
Decoding Clonal Hematopoiesis: Emerging Themes and Novel Mechanistic Insights


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Early Evidence for Clonal Expansion
3. Identifying the Driver of Clonal Expansion: The Genomic Revolution
4. Cell of Origin
5. Mechanisms of Clonal Expansion
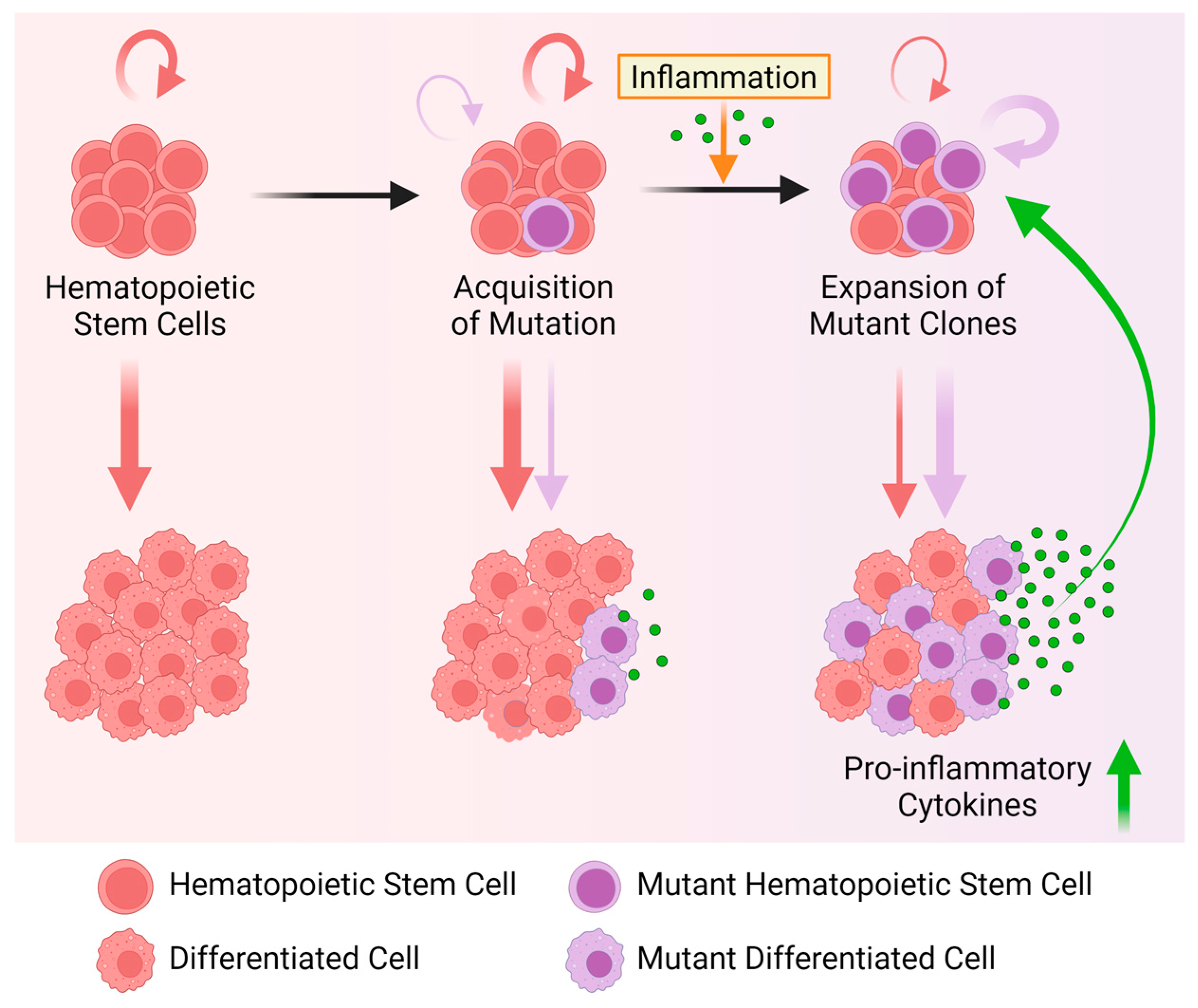
6. Inflammation and Clonal Hematopoiesis
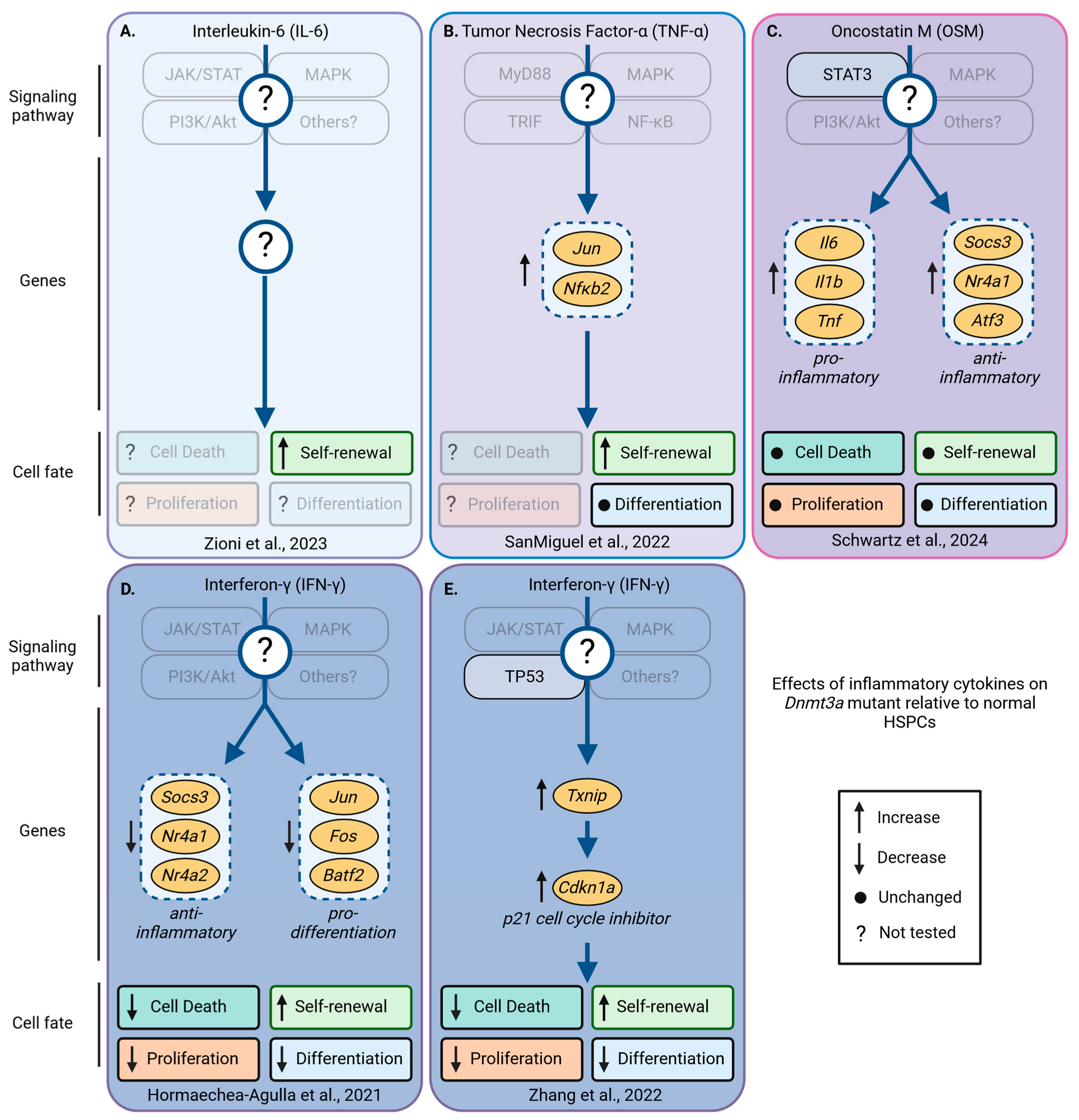
6.1. Mutant Mature Myeloid Cells Produce Aberrant Levels of Inflammatory Cytokines
6.2. Changes in the Adaptive Immune System
6.3. An Anti-Inflammatory Program Protects Mutant HSPCs
7. Changes in Normal and Mutant HSC Fate Decisions
7.1. Improved Survival of Mutant Clones
7.2. Changes in Proliferation of Mutant Clones
7.3. Self-Renewal and Differentiation
8. DNA Methylation
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jagannathan-Bogdan, M.; Zon, L.I. Hematopoiesis. Development 2013, 140, 2463–2467. [Google Scholar] [CrossRef] [PubMed]
- de Haan, G.; Lazare, S.S. Aging of hematopoietic stem cells. Blood 2018, 131, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Mitchell, E.; Chapman, M.S.; Williams, N.; Dawson, K.J.; Mende, N.; Calderbank, E.F.; Jung, H.; Mitchell, T.; Coorens, T.H.H.; Spencer, D.H.; et al. Clonal dynamics of haematopoiesis across the human lifespan. Nature 2022, 606, 343–350. [Google Scholar]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [PubMed]
- Shlush, L.I. Age-related clonal hematopoiesis. Blood 2018, 131, 496–504. [Google Scholar] [PubMed]
- Trowbridge, J.J.; Starczynowski, D.T. Innate immune pathways and inflammation in hematopoietic aging, clonal hematopoiesis, and MDS. J. Exp. Med. 2021, 218, e20201544. [Google Scholar] [CrossRef]
- Warren, J.T.; Link, D.C. Clonal hematopoiesis and risk for hematologic malignancy. Blood 2020, 136, 1599–1605. [Google Scholar]
- von Beck, K.; von Beck, T.; Ferrell, P.B.; Bick, A.G.; Kishtagari, A. Lymphoid clonal hematopoiesis: Implications for malignancy, immunity, and treatment. Blood Cancer J. 2023, 13, 5. [Google Scholar] [CrossRef]
- Vlasschaert, C.; Lanktree, M.B.; Rauh, M.J.; Kelly, T.N.; Natarajan, P. Clonal haematopoiesis, ageing and kidney disease. Nat. Rev. Nephrol. 2024, 20, 161–174. [Google Scholar] [CrossRef]
- Mendez, L.M.; Patnaik, M.M. Clonal Hematopoiesis: Origins and determinants of evolution. Leuk. Res. 2023, 129, 107076. [Google Scholar] [CrossRef] [PubMed]
- Fabre, M.A.; de Almeida, J.G.; Fiorillo, E.; Mitchell, E.; Damaskou, A.; Rak, J.; Orru, V.; Marongiu, M.; Chapman, M.S.; Vijayabaskar, M.S.; et al. The longitudinal dynamics and natural history of clonal haematopoiesis. Nature 2022, 606, 335–342. [Google Scholar]
- Bolton, K.L.; Koh, Y.; Foote, M.B.; Im, H.; Jee, J.; Sun, C.H.; Safonov, A.; Ptashkin, R.; Moon, J.H.; Lee, J.Y.; et al. Clonal hematopoiesis is associated with risk of severe COVID-19. Nat. Commun. 2021, 12, 5975. [Google Scholar]
- Lyon, M.F. Sex chromatin and gene action in the mammalian X-chromosome. Am. J. Hum. Genet. 1962, 14, 135–148. [Google Scholar]
- Beutler, E.; Yeh, M.; Fairbanks, V.F. The normal human female as a mosaic of X-chromosome activity: Studies using the gene for C-6-PD-deficiency as a marker. Proc. Natl. Acad. Sci. USA 1962, 48, 9–16. [Google Scholar]
- Linder, D.; Gartler, S.M. Glucose-6-phosphate dehydrogenase mosaicism: Utilization as a cell marker in the study of leiomyomas. Science 1965, 150, 67–69. [Google Scholar]
- Fialkow, P.J.; Gartler, S.M.; Yoshida, A. Clonal origin of chronic myelocytic leukemia in man. Proc. Natl. Acad. Sci. USA 1967, 58, 1468–1471. [Google Scholar]
- Allen, R.C.; Zoghbi, H.Y.; Moseley, A.B.; Rosenblatt, H.M.; Belmont, J.W. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am. J. Hum. Genet. 1992, 51, 1229–1239. [Google Scholar] [PubMed]
- Gale, R.E.; Wheadon, H.; Boulos, P.; Linch, D.C. Tissue specificity of X-chromosome inactivation patterns. Blood 1994, 83, 2899–2905. [Google Scholar] [PubMed]
- Champion, K.M.; Gilbert, J.G.; Asimakopoulos, F.A.; Hinshelwood, S.; Green, A.R. Clonal haemopoiesis in normal elderly women: Implications for the myeloproliferative disorders and myelodysplastic syndromes. Br. J. Haematol. 1997, 97, 920–926. [Google Scholar]
- Fey, M.F.; Liechti-Gallati, S.; von Rohr, A.; Borisch, B.; Theilkäs, L.; Schneider, V.; Oestreicher, M.; Nagel, S.; Ziemiecki, A.; Tobler, A. Clonality and X-inactivation patterns in hematopoietic cell populations detected by the highly informative M27 beta DNA probe. Blood 1994, 83, 931–938. [Google Scholar] [PubMed]
- Ayachi, S.; Buscarlet, M.; Busque, L. 60 Years of clonal hematopoiesis research: From X-chromosome inactivation studies to the identification of driver mutations. Exp. Hematol. 2020, 83, 2–11. [Google Scholar] [PubMed]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [PubMed]
- Min, K.D.; Polizio, A.H.; Kour, A.; Thel, M.C.; Walsh, K. Experimental ASXL1-Mediated Clonal Hematopoiesis Promotes Inflammation and Accelerates Heart Failure. J. Am. Heart Assoc. 2022, 11, e026154. [Google Scholar]
- Jongen-Lavrencic, M.; Grob, T.; Hanekamp, D.; Kavelaars, F.G.; Al Hinai, A.; Zeilemaker, A.; Erpelinck-Verschueren, C.A.J.; Gradowska, P.L.; Meijer, R.; Cloos, J.; et al. Molecular Minimal Residual Disease in Acute Myeloid Leukemia. N. Engl. J. Med. 2018, 378, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Pløen, G.G.; Nederby, L.; Guldberg, P.; Hansen, M.; Ebbesen, L.H.; Jensen, U.B.; Hokland, P.; Aggerholm, A. Persistence of DNMT3A mutations at long-term remission in adult patients with AML. Br. J. Haematol. 2014, 167, 478–486. [Google Scholar] [PubMed]
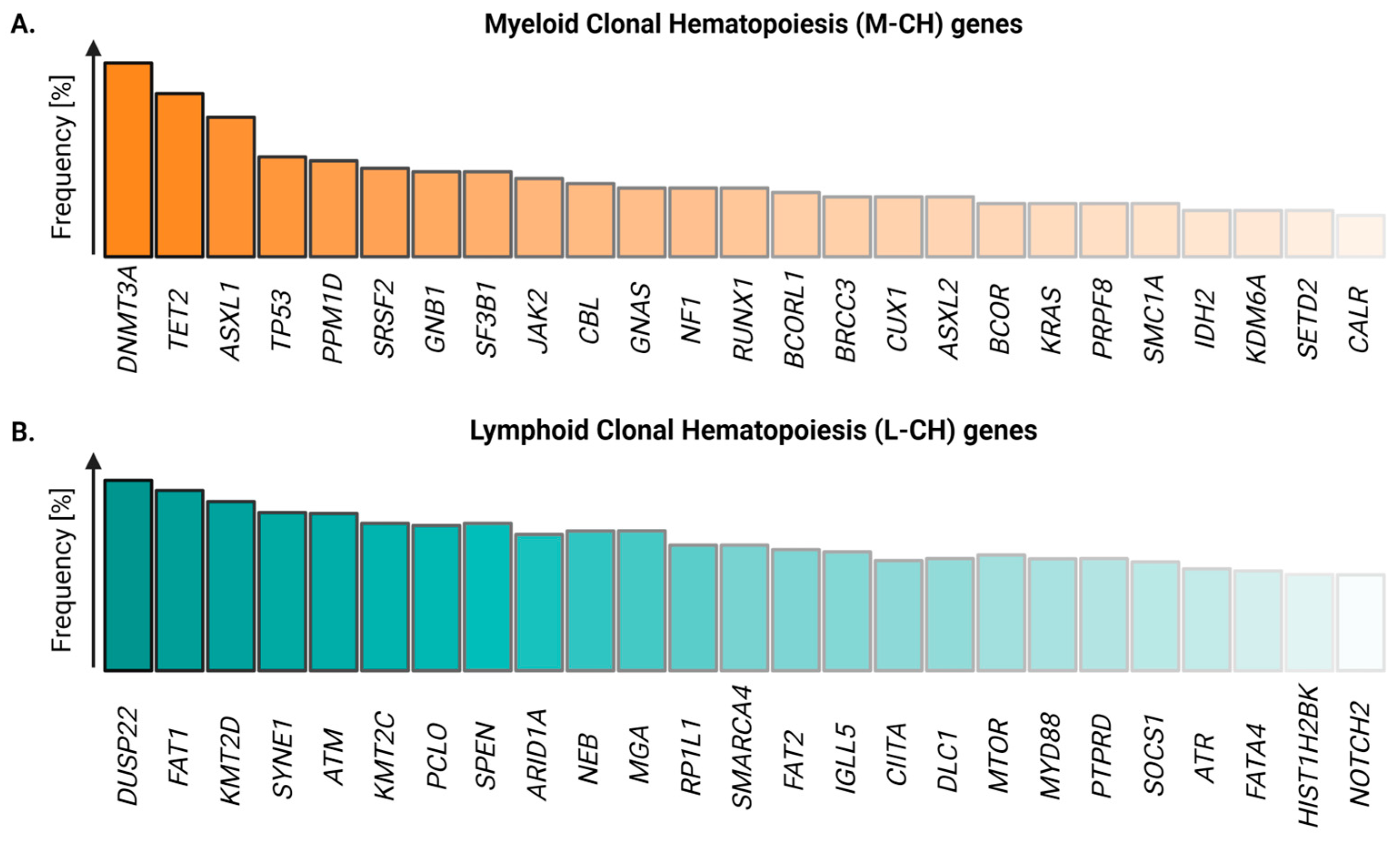
- Niroula, A.; Sekar, A.; Murakami, M.A.; Trinder, M.; Agrawal, M.; Wong, W.J.; Bick, A.G.; Uddin, M.M.; Gibson, C.J.; Griffin, G.K.; et al. Distinction of lymphoid and myeloid clonal hematopoiesis. Nat. Med. 2021, 27, 1921–1927. [Google Scholar] [PubMed]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef]
- Young, A.L.; Challen, G.A.; Birmann, B.M.; Druley, T.E. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat. Commun. 2016, 7, 12484. [Google Scholar]
- Miller, P.G.; Qiao, D.; Rojas-Quintero, J.; Honigberg, M.C.; Sperling, A.S.; Gibson, C.J.; Bick, A.G.; Niroula, A.; McConkey, M.E.; Sandoval, B.; et al. Association of clonal hematopoiesis with chronic obstructive pulmonary disease. Blood 2022, 139, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [PubMed]
- Acuna-Hidalgo, R.; Sengul, H.; Steehouwer, M.; van de Vorst, M.; Vermeulen, S.H.; Kiemeney, L.; Veltman, J.A.; Gilissen, C.; Hoischen, A. Ultra-sensitive Sequencing Identifies High Prevalence of Clonal Hematopoiesis-Associated Mutations throughout Adult Life. Am. J. Hum. Genet. 2017, 101, 50–64. [Google Scholar] [PubMed]
- Uddin, M.M.; Zhou, Y.; Bick, A.G.; Burugula, B.B.; Jaiswal, S.; Desai, P.; Honigberg, M.C.; Love, S.A.; Barac, A.; Hayden, K.M.; et al. Longitudinal profiling of clonal hematopoiesis provides insight into clonal dynamics. Immun. Ageing 2022, 19, 23. [Google Scholar] [PubMed]
- Robertson, N.A.; Latorre-Crespo, E.; Terradas-Terradas, M.; Lemos-Portela, J.; Purcell, A.C.; Livesey, B.J.; Hillary, R.F.; Murphy, L.; Fawkes, A.; MacGillivray, L.; et al. Longitudinal dynamics of clonal hematopoiesis identifies gene-specific fitness effects. Nat. Med. 2022, 28, 1439–1446. [Google Scholar] [PubMed]
- Andersen, M.A.; Bjerrum, O.W.; Ranjan, A.; Skov, V.; Kruse, T.A.; Thomassen, M.; Skytthe, A.; Hasselbalch, H.C.; Christensen, K. Myeloproliferative Neoplasms in Danish Twins. Acta Haematol. 2018, 139, 195–198. [Google Scholar] [PubMed]
- Kohnke, T.; Majeti, R. Clonal Hematopoiesis: From Mechanisms to Clinical Intervention. Cancer Discov. 2021, 11, 2987–2997. [Google Scholar] [PubMed]
- Lee-Six, H.; Øbro, N.F.; Shepherd, M.S.; Grossmann, S.; Dawson, K.; Belmonte, M.; Osborne, R.J.; Huntly, B.J.P.; Martincorena, I.; Anderson, E.; et al. Population dynamics of normal human blood inferred from somatic mutations. Nature 2018, 561, 473–478. [Google Scholar]
- Calvanese, V.; Capellera-Garcia, S.; Ma, F.; Fares, I.; Liebscher, S.; Ng, E.S.; Ekstrand, S.; Aguadé-Gorgorió, J.; Vavilina, A.; Lefaudeux, D.; et al. Mapping human haematopoietic stem cells from haemogenic endothelium to birth. Nature 2022, 604, 534–540. [Google Scholar]
- Williams, N.; Lee, J.; Mitchell, E.; Moore, L.; Baxter, E.J.; Hewinson, J.; Dawson, K.J.; Menzies, A.; Godfrey, A.L.; Green, A.R.; et al. Life histories of myeloproliferative neoplasms inferred from phylogenies. Nature 2022, 602, 162–168. [Google Scholar]
- Fabre, M.A.; McKerrell, T.; Zwiebel, M.; Vijayabaskar, M.S.; Park, N.; Wells, P.M.; Rad, R.; Deloukas, P.; Small, K.; Steves, C.J.; et al. Concordance for clonal hematopoiesis is limited in elderly twins. Blood 2020, 135, 269–273. [Google Scholar]
- Behjati, S.; Huch, M.; van Boxtel, R.; Karthaus, W.; Wedge, D.C.; Tamuri, A.U.; Martincorena, I.; Petljak, M.; Alexandrov, L.B.; Gundem, G.; et al. Genome sequencing of normal cells reveals developmental lineages and mutational processes. Nature 2014, 513, 422–425. [Google Scholar] [PubMed]
- Welch, J.S.; Ley, T.J.; Link, D.C.; Miller, C.A.; Larson, D.E.; Koboldt, D.C.; Wartman, L.D.; Lamprecht, T.L.; Liu, F.; Xia, J.; et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012, 150, 264–278. [Google Scholar] [PubMed]
- Tovy, A.; Reyes, J.M.; Gundry, M.C.; Brunetti, L.; Lee-Six, H.; Petljak, M.; Park, H.J.; Guzman, A.G.; Rosas, C.; Jeffries, A.R.; et al. Tissue-Biased Expansion of DNMT3A-Mutant Clones in a Mosaic Individual Is Associated with Conserved Epigenetic Erosion. Cell Stem Cell 2020, 27, 326–335.e4. [Google Scholar] [PubMed]
- Waldvogel, S.M.; Posey, J.E.; Goodell, M.A. Human embryonic genetic mosaicism and its effects on development and disease. Nat. Rev. Genet. 2024. ahead of print. [Google Scholar]
- Dykstra, B.; Kent, D.; Bowie, M.; McCaffrey, L.; Hamilton, M.; Lyons, K.; Lee, S.J.; Brinkman, R.; Eaves, C. Long-term propagation of distinct hematopoietic differentiation programs in vivo. Cell Stem Cell 2007, 1, 218–229. [Google Scholar]
- Sieburg, H.B.; Cho, R.H.; Dykstra, B.; Uchida, N.; Eaves, C.J.; Muller-Sieburg, C.E. The hematopoietic stem compartment consists of a limited number of discrete stem cell subsets. Blood 2006, 107, 2311–2316. [Google Scholar]
- Jurecic, R. Hematopoietic Stem Cell Heterogeneity. Adv. Exp. Med. Biol. 2019, 1169, 195–211. [Google Scholar]
- Challen, G.A.; Boles, N.C.; Chambers, S.M.; Goodell, M.A. Distinct hematopoietic stem cell subtypes are differentially regulated by TGF-beta1. Cell Stem Cell 2010, 6, 265–278. [Google Scholar]
- Zink, F.; Stacey, S.N.; Norddahl, G.L.; Frigge, M.L.; Magnusson, O.T.; Jonsdottir, I.; Thorgeirsson, T.E.; Sigurdsson, A.; Gudjonsson, S.A.; Gudmundsson, J.; et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 2017, 130, 742–752. [Google Scholar]
- Bowman, R.L.; Busque, L.; Levine, R.L. Clonal Hematopoiesis and Evolution to Hematopoietic Malignancies. Cell Stem Cell 2018, 22, 157–170. [Google Scholar] [PubMed]
- Challen, G.A.; Goodell, M.A. Clonal hematopoiesis: Mechanisms driving dominance of stem cell clones. Blood 2020, 136, 1590–1598. [Google Scholar] [PubMed]
- Avagyan, S.; Zon, L.I. Clonal hematopoiesis and inflammation—The perpetual cycle. Trends Cell Biol. 2023, 33, 695–707. [Google Scholar] [PubMed]
- Florez, M.A.; Tran, B.T.; Wathan, T.K.; DeGregori, J.; Pietras, E.M.; King, K.Y. Clonal hematopoiesis: Mutation-specific adaptation to environmental change. Cell Stem Cell 2022, 29, 882–904. [Google Scholar] [PubMed]
- Chen, S.; Benbarche, S.; Abdel-Wahab, O. Splicing factor mutations in hematologic malignancies. Blood 2021, 138, 599–612. [Google Scholar]
- Chen, C.W.; Zhang, L.; Dutta, R.; Niroula, A.; Miller, P.G.; Gibson, C.J.; Bick, A.G.; Reyes, J.M.; Lee, Y.T.; Tovy, A.; et al. SRCAP mutations drive clonal hematopoiesis through epigenetic and DNA repair dysregulation. Cell Stem Cell 2023, 30, 1503–1519.e8. [Google Scholar] [PubMed]
- Lin, S.; Zhao, R.; Xiao, Y.; Li, P. Mechanisms determining the fate of hematopoietic stem cells. Stem Cell Investig. 2015, 2, 10. [Google Scholar] [PubMed]
- Lee, Y.; Decker, M.; Lee, H.; Ding, L. Extrinsic regulation of hematopoietic stem cells in development, homeostasis and diseases. Wiley Interdiscip. Rev. Dev. Biol. 2017, 6, e279. [Google Scholar] [CrossRef]
- Baldridge, M.T.; King, K.Y.; Boles, N.C.; Weksberg, D.C.; Goodell, M.A. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature 2010, 465, 793–797. [Google Scholar]
- Essers, M.A.; Offner, S.; Blanco-Bose, W.E.; Waibler, Z.; Kalinke, U.; Duchosal, M.A.; Trumpp, A. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature 2009, 458, 904–908. [Google Scholar]
- Wilson, A.; Laurenti, E.; Oser, G.; van der Wath, R.C.; Blanco-Bose, W.; Jaworski, M.; Offner, S.; Dunant, C.F.; Eshkind, L.; Bockamp, E.; et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell 2008, 135, 1118–1129. [Google Scholar] [CrossRef]
- Bogeska, R.; Mikecin, A.M.; Kaschutnig, P.; Fawaz, M.; Büchler-Schäff, M.; Le, D.; Ganuza, M.; Vollmer, A.; Paffenholz, S.V.; Asada, N.; et al. Inflammatory exposure drives long-lived impairment of hematopoietic stem cell self-renewal activity and accelerated aging. Cell Stem Cell 2022, 29, 1273–1284.e8. [Google Scholar] [PubMed]
- Michaud, M.; Balardy, L.; Moulis, G.; Gaudin, C.; Peyrot, C.; Vellas, B.; Cesari, M.; Nourhashemi, F. Proinflammatory cytokines, aging, and age-related diseases. J. Am. Med. Dir. Assoc. 2013, 14, 877–882. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, W.; Yi, L.; Zhao, M.; Li, P.-Y.; Fu, M.-H.; Lin, R.; Zhu, Y.-M.; Li, J.-F.; Yang, W.-P.; et al. The impact of age and number of mutations on the size of clonal hematopoiesis. Proc. Natl. Acad. Sci. USA 2024, 121, e2319364121. [Google Scholar]
- Bick, A.G.; Weinstock, J.S.; Nandakumar, S.K.; Fulco, C.P.; Bao, E.L.; Zekavat, S.M.; Szeto, M.D.; Liao, X.; Leventhal, M.J.; Nasser, J.; et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature 2020, 586, 763–768. [Google Scholar]
- Busque, L.; Sun, M.; Buscarlet, M.; Ayachi, S.; Zada, Y.F.; Provost, S.; Bourgoin, V.; Mollica, L.; Meisel, M.; Hinterleitner, R.; et al. High-sensitivity C-reactive protein is associated with clonal hematopoiesis of indeterminate potential. Blood Adv. 2020, 4, 2430–2438. [Google Scholar] [PubMed]
- Weeks, L.D.; Ebert, B.L. Causes and consequences of clonal hematopoiesis. Blood 2023, 142, 2235–2246. [Google Scholar] [PubMed]
- Seyfried, A.N.; Maloney, J.M.; MacNamara, K.C. Macrophages Orchestrate Hematopoietic Programs and Regulate HSC Function during Inflammatory Stress. Front. Immunol. 2020, 11, 1499. [Google Scholar]
- Fuster, J.J.; MacLauchlan, S.; Zuriaga, M.A.; Polackal, M.N.; Ostriker, A.C.; Chakraborty, R.; Wu, C.L.; Sano, S.; Muralidharan, S.; Rius, C.; et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017, 355, 842–847. [Google Scholar] [CrossRef]
- Yura, Y.; Miura-Yura, E.; Katanasaka, Y.; Min, K.D.; Chavkin, N.; Polizio, A.H.; Ogawa, H.; Horitani, K.; Doviak, H.; Evans, M.A.; et al. The Cancer Therapy-Related Clonal Hematopoiesis Driver Gene Ppm1d Promotes Inflammation and Non-Ischemic Heart Failure in Mice. Circ. Res. 2021, 129, 684–698. [Google Scholar] [CrossRef]
- Avagyan, S.; Henninger, J.E.; Mannherz, W.P.; Mistry, M.; Yoon, J.; Yang, S.; Weber, M.C.; Moore, J.L.; Zon, L.I. Resistance to inflammation underlies enhanced fitness in clonal hematopoiesis. Science 2021, 374, 768–772. [Google Scholar] [CrossRef] [PubMed]
- Quin, C.; DeJong, E.N.; Cook, E.K.; Luo, Y.Z.; Vlasschaert, C.; Sadh, S.; McNaughton, A.J.; Buttigieg, M.M.; Breznik, J.A.; Kennedy, A.E.; et al. Neutrophil-mediated innate immune resistance to bacterial pneumonia is dependent on Tet2 function. J. Clin. Investig. 2024, 134, e171002. [Google Scholar] [CrossRef]
- Bellissimo, D.C.; Chen, C.H.; Zhu, Q.; Bagga, S.; Lee, C.T.; He, B.; Wertheim, G.B.; Jordan, M.; Tan, K.; Worthen, G.S.; et al. Runx1 negatively regulates inflammatory cytokine production by neutrophils in response to Toll-like receptor signaling. Blood Adv. 2020, 4, 1145–1158. [Google Scholar]
- Leoni, C.; Montagner, S.; Rinaldi, A.; Bertoni, F.; Polletti, S.; Balestrieri, C.; Monticelli, S. Dnmt3a restrains mast cell inflammatory responses. Proc. Natl. Acad. Sci. USA 2017, 114, E1490–E1499. [Google Scholar] [CrossRef]
- Reszka, E.; Jabłońska, E.; Wieczorek, E.; Valent, P.; Arock, M.; Nilsson, G.; Nedoszytko, B.; Niedoszytko, M. Epigenetic Changes in Neoplastic Mast Cells and Potential Impact in Mastocytosis. Int. J. Mol. Sci. 2021, 22, 2964. [Google Scholar] [CrossRef] [PubMed]
- Traina, F.; Visconte, V.; Jankowska, A.M.; Makishima, H.; O’Keefe, C.L.; Elson, P.; Han, Y.; Hsieh, F.H.; Sekeres, M.A.; Mali, R.S.; et al. Single nucleotide polymorphism array lesions, TET2, DNMT3A, ASXL1 and CBL mutations are present in systemic mastocytosis. PLoS ONE 2012, 7, e43090. [Google Scholar] [CrossRef]
- Wattrus, S.J.; Smith, M.L.; Rodrigues, C.P.; Hagedorn, E.J.; Kim, J.W.; Budnik, B.; Zon, L.I. Quality assurance of hematopoietic stem cells by macrophages determines stem cell clonality. Science 2022, 377, 1413–1419. [Google Scholar] [CrossRef]
- Asada, S.; Kitamura, T. Clonal hematopoiesis and associated diseases: A review of recent findings. Cancer Sci. 2021, 112, 3962–3971. [Google Scholar] [CrossRef] [PubMed]
- Kersh, E.N.; Fitzpatrick, D.R.; Murali-Krishna, K.; Shires, J.; Speck, S.H.; Boss, J.M.; Ahmed, R. Rapid demethylation of the IFN-gamma gene occurs in memory but not naive CD8 T cells. J. Immunol. 2006, 176, 4083–4093. [Google Scholar] [CrossRef]
- Makar, K.W.; Pérez-Melgosa, M.; Shnyreva, M.; Weaver, W.M.; Fitzpatrick, D.R.; Wilson, C.B. Active recruitment of DNA methyltransferases regulates interleukin 4 in thymocytes and T cells. Nat. Immunol. 2003, 4, 1183–1190. [Google Scholar]
- Bruniquel, D.; Schwartz, R.H. Selective, stable demethylation of the interleukin-2 gene enhances transcription by an active process. Nat. Immunol. 2003, 4, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Leoni, C.; Vincenzetti, L.; Emming, S.; Monticelli, S. Epigenetics of T lymphocytes in health and disease. Swiss Med. Wkly. 2015, 145, w14191. [Google Scholar] [CrossRef] [PubMed]
- Biran, A.; Yin, S.; Kretzmer, H.; Hacken, E.T.; Parvin, S.; Lucas, F.; Uduman, M.; Gutierrez, C.; Dangle, N.; Billington, L.; et al. Activation of Notch and Myc Signaling via B-cell-Restricted Depletion of Dnmt3a Generates a Consistent Murine Model of Chronic Lymphocytic Leukemia. Cancer Res. 2021, 81, 6117–6130. [Google Scholar] [PubMed]
- Barwick, B.G.; Scharer, C.D.; Martinez, R.J.; Price, M.J.; Wein, A.N.; Haines, R.R.; Bally, A.P.R.; Kohlmeier, J.E.; Boss, J.M. B cell activation and plasma cell differentiation are inhibited by de novo DNA methylation. Nat. Commun. 2018, 9, 1900. [Google Scholar] [PubMed]
- Dominguez, P.M.; Ghamlouch, H.; Rosikiewicz, W.; Kumar, P.; Béguelin, W.; Fontán, L.; Rivas, M.A.; Pawlikowska, P.; Armand, M.; Mouly, E.; et al. TET2 Deficiency Causes Germinal Center Hyperplasia, Impairs Plasma Cell Differentiation, and Promotes B-cell Lymphomagenesis. Cancer Discov. 2018, 8, 1632–1653. [Google Scholar] [PubMed]
- Dennis, E.; Murach, M.; Blackburn, C.M.R.; Marshall, M.; Root, K.; Pattarabanjird, T.; Deroissart, J.; Erickson, L.D.; Binder, C.J.; Bekiranov, S.; et al. Loss of TET2 increases B-1 cell number and IgM production while limiting CDR3 diversity. Front. Immunol. 2024, 15, 1380641. [Google Scholar] [CrossRef] [PubMed]
- Prinzing, B.; Zebley, C.C.; Petersen, C.T.; Fan, Y.; Anido, A.A.; Yi, Z.; Nguyen, P.; Houke, H.; Bell, M.; Haydar, D.; et al. Deleting DNMT3A in CAR T cells prevents exhaustion and enhances antitumor activity. Sci. Transl. Med. 2021, 13, eabh0272. [Google Scholar] [PubMed]
- Fraietta, J.A.; Nobles, C.L.; Sammons, M.A.; Lundh, S.; Carty, S.A.; Reich, T.J.; Cogdill, A.P.; Morrissette, J.J.D.; DeNizio, J.E.; Reddy, S.; et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 2018, 558, 307–312. [Google Scholar]
- Matatall, K.A.; Jeong, M.; Chen, S.; Sun, D.; Chen, F.; Mo, Q.; Kimmel, M.; King, K.Y. Chronic Infection Depletes Hematopoietic Stem Cells through Stress-Induced Terminal Differentiation. Cell Rep. 2016, 17, 2584–2595. [Google Scholar]
- Schwartz, L.S.; Young, K.A.; Stearns, T.M.; Boyer, N.; Mujica, K.D.; Trowbridge, J.J. Transcriptional and functional consequences of Oncostatin M signaling on young Dnmt3a-mutant hematopoietic stem cells. Exp. Hematol. 2024, 130, 104131. [Google Scholar] [CrossRef]
- McClatchy, J.; Strogantsev, R.; Wolfe, E.; Lin, H.Y.; Mohammadhosseini, M.; Davis, B.A.; Eden, C.; Goldman, D.; Fleming, W.H.; Conley, P.; et al. Clonal hematopoiesis related TET2 loss-of-function impedes IL1β-mediated epigenetic reprogramming in hematopoietic stem and progenitor cells. Nat. Commun. 2023, 14, 8102. [Google Scholar] [PubMed]
- Hormaechea-Agulla, D.; Matatall, K.A.; Le, D.T.; Kain, B.; Long, X.; Kus, P.; Jaksik, R.; Challen, G.A.; Kimmel, M.; King, K.Y. Chronic infection drives Dnmt3a-loss-of-function clonal hematopoiesis via IFNγ signaling. Cell Stem Cell 2021, 28, 1428–1442.e6. [Google Scholar] [PubMed]
- Liu, Y.; Chen, Y.; Deng, X.; Zhou, J. ATF3 Prevents Stress-Induced Hematopoietic Stem Cell Exhaustion. Front. Cell Dev. Biol. 2020, 8, 585771. [Google Scholar] [CrossRef] [PubMed]
- Ushiki, T.; Huntington, N.D.; Glaser, S.P.; Kiu, H.; Georgiou, A.; Zhang, J.G.; Metcalf, D.; Nicola, N.A.; Roberts, A.W.; Alexander, W.S. Rapid Inflammation in Mice Lacking Both SOCS1 and SOCS3 in Hematopoietic Cells. PLoS ONE 2016, 11, e0162111. [Google Scholar]
- Jakobsen, N.A.; Turkalj, S.; Zeng, A.G.X.; Stoilova, B.; Metzner, M.; Rahmig, S.; Nagree, M.S.; Shah, S.; Moore, R.; Usukhbayar, B.; et al. Selective advantage of mutant stem cells in human clonal hematopoiesis is associated with attenuated response to inflammation and aging. Cell Stem Cell 2024. ahead of print. [Google Scholar]
- Fujino, T.; Goyama, S.; Sugiura, Y.; Inoue, D.; Asada, S.; Yamasaki, S.; Matsumoto, A.; Yamaguchi, K.; Isobe, Y.; Tsuchiya, A.; et al. Mutant ASXL1 induces age-related expansion of phenotypic hematopoietic stem cells through activation of Akt/mTOR pathway. Nat. Commun. 2021, 12, 1826. [Google Scholar] [PubMed]
- Zioni, N.; Bercovich, A.A.; Chapal-Ilani, N.; Bacharach, T.; Rappoport, N.; Solomon, A.; Avraham, R.; Kopitman, E.; Porat, Z.; Sacma, M.; et al. Inflammatory signals from fatty bone marrow support DNMT3A driven clonal hematopoiesis. Nat. Commun. 2023, 14, 2070. [Google Scholar] [CrossRef]
- San Miguel, J.M.; Eudy, E.; Loberg, M.A.; Young, K.A.; Mistry, J.J.; Mujica, K.D.; Schwartz, L.S.; Stearns, T.M.; Challen, G.A.; Trowbridge, J.J. Distinct Tumor Necrosis Factor Alpha Receptors Dictate Stem Cell Fitness versus Lineage Output in Dnmt3a-Mutant Clonal Hematopoiesis. Cancer Discov. 2022, 12, 2763–2773. [Google Scholar]
- Zhang, C.R.; Ostrander, E.L.; Kukhar, O.; Mallaney, C.; Sun, J.; Haussler, E.; Celik, H.; Koh, W.K.; King, K.Y.; Gontarz, P.; et al. Txnip Enhances Fitness of Dnmt3a-Mutant Hematopoietic Stem Cells via p21. Blood Cancer Discov. 2022, 3, 220–239. [Google Scholar] [CrossRef]
- Cai, Z.; Kotzin, J.J.; Ramdas, B.; Chen, S.; Nelanuthala, S.; Palam, L.R.; Pandey, R.; Mali, R.S.; Liu, Y.; Kelley, M.R.; et al. Inhibition of Inflammatory Signaling in Tet2 Mutant Preleukemic Cells Mitigates Stress-Induced Abnormalities and Clonal Hematopoiesis. Cell Stem Cell 2018, 23, 833–849.e5. [Google Scholar]
- Abegunde, S.O.; Buckstein, R.; Wells, R.A.; Rauh, M.J. An inflammatory environment containing TNFalpha favors Tet2-mutant clonal hematopoiesis. Exp. Hematol. 2018, 59, 60–65. [Google Scholar]
- Caiado, F.; Kovtonyuk, L.V.; Gonullu, N.G.; Fullin, J.; Boettcher, S.; Manz, M.G. Aging drives Tet2+/− clonal hematopoiesis via IL-1 signaling. Blood 2023, 141, 886–903. [Google Scholar] [PubMed]
- Xie, J.; Sheng, M.; Rong, S.; Zhou, D.; Wang, C.; Wu, W.; Huang, J.; Sun, Y.; Wang, Y.; Chen, P.; et al. STING activation in TET2-mutated hematopoietic stem/progenitor cells contributes to the increased self-renewal and neoplastic transformation. Leukemia 2023, 37, 2457–2467. [Google Scholar]
- Hsu, J.I.; Dayaram, T.; Tovy, A.; De Braekeleer, E.; Jeong, M.; Wang, F.; Zhang, J.; Heffernan, T.P.; Gera, S.; Kovacs, J.J.; et al. PPM1D Mutations Drive Clonal Hematopoiesis in Response to Cytotoxic Chemotherapy. Cell Stem Cell 2018, 23, 700–713.e6. [Google Scholar] [CrossRef]
- Bondar, T.; Medzhitov, R. p53-mediated hematopoietic stem and progenitor cell competition. Cell Stem Cell 2010, 6, 309–322. [Google Scholar] [CrossRef]
- Kowalczyk, M.S.; Tirosh, I.; Heckl, D.; Rao, T.N.; Dixit, A.; Haas, B.J.; Schneider, R.K.; Wagers, A.J.; Ebert, B.L.; Regev, A. Single-cell RNA-seq reveals changes in cell cycle and differentiation programs upon aging of hematopoietic stem cells. Genome Res. 2015, 25, 1860–1872. [Google Scholar]
- Challen, G.A.; Sun, D.; Jeong, M.; Luo, M.; Jelinek, J.; Berg, J.S.; Bock, C.; Vasanthakumar, A.; Gu, H.; Xi, Y.; et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet. 2011, 44, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Moran-Crusio, K.; Reavie, L.; Shih, A.; Abdel-Wahab, O.; Ndiaye-Lobry, D.; Lobry, C.; Figueroa, M.E.; Vasanthakumar, A.; Patel, J.; Zhao, X.; et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 2011, 20, 11–24. [Google Scholar] [PubMed]
- Ostrander, E.L.; Kramer, A.C.; Mallaney, C.; Celik, H.; Koh, W.K.; Fairchild, J.; Haussler, E.; Zhang, C.R.C.; Challen, G.A. Divergent Effects of Dnmt3a and Tet2 Mutations on Hematopoietic Progenitor Cell Fitness. Stem Cell Rep. 2020, 14, 551–560. [Google Scholar]
- Jeong, M.; Park, H.J.; Celik, H.; Ostrander, E.L.; Reyes, J.M.; Guzman, A.; Rodriguez, B.; Lei, Y.; Lee, Y.; Ding, L.; et al. Loss of Dnmt3a Immortalizes Hematopoietic Stem Cells In Vivo. Cell Rep. 2018, 23, 1–10. [Google Scholar]
- Ludwig, L.S.; Lareau, C.A.; Ulirsch, J.C.; Christian, E.; Muus, C.; Li, L.H.; Pelka, K.; Ge, W.; Oren, Y.; Brack, A.; et al. Lineage Tracing in Humans Enabled by Mitochondrial Mutations and Single-Cell Genomics. Cell 2019, 176, 1325–1339.e22. [Google Scholar] [PubMed]
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation 2011, 123, 2145–2156. [Google Scholar] [PubMed]
- Wiehle, L.; Raddatz, G.; Musch, T.; Dawlaty, M.M.; Jaenisch, R.; Lyko, F.; Breiling, A. Tet1 and Tet2 Protect DNA Methylation Canyons against Hypermethylation. Mol. Cell Biol. 2016, 36, 452–461. [Google Scholar] [PubMed]
- Momparler, R.L.; Bovenzi, V. DNA methylation and cancer. J. Cell Physiol. 2000, 183, 145–154. [Google Scholar] [PubMed]
- Liao, M.; Chen, R.; Yang, Y.; He, H.; Xu, L.; Jiang, Y.; Guo, Z.; He, W.; Jiang, H.; Wang, J. Aging-elevated inflammation promotes DNMT3A R878H-driven clonal hematopoiesis. Acta Pharm. Sin. B 2022, 12, 678–691. [Google Scholar]
- Wang, Y.; Sano, S.; Yura, Y.; Ke, Z.; Sano, M.; Oshima, K.; Ogawa, H.; Horitani, K.; Min, K.D.; Miura-Yura, E.; et al. Tet2-mediated clonal hematopoiesis in nonconditioned mice accelerates age-associated cardiac dysfunction. JCI Insight 2020, 5, e135204. [Google Scholar] [PubMed]
- Li, Z.; Cai, X.; Cai, C.L.; Wang, J.; Zhang, W.; Petersen, B.E.; Yang, F.C.; Xu, M. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 2011, 118, 4509–4518. [Google Scholar] [PubMed]
- Nagase, R.; Inoue, D.; Pastore, A.; Fujino, T.; Hou, H.A.; Yamasaki, N.; Goyama, S.; Saika, M.; Kanai, A.; Sera, Y.; et al. Expression of mutant Asxl1 perturbs hematopoiesis and promotes susceptibility to leukemic transformation. J. Exp. Med. 2018, 215, 1729–1747. [Google Scholar]
- Stadler, M.B.; Murr, R.; Burger, L.; Ivanek, R.; Lienert, F.; Schöler, A.; van Nimwegen, E.; Wirbelauer, C.; Oakeley, E.J.; Gaidatzis, D.; et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 2011, 480, 490–495. [Google Scholar] [CrossRef]
- Mitsumori, R.; Sakaguchi, K.; Shigemizu, D.; Mori, T.; Akiyama, S.; Ozaki, K.; Niida, S.; Shimoda, N. Lower DNA methylation levels in CpG island shores of CR1, CLU, and PICALM in the blood of Japanese Alzheimer’s disease patients. PLoS ONE 2020, 15, e0239196. [Google Scholar] [CrossRef]
- Edgar, R.; Tan, P.P.; Portales-Casamar, E.; Pavlidis, P. Meta-analysis of human methylomes reveals stably methylated sequences surrounding CpG islands associated with high gene expression. Epigenetics Chromatin 2014, 7, 28. [Google Scholar] [PubMed]
- Jeong, M.; Sun, D.; Luo, M.; Huang, Y.; Challen, G.A.; Rodriguez, B.; Zhang, X.; Chavez, L.; Wang, H.; Hannah, R.; et al. Large conserved domains of low DNA methylation maintained by Dnmt3a. Nat. Genet. 2014, 46, 17–23. [Google Scholar]
- Liu, Y.V.; Bassal, M.A.; Jayasinghe, M.K.; Lin, Q.X.X.; Wu, C.-S.; Tang, J.P.; Kwon, J.; Zhou, Q.; Tan, H.K.; Ebralidze, A.K.; et al. Targeted intragenic demethylation initiates chromatin rewiring for gene activation. bioRxiv 2024. bioRxiv:2020.07.16.205922. [Google Scholar]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes. Dev. 2011, 25, 1010–1022. [Google Scholar] [PubMed]
- Strichman-Almashanu, L.Z.; Lee, R.S.; Onyango, P.O.; Perlman, E.; Flam, F.; Frieman, M.B.; Feinberg, A.P. A genome-wide screen for normally methylated human CpG islands that can identify novel imprinted genes. Genome Res. 2002, 12, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Trino, S.; Zoppoli, P.; Carella, A.M.; Laurenzana, I.; Weisz, A.; Memoli, D.; Calice, G.; La Rocca, F.; Simeon, V.; Savino, L.; et al. DNA methylation dynamic of bone marrow hematopoietic stem cells after allogeneic transplantation. Stem Cell Res. Ther. 2019, 10, 138. [Google Scholar] [PubMed]
- Cypris, O.; Božić, T.; Wagner, W. Chicken or Egg: Is Clonal Hematopoiesis Primarily Caused by Genetic or Epigenetic Aberrations? Front. Genet. 2019, 10, 785. [Google Scholar]
- Zhang, X.; Su, J.; Jeong, M.; Ko, M.; Huang, Y.; Park, H.J.; Guzman, A.; Lei, Y.; Huang, Y.H.; Rao, A.; et al. DNMT3A and TET2 compete and cooperate to repress lineage-specific transcription factors in hematopoietic stem cells. Nat. Genet. 2016, 48, 1014–1023. [Google Scholar]
- Bergstedt, J.; Azzou, S.A.K.; Tsuo, K.; Jaquaniello, A.; Urrutia, A.; Rotival, M.; Lin, D.T.S.; MacIsaac, J.L.; Kobor, M.S.; Albert, M.L.; et al. The immune factors driving DNA methylation variation in human blood. Nat. Commun. 2022, 13, 5895. [Google Scholar]
- Refn, M.R.; Andersen, M.M.; Kampmann, M.-L.; Tfelt-Hansen, J.; Sørensen, E.; Larsen, M.H.; Morling, N.; Børsting, C.; Pereira, V. Longitudinal changes and variation in human DNA methylation analysed with the Illumina MethylationEPIC BeadChip assay and their implications on forensic age prediction. Sci. Rep. 2023, 13, 21658. [Google Scholar]
- Aanes, H.; Bleka, Ø.; Dahlberg, P.S.; Carm, K.T.; Lehtimäki, T.; Raitakari, O.; Kähönen, M.; Hurme, M.; Rolseth, V. A new blood based epigenetic age predictor for adolescents and young adults. Sci. Rep. 2023, 13, 2303. [Google Scholar] [PubMed]
- Salameh, Y.; Bejaoui, Y.; El Hajj, N. DNA Methylation Biomarkers in Aging and Age-Related Diseases. Front. Genet. 2020, 11, 171. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.H.; Marioni, R.E.; Colicino, E.; Peters, M.J.; Ward-Caviness, C.K.; Tsai, P.C.; Roetker, N.S.; Just, A.C.; Demerath, E.W.; Guan, W.; et al. DNA methylation-based measures of biological age: Meta-analysis predicting time to death. Aging 2016, 8, 1844–1865. [Google Scholar] [PubMed]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.B.; Gao, Y.; et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Bergsma, T.; Rogaeva, E. DNA Methylation Clocks and Their Predictive Capacity for Aging Phenotypes and Healthspan. Neurosci. Insights 2020, 15, 2633105520942221. [Google Scholar] [PubMed]
- Nachun, D.; Lu, A.T.; Bick, A.G.; Natarajan, P.; Weinstock, J.; Szeto, M.D.; Kathiresan, S.; Abecasis, G.; Taylor, K.D.; Guo, X.; et al. Clonal hematopoiesis associated with epigenetic aging and clinical outcomes. Aging Cell 2021, 20, e13366. [Google Scholar] [CrossRef] [PubMed]
- Feldkamp, J.D.; Vetter, V.M.; Arends, C.M.; Lang, T.J.L.; Bullinger, L.; Damm, F.; Demuth, I.; Frick, M. Clonal hematopoiesis of indeterminate potential-related epigenetic age acceleration correlates with clonal hematopoiesis of indeterminate potential clone size in patients with high morbidity. Haematologica 2022, 107, 1703–1708. [Google Scholar] [CrossRef] [PubMed]
- Soerensen, M.; Tulstrup, M.; Hansen, J.W.; Weischenfeldt, J.; Grønbæk, K.; Christensen, K. Clonal Hematopoiesis and Epigenetic Age Acceleration in Elderly Danish Twins. HemaSphere 2022, 6, e768. [Google Scholar] [PubMed]
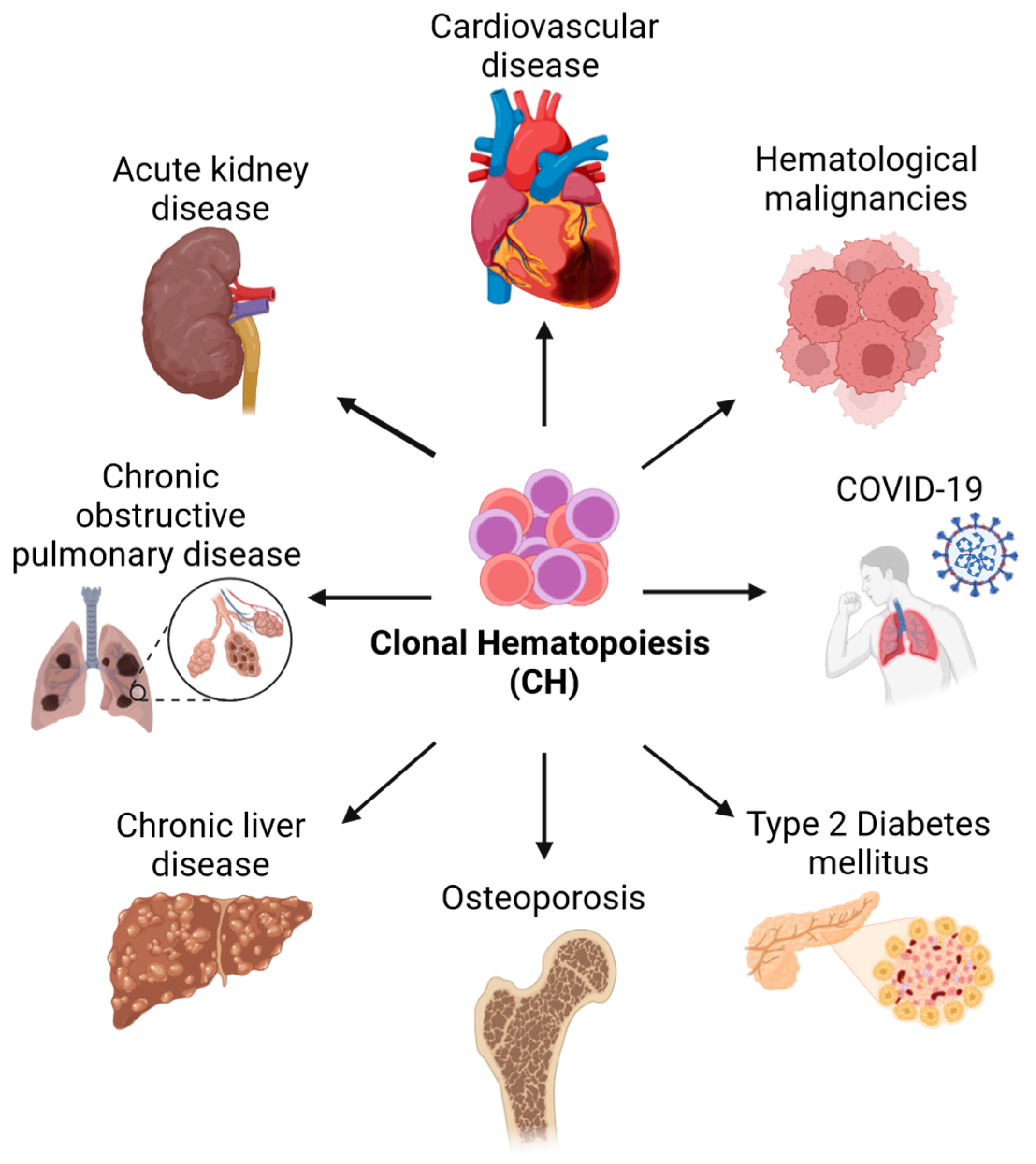
- Lin, A.E.; Rauch, P.J.; Jaiswal, S.; Ebert, B.L. Clonal Hematopoiesis: Confluence of Malignant and Nonmalignant Diseases. Annu. Rev. Cancer Biol. 2022, 6, 187–200. [Google Scholar] [CrossRef]
- Di Cesare, M.; Perel, P.; Taylor, S.; Kabudula, C.; Bixby, H.; Gaziano, T.A.; McGhie, D.V.; Mwangi, J.; Pervan, B.; Narula, J.; et al. The Heart of the World. Glob. Heart 2024, 19, 11. [Google Scholar]
- Deng, J.; Fleming, J.B. Inflammation and Myeloid Cells in Cancer Progression and Metastasis. Front. Cell Dev. Biol. 2021, 9, 759691. [Google Scholar]
- Zhang, C.R.C.; Nix, D.; Gregory, M.; Ciorba, M.A.; Ostrander, E.L.; Newberry, R.D.; Spencer, D.H.; Challen, G.A. Inflammatory cytokines promote clonal hematopoiesis with specific mutations in ulcerative colitis patients. Exp. Hematol. 2019, 80, 36–41.e3. [Google Scholar]
- Shen, Q.; Zhang, Q.; Shi, Y.; Shi, Q.; Jiang, Y.; Gu, Y.; Li, Z.; Li, X.; Zhao, K.; Wang, C.; et al. Tet2 promotes pathogen infection-induced myelopoiesis through mRNA oxidation. Nature 2018, 554, 123–127. [Google Scholar]
- Levin, M.G.; Nakao, T.; Zekavat, S.M.; Koyama, S.; Bick, A.G.; Niroula, A.; Ebert, B.; Damrauer, S.M.; Natarajan, P. Genetics of smoking and risk of clonal hematopoiesis. Sci. Rep. 2022, 12, 7248. [Google Scholar]
- Weinstock, J.S.; Gopakumar, J.; Burugula, B.B.; Uddin, M.M.; Jahn, N.; Belk, J.A.; Bouzid, H.; Daniel, B.; Miao, Z.; Ly, N.; et al. Aberrant activation of TCL1A promotes stem cell expansion in clonal haematopoiesis. Nature 2023, 616, 755–763. [Google Scholar] [CrossRef]
- Girotra, M.; Trachsel, V.; Roch, A.; Lutolf, M.P. In Vivo Pre-Instructed HSCs Robustly Execute Asymmetric Cell Divisions In Vitro. Int. J. Mol. Sci. 2020, 21, 8225. [Google Scholar] [CrossRef]
- Wehling, A.; Loeffler, D.; Zhang, Y.; Kull, T.; Donato, C.; Szczerba, B.; Ortega, G.C.; Lee, M.; Moor, A.; Göttgens, B.; et al. Combining single-cell tracking and omics improves blood stem cell fate regulator identification. Blood 2022, 140, 1482–1495. [Google Scholar]
- Loeffler, D.; Schneiter, F.; Wang, W.; Wehling, A.; Kull, T.; Lengerke, C.; Manz, M.G.; Schroeder, T. Asymmetric organelle inheritance predicts human blood stem cell fate. Blood 2022, 139, 2011–2023. [Google Scholar] [CrossRef]
- Loeffler, D.; Wehling, A.; Schneiter, F.; Zhang, Y.; Müller-Bötticher, N.; Hoppe, P.S.; Hilsenbeck, O.; Kokkaliaris, K.D.; Endele, M.; Schroeder, T. Asymmetric lysosome inheritance predicts activation of haematopoietic stem cells. Nature 2019, 573, 426–429. [Google Scholar]
- Nunes, J.; Loeffler, D. Asymmetric cell division of hematopoietic stem cells: Recent advances, emerging concepts, and future perspectives. Front. Hematol. 2024, 3, 1373554. [Google Scholar] [CrossRef]
- Marchetti, F.; Cardoso, R.; Chen, C.L.; Douglas, G.R.; Elloway, J.; Escobar, P.A.; Harper, T.; Heflich, R.H.; Kidd, D.; Lynch, A.M.; et al. Error-corrected next generation sequencing—Promises and challenges for genotoxicity and cancer risk assessment. Mutat. Res. Rev. Mutat. Res. 2023, 792, 108466. [Google Scholar] [PubMed]
- Chan, I.C.C.; Panchot, A.; Schmidt, E.; McNulty, S.; Wiley, B.J.; Liu, J.; Turner, K.; Moukarzel, L.; Wong, W.S.W.; Tran, D.; et al. ArCH: Improving the performance of clonal hematopoiesis variant calling and interpretation. Bioinformatics 2024, 40, btae121. [Google Scholar]
- Chen, C.; Wang, J.; Pan, D.; Wang, X.; Xu, Y.; Yan, J.; Wang, L.; Yang, X.; Yang, M.; Liu, G.P. Applications of multi-omics analysis in human diseases. MedComm 2023, 4, e315. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pendse, S.; Loeffler, D. Decoding Clonal Hematopoiesis: Emerging Themes and Novel Mechanistic Insights. Cancers 2024, 16, 2634. https://doi.org/10.3390/cancers16152634
Pendse S, Loeffler D. Decoding Clonal Hematopoiesis: Emerging Themes and Novel Mechanistic Insights. Cancers. 2024; 16(15):2634. https://doi.org/10.3390/cancers16152634
Chicago/Turabian StylePendse, Shalmali, and Dirk Loeffler. 2024. "Decoding Clonal Hematopoiesis: Emerging Themes and Novel Mechanistic Insights" Cancers 16, no. 15: 2634. https://doi.org/10.3390/cancers16152634
APA StylePendse, S., & Loeffler, D. (2024). Decoding Clonal Hematopoiesis: Emerging Themes and Novel Mechanistic Insights. Cancers, 16(15), 2634. https://doi.org/10.3390/cancers16152634