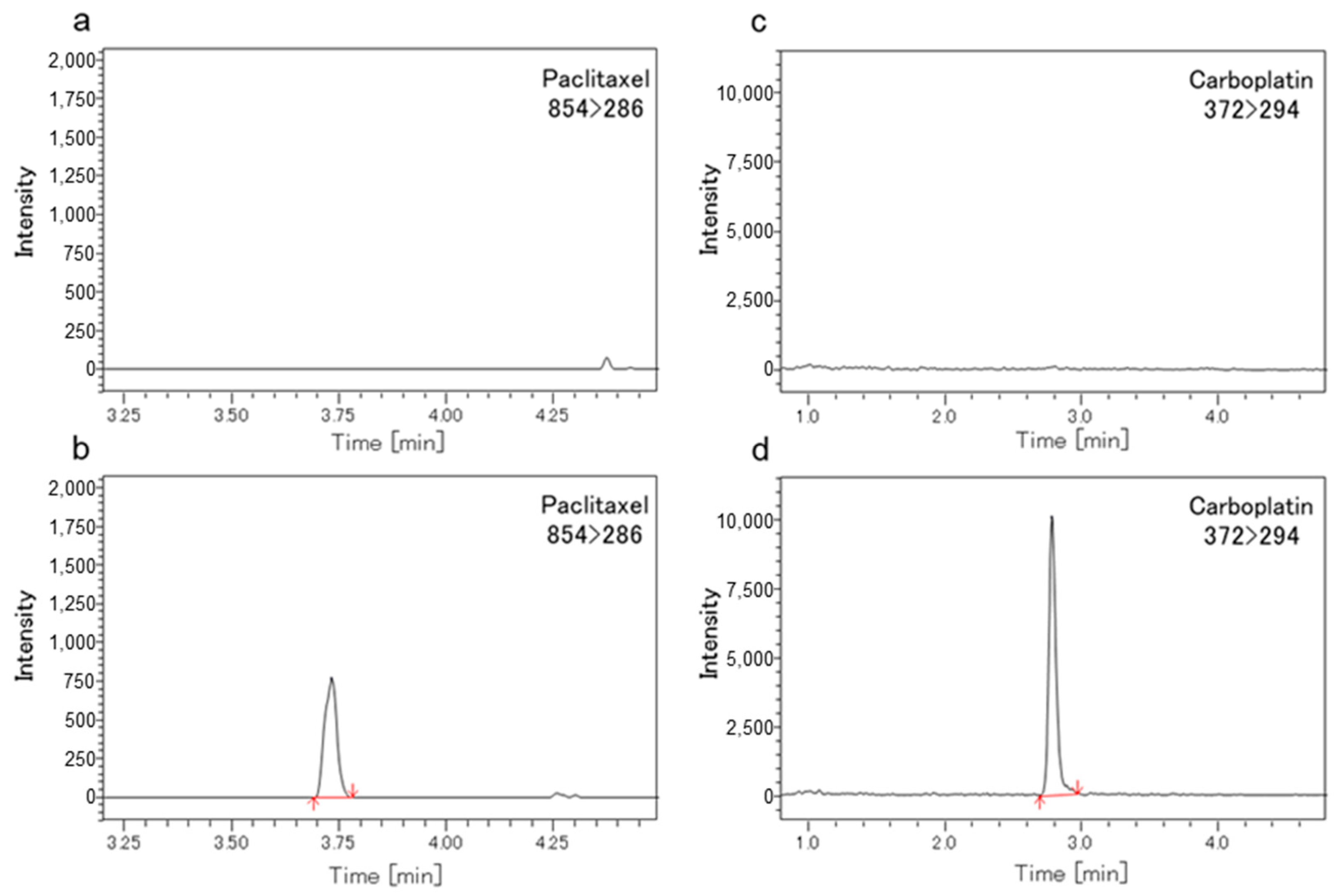
3.3. Methods Validation of LC-MS/MS
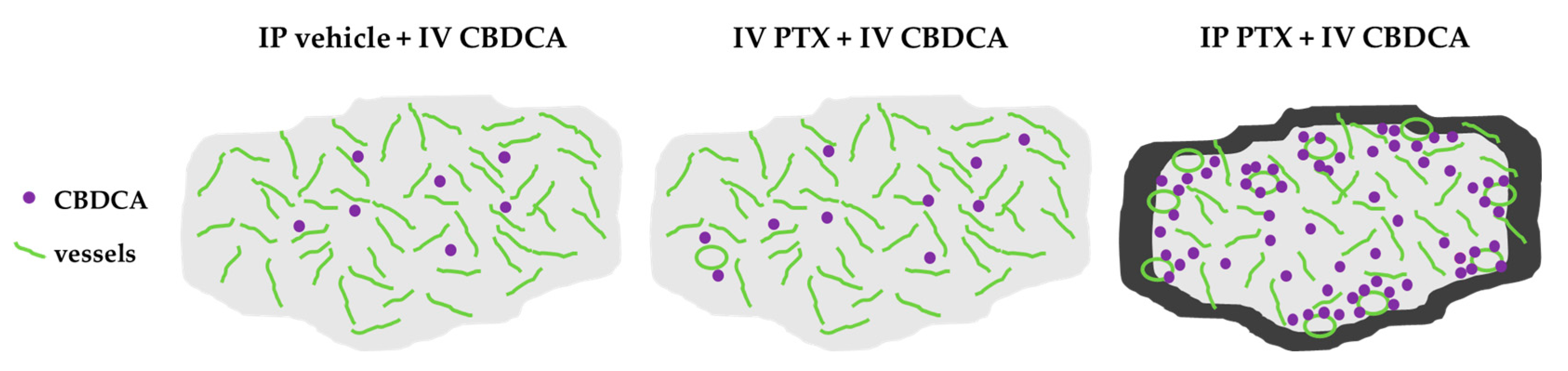
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Yarema, R.; Ohorchak, M.; Hyrya, P.; Kovalchuk, Y.; Safiyan, V.; Karelin, I.; Ferneza, S.; Fetsych, M.; Matusyak, M.; Oliynyk, Y.; et al. Gastric cancer with peritoneal metastases: Efficiency of standard treatment methods. World J. Gastrointest. Oncol. 2020, 12, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, R.; Fujitani, K.; Sakamaki, K.; Ando, M.; Ito, Y.; Tanizawa, Y.; Yamada, T.; Hirao, M.; Yamada, M.; Hihara, J.; et al. Survival analysis of a prospective multicenter observational study on surgical palliation among patients with malignant bowel obstruction caused by peritoneal dissemination of gastric cancer. Gastric Cancer 2022, 25, 422–429. [Google Scholar] [CrossRef]
- Yoo, C.H.; Noh, S.H.; Shin, D.W.; Choi, S.H.; Min, J.S. Recurrence following curative resection for gastric carcinoma. Br. J. Surg. 2000, 87, 236–242. [Google Scholar] [CrossRef]
- Roviello, F.; Marrelli, D.; de Manzoni, G.; Morgagni, P.; Di Leo, A.; Saragoni, L.; De Stefano, A. Prospective study of peritoneal recurrence after curative surgery for gastric cancer. Br. J. Surg. 2003, 90, 1113–1119. [Google Scholar] [CrossRef]
- Bilici, A.; Selcukbiricik, F. Prognostic significance of the recurrence pattern and risk factors for recurrence in patients with proximal gastric cancer who underwent curative gastrectomy. Tumour Biol. 2015, 36, 6191–6199. [Google Scholar] [CrossRef]
- Koizumi, W.; Narahara, H.; Hara, T.; Takagane, A.; Akiya, T.; Takagi, M.; Miyashita, K.; Nishizaki, T.; Kobayashi, O.; Takiyama, W.; et al. S-1 plus cisplatin versus S-1 alone for first-line treatment of advanced gastric cancer (SPIRITS trial): A phase III trial. Lancet Oncol. 2008, 9, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.K.; Kang, W.K.; Shin, D.B.; Chen, J.; Xiong, J.; Wang, J.; Lichinitser, M.; Guan, Z.; Khasanov, R.; Zheng, L.; et al. Capecitabine/cisplatin versus 5-fluorouracil/cisplatin as first-line therapy in patients with advanced gastric cancer: A randomised phase III noninferiority trial. Ann. Oncol. 2009, 20, 666–673. [Google Scholar] [CrossRef]
- Fujitani, K.; Yang, H.K.; Mizusawa, J.; Kim, Y.W.; Terashima, M.; Han, S.U.; Iwasaki, Y.; Hyung, W.J.; Takagane, A.; Park, D.J.; et al. Gastrectomy plus chemotherapy versus chemotherapy alone for advanced gastric cancer with a single non-curable factor (REGATTA): A phase 3, randomised controlled trial. Lancet Oncol. 2016, 17, 309–318. [Google Scholar] [CrossRef]
- Shirao, K.; Boku, N.; Yamada, Y.; Yamaguchi, K.; Doi, T.; Goto, M.; Nasu, J.; Denda, T.; Hamamoto, Y.; Takashima, A.; et al. Randomized Phase III study of 5-fluorouracil continuous infusion vs. sequential methotrexate and 5-fluorouracil therapy in far advanced gastric cancer with peritoneal metastasis (JCOG0106). Jpn. J. Clin. Oncol. 2013, 43, 972–980. [Google Scholar] [CrossRef]
- Nakajima, T.K.; Yamaguchi, K.; Boku, N.; Hyodo, I.; Mizusawa, J.; Hara, H.; Nishina, T.; Sakamoto, T.; Shitara, K.; Shinozaki, K.; et al. Randomized phase II/III study of 5-fluorouracil/l-leucovorin versus 5-fluorouracil/l-leucovorin plus paclitaxel administered to patients with severe peritoneal metastases of gastric cancer (JCOG1108/WJOG7312G). Gastric Cancer. 2020, 23, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Jacquet, P.; Sugarbaker, P.H. Peritoneal-plasma barrier. Cancer Treat Res. 1996, 82, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Dedrick, R.L.; Myers, C.E.; Bungay, P.M.; DeVita, V.T., Jr. Pharmacokinetic rationale for peritoneal drug administration in the treatment of ovarian cancer. Cancer Treat Rep. 1978, 62, 1–11. [Google Scholar] [PubMed]
- Los, G.; Mutsaers, P.H.; van der Vijgh, W.J.; Baldew, G.S.; de Graaf, P.W.; McVie, J.G. Direct diffusion of cis-diamminedichloroplatinum(II) in intraperitoneal rat tumors after intraperitoneal chemotherapy: A comparison with systemic chemotherapy. Cancer Res. 1989, 49, 3380–3384. [Google Scholar] [PubMed]
- Flessner, M.F.; Fenstermacher, J.D.; Blasberg, R.G.; Dedrick, R.L. Peritoneal absorption of macromolecules studied by quantitative autoradiography. Am. J. Physiol. 1985, 248, H26–H32. [Google Scholar] [CrossRef]
- Kamei, T.; Kitayama, J.; Yamaguchi, H.; Soma, D.; Emoto, S.; Konno, T.; Ishihara, K.; Ishigami, H.; Kaisaki, S.; Nagawa, H. Spatial distribution of intraperitoneally administrated paclitaxel nanoparticles solubilized with poly (2-methacryloxyethyl phosphorylcholine-co n-butyl methacrylate) in peritoneal metastatic nodules. Cancer Sci. 2011, 102, 200–205. [Google Scholar] [CrossRef]
- Markman, M.; Rowinsky, E.; Hakes, T.; Reichman, B.; Jones, W.; Lewis, J.L., Jr.; Rubin, S.; Curtin, J.; Barakat, R.; Phillips, M. Phase I trial of intraperitoneal taxol: A Gynecoloic Oncology Group study. J. Clin. Oncol. 1992, 10, 1485–1491. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.K.; Bundy, B.; Wenzel, L.; Huang, H.Q.; Baergen, R.; Lele, S.; Copeland, L.J.; Walker, J.L.; Burger, R.A. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N. Engl. J. Med. 2006, 354, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Rufián, S.; Muñoz-Casares, F.C.; Briceño, J.; Díaz, C.J.; Rubio, M.J.; Ortega, R.; Ciria, R.; Morillo, M.; Aranda, E.; Muntané, J. Radical surgery-peritonectomy and intraoperative intraperitoneal chemotherapy for the treatment of peritoneal carcinomatosis in recurrent or primary ovarian cancer. J. Surg. Oncol. 2006, 94, 316–324. [Google Scholar] [CrossRef]
- Yang, S.; Feng, R.; Pan, Z.C.; Jiang, T.; Xu, Q.; Chen, Q. A Comparison of Intravenous plus Intraperitoneal Chemotherapy with Intravenous Chemotherapy Alone for the Treatment of Gastric Cancer: A Meta-Analysis. Sci. Rep. 2015, 5, 12538. [Google Scholar] [CrossRef]
- Ishigami, H.; Fujiwara, Y.; Fukushima, R.; Nashimoto, A.; Yabusaki, H.; Imano, M.; Imamoto, H.; Kodera, Y.; Uenosono, Y.; Amagai, K. Phase III Trial Comparing Intraperitoneal and Intravenous Paclitaxel Plus S-1 Versus Cisplatin Plus S-1 in Patients With Gastric Cancer With Peritoneal Metastasis: PHOENIX-GC Trial. J. Clin. Oncol. 2018, 36, 1922–1929. [Google Scholar] [CrossRef] [PubMed]
- Hosie, K.; Gilbert, J.A.; Kerr, D.; Brown, C.B.; Peers, E.M. Fluid dynamics in man of an intraperitoneal drug delivery solution: 4% icodextrin. Drug Deliv. 2001, 8, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, F.; Marchettini, P.; Stuart, O.A.; Sugarbaker, P.H. Pharmacokinetics and tissue distribution of intraperitoneal paclitaxel with different carrier solutions. Cancer Chemother. Pharmacol. 2003, 52, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Bregoli, L.; Movia, D.; Gavigan-Imedio, J.D.; Lysaght, J.; Reynolds, J.; Prina-Mello, A. Nanomedicine applied to translational oncology: A future perspective on cancer treatment. Nanomedicine 2016, 12, 81–103. [Google Scholar] [CrossRef]
- Yamashita, K.; Tsunoda, S.; Gunji, S.; Murakami, T.; Suzuki, T.; Tabata, Y.; Sakai, Y. Intraperitoneal chemotherapy for peritoneal metastases using sustained release formula of cisplatin-incorporated gelatin hydrogel granules. Surg. Today 2019, 49, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Nagao, S.; Fujiwara, K.; Yamamoto, K.; Tanabe, H.; Okamoto, A.; Takehara, K.; Saito, M.; Fujiwara, H.; Tan, D.S.P.; Yamaguchi, S.; et al. Intraperitoneal Carboplatin for Ovarian Cancer—A Phase 2/3 Trial. NEJM Evid. 2023, 2, EVIDoa2200225. [Google Scholar] [CrossRef] [PubMed]
- Sako, A.; Kitayama, J.; Koyama, H.; Ueno, H.; Uchida, H.; Hamada, H.; Nagawa, H. Transduction of soluble Flt-1 gene to peritoneal mesothelial cells can effectively suppress peritoneal metastasis of gastric cancer. Cancer Res. 2004, 64, 3624–3628. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Kimura, N.; Kaneda, Y.; Ohzawa, H.; Miyato, H.; Yamaguchi, H.; Lefor, A.K.; Nagai, R.; Sata, N.; Kitayama, J.; et al. Novel Drug Delivery Method Targeting Para-Aortic Lymph Nodes by Retrograde Infusion of Paclitaxel into Pigs’ Thoracic Duct. Cancers 2022, 14, 3753. [Google Scholar] [CrossRef]
- Francis, P.; Rowinsky, E.; Schneider, J.; Hakes, T.; Hoskins, W.; Markman, M. Phase I feasibility and pharmacologic study of weekly intraperitoneal paclitaxel: A Gynecologic Oncology Group pilot Study. J. Clin. Oncol. 1995, 13, 2961–2967. [Google Scholar] [CrossRef]
- Soma, D.; Kitayama, J.; Ishigami, H.; Kaisaki, S.; Nagawa, H. Different tissue distribution of paclitaxel with intravenous and intraperitoneal administration. J. Surg. Res. 2009, 155, 142–146. [Google Scholar] [CrossRef]
- Emoto, S.; Yamaguchi, H.; Kamei, T.; Ishigami, H.; Suhara, T.; Suzuki, Y.; Ito, T.; Kitayama, J.; Watanabe, T. Intraperitoneal administration of cisplatin via an in situ cross-linkable hyaluronic acid-based hydrogel for peritoneal dissemination of gastric cancer. Surg. Today 2014, 44, 919–926. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.; Guo, J.; Holleran, J.L.; Kiesel, B.; Taylor, S.; Christner, S.; Parise, R.A.; Miller, B.M.; Ivy, S.P.; Chu, E. Evaluation of the pharmacokinetic drug-drug interaction potential of iohexol, a renal filtration marker. Cancer Chemother. Pharmacol. 2020, 86, 535–545. [Google Scholar] [CrossRef]
- Jarvis, I.W.H.; Meczes, E.L.; Thomas, H.D.; Edmondson, R.J.; Veal, G.J.; Boddy, A.V.; Ottley, C.J.; Pearson, D.G.; Tilby, M.J. Therapy-induced carboplatin-DNA adduct levels in human ovarian tumours in relation to assessment of adduct measurement in mouse tissues. Biochem. Pharmacol. 2012, 83, 69–77. [Google Scholar] [CrossRef]
- Kitayama, J.; Emoto, S.; Yamaguchi, H.; Ishigami, H.; Watanabe, T. Intraperitoneal paclitaxel induces regression of peritoneal metastasis partly by destruction of peripheral microvessels. Cancer Chemother. Pharmacol. 2014, 73, 605–612. [Google Scholar] [CrossRef] [PubMed]
- El-Tanani, M.; Rabbani, S.A.; Babiker, R.; Rangraze, I.; Kapre, S.; Palakurthi, S.S.; Alnuqaydan, A.M.; Aljabali, A.A.; Rizzo, M.; El-Tanani, Y.; et al. Unraveling the tumor microenvironment: Insights into cancer metastasis and therapeutic strategies. Cancer Lett. 2024, 591, 216894. [Google Scholar] [CrossRef]
- Glehen, O.; Gilly, F.N.; Arvieux, C.; Cotte, E.; Boutitie, F.; Mansvelt, B.; Bereder, J.M.; Lorimier, G.; Quenet, F.; Elias, D.; et al. Peritoneal carcinomatosis from gastric cancer: A multi-institutional study of 159 patients treated by cytoreductive surgery combined with perioperative intraperitoneal chemotherapy. Ann. Surg. Oncol. 2010, 17, 2370–2377. [Google Scholar] [CrossRef]
- Rudloff, U.; Langan, R.C.; Mullinax, J.E.; Beane, J.D.; Steinberg, S.M.; Beresnev, T.; Webb, C.C.; Walker, M.; Toomey, M.A.; Schrump, D.; et al. Impact of maximal cytoreductive surgery plus regional heated intraperitoneal chemotherapy (HIPEC) on outcome of patients with peritoneal carcinomatosis of gastric origin: Results of the GYMSSA trial. J. Surg. Oncol. 2014, 110, 275–284. [Google Scholar] [CrossRef]
- Yonemura, Y.; Kawamura, T.; Bandou, E.; Takahashi, S.; Sawa, T.; Matsuki, N. Treatment of peritoneal dissemination from gastric cancer by peritonectomy and chemohyperthermic peritoneal perfusion. Br. J. Surg. 2005, 92, 370–375. [Google Scholar] [CrossRef]
- Murono, K.; Nozawa, H.; Nagata, H.; Ishimaru, K.; Sonoda, H.; Emoto, S.; Kaneko, M.; Sasaki, K.; Otani, K.; Kawai, K. Efficacy of intraperitoneally administered paclitaxel for colorectal cancer with peritoneal metastases. Int. J. Color. Dis. 2020, 35, 1945–1949. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tamura, K.; Kimura, N.; Ohzawa, H.; Miyato, H.; Sata, N.; Koyanagi, T.; Saga, Y.; Takei, Y.; Fujiwara, H.; Nagai, R.; et al. Optimizing Timing of Intraperitoneal Chemotherapy to Enhance Intravenous Carboplatin Concentration. Cancers 2024, 16, 2841. https://doi.org/10.3390/cancers16162841
Tamura K, Kimura N, Ohzawa H, Miyato H, Sata N, Koyanagi T, Saga Y, Takei Y, Fujiwara H, Nagai R, et al. Optimizing Timing of Intraperitoneal Chemotherapy to Enhance Intravenous Carboplatin Concentration. Cancers. 2024; 16(16):2841. https://doi.org/10.3390/cancers16162841
Chicago/Turabian StyleTamura, Kohei, Natsuka Kimura, Hideyuki Ohzawa, Hideyo Miyato, Naohiro Sata, Takahiro Koyanagi, Yasushi Saga, Yuji Takei, Hiroyuki Fujiwara, Ryozo Nagai, and et al. 2024. "Optimizing Timing of Intraperitoneal Chemotherapy to Enhance Intravenous Carboplatin Concentration" Cancers 16, no. 16: 2841. https://doi.org/10.3390/cancers16162841
APA StyleTamura, K., Kimura, N., Ohzawa, H., Miyato, H., Sata, N., Koyanagi, T., Saga, Y., Takei, Y., Fujiwara, H., Nagai, R., Kitayama, J., & Aizawa, K. (2024). Optimizing Timing of Intraperitoneal Chemotherapy to Enhance Intravenous Carboplatin Concentration. Cancers, 16(16), 2841. https://doi.org/10.3390/cancers16162841