1. Introduction
Medulloblastoma (MB) is the most common malignant embryonal brain tumor in children, with about 500 new cases in the United States per year [
1,
2]. MB tumors arise in the cerebellum and can grow rapidly, leading to cerebellar dysfunction and disruptions to cerebrospinal fluid flow [
1,
2]. This manifests in children as problems with gait, coordination, nausea, vomiting, and obstructive hydrocephalus [
1,
2]. The standard treatment paradigm for MB begins with surgical resection followed by radiation and/or chemotherapy, with specific regimens determined by a patient’s risk of progressive disease [
1,
2,
3]. Historically, risk assessment relies on factors such as the patient’s age, the extent of tumor resection, and presence of metastasis [
1,
2,
3]. While this stratification method yields positive outcomes for many patients, approximately one-third of high-risk patients still experience progressive or relapsed disease [
1,
2]. The limited stratification of patients into risk groups often overlooks the high intratumoral heterogeneity characteristic of MB [
3,
4]. Advancements in molecular profiling have led to the categorization of MB into distinct molecular subgroups: WNT-activated, SHH-activated, and non-WNT/non-SHH (consisting of Group 3/4) [
3,
5,
6,
7]. Each subgroup exhibits unique genomic landscapes and dysregulated molecular pathways, with the latter being notably more aggressive and associated with unfavorable treatment outcomes [
3,
5,
6]. While this subgrouping has revolutionized the standard of care for patients with MB, relapsed disease remains a significant therapeutic challenge. Dominant-negative TP53 mutation and c-MYC amplification are frequently observed genomic alterations in relapsed disease and indicate aggressive disease behavior [
4,
8]. These tumors are generally refractory to traditional treatment modalities, regardless of the initial subgroup at diagnosis.
Early-phase clinical trials for patients with progressive or relapsed MB have evaluated the efficacy of FDA-approved immune checkpoint inhibitors, such as anti-PD-(L)1, which aim to enhance T-cell recognition and elimination of tumor cells (NCT02359565 and NCT03130959). While these immune checkpoint inhibitors have demonstrated efficacy across a variety of solid and hematological malignancies, they have not yielded success in treating MB patients [
9,
10]. The cold tumor microenvironment of MB, characterized by a lack of infiltrating T-cells and scarcity of PD-L1 expression, renders many of these immune checkpoint inhibitors ineffective [
11,
12,
13]. However, the exact mechanisms behind poor T-cell infiltration have yet to be fully elucidated. Finding ways to enhance T-cell infiltration, activation, and persistence in the tumor can shift these tumor microenvironments from ‘cold’ to ‘hot’ and render them more responsive to immune checkpoint modulation [
10].
Despite the scarcity of T-cells, tumor-associated macrophages (TAMs) have emerged as key cellular players in MB, comprising both tissue-resident microglia and bone marrow-derived macrophages (BMDMs) [
14]. TAMs are remarkably plastic and adapt quickly to their surroundings. One possible mechanism that TAMs use to suppress T-cells is V-domain Ig Suppressor of T-cell Activation (VISTA), a novel B7 family immune checkpoint regulator predominantly expressed on hematopoietic cells, with enriched expression on myeloid cells, including microglia and macrophages [
15,
16,
17]. This molecule has been implicated in suppressing T-cell activation, promoting T-cell and myeloid quiescence, and reprogramming myeloid cells towards an anti-inflammatory pro-tumoral state [
15,
18,
19,
20,
21]. There has been an increased effort to target VISTA in solid tumors to activate the immune response against cancer cells [
20,
21,
22,
23,
24,
25]. Thus far, multiple preclinical models have shown that blocking VISTA deters tumor growth and improves T-cell effector function against tumors, including brain tumors such as gliomas [
22,
26,
27,
28,
29]. Although VISTA-blocking antibodies are currently in Phase I clinical trials for solid tumor malignancies, this treatment remains untried in patients with brain tumors, and the effects of blocking VISTA in MB have not yet been explored.
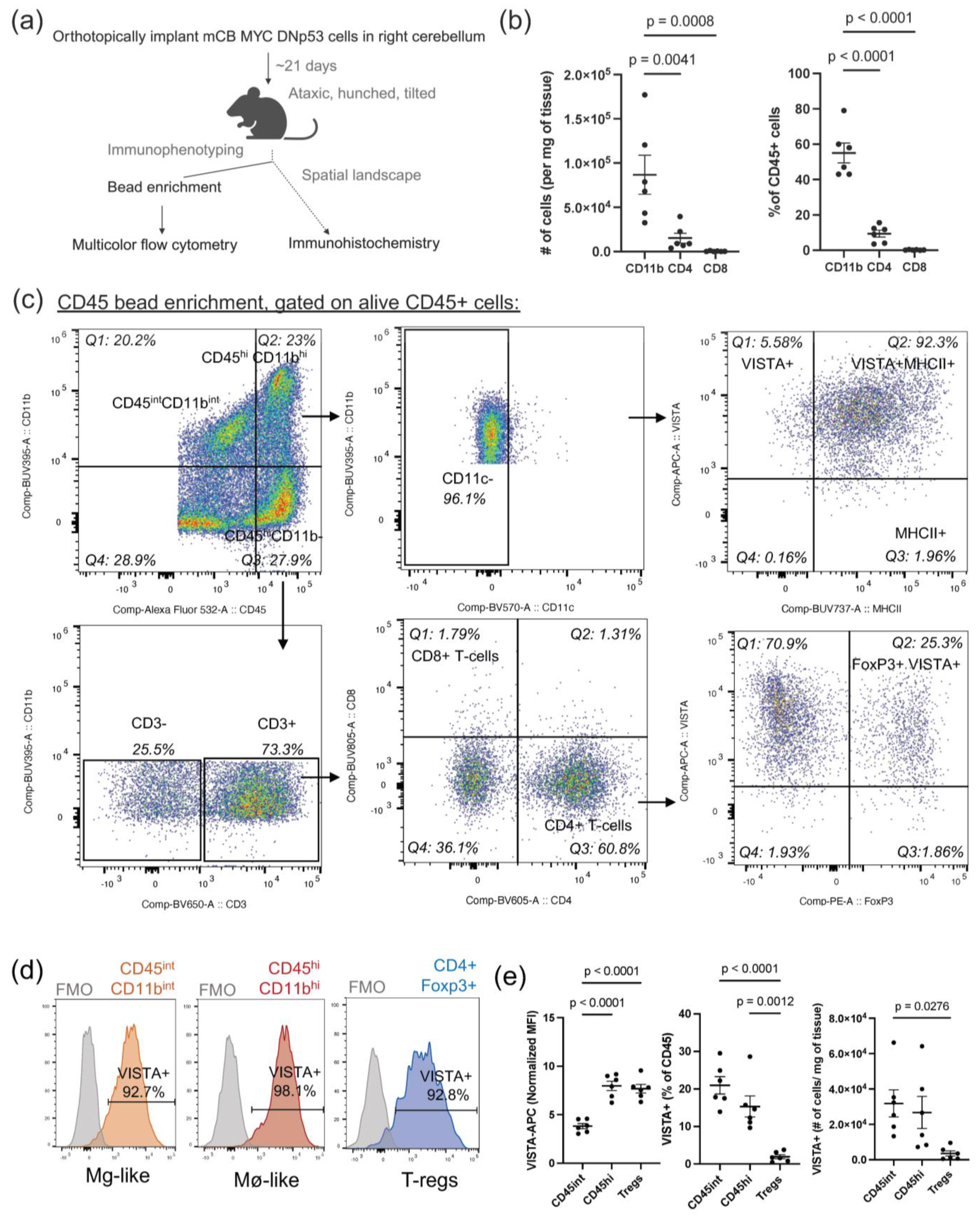
Utilizing our previously established orthotopic syngeneic mouse model of MB harboring both a dominant-negative TP53 mutation and MYC overexpression, which lend it a clinically aggressive and anaplastic phenotype [
30], we aimed to assess the extent to which the immune landscape resembles that of human MB. Additionally, we delved into the immunosuppressive mechanisms operating within this context. Our studies encompassed the characterization of myeloid polarization states and the immune checkpoints they express. Moreover, we determined the specific cellular and spatial patterns of VISTA expression within the tumor microenvironment. To our knowledge, this is the first study to set the stage for VISTA as an important mediator of immune suppression in MB and its potential as a valuable therapeutic target within this tumor microenvironment.
2. Materials and Methods
2.1. Cell Culture
All cells were maintained in a 37 °C humidified 5% CO
2 incubator and manipulated in a BSL-2 certified tissue culture hood. A previously established mouse medulloblastoma cell line (mCB DNp53 MYC) which contains dominant-negative
TP53,
c-MYC, and an EGFP reporter, was used; this cell line originated in MD, USA, and was provided by Allison M. Martin [
30]. Cells were grown in EF medium composed of DMEM/F12 (1:1) (+) L-Glutamine (+) 15 mM HEPES (Gibco, Waltham, MA, USA) supplemented with 2% B27 supplement, 5 μg/mL heparin, 20 ng/mL EGF, and 20 ng/mL FGF. Additionally, the human cell lines D283-MED and D425-MED were grown in MEM medium composed of MEM (+) L-Glutamine (Gibco) supplemented with 10% FBS, 1% sodium pyruvate, 1% MEM non-essential amino acids, and 1% penicillin/streptomycin. D283-MED was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and originated from NC, USA. D425-MED was a gift from Charles Eberhart and also originated in NC, USA. All cell lines were PCR-tested for mycoplasma every 6 months, and STR testing was performed annually on the human cell lines.
2.2. Animal Models
All animal experiments were conducted under the approval of the Albert Einstein College of Medicine Institute for Animal Care and Use Committee (IACUC, Bronx, NY, USA) protocol no. 00001069. C57BL/6J mice were obtained from Jackson Laboratories (Bar Harbor, ME, USA). Six-to-eight-week-old male and female mice were housed in a pathogen-free facility with access to standard chow and water. All surgical procedures were carried out in a BSL-2 certified laminar flow hood in the animal facility. To prepare the cells for implantation, mCB DNp53 MYC cells were collected, treated with StemPro Accutase Cell Dissociation reagent (LifeTech, Shenzhen, China), and resuspended in EF medium to yield a solution containing 100 cells per microliter (cells/µL). The mice were sedated with ketamine (100 mg/kg), and the posterior part of the head was shaved, sterilized, and locally anesthetized with 0.25% bupivacaine. A 1 cm midline craniocaudal incision was made in the scalp, exposing the lambdoid suture. An 18-gauge needle was used to drill a burr hole in the right cerebellum, positioned 1 mm posterior and 1 mm lateral to the lambda. Intracranial implantation of 300 mCB DNp53 MYC cells (3 µL) was performed by slowly injecting the cells with a 25-gauge Hamilton Syringe equipped with a sterile cut pipette tip to ensure the appropriate depth of 2 mm into the cerebellum, as previously described [
30]. Sham control mice underwent an identical procedure but instead received 3 µL of EF medium. The incision was closed with surgical staples. Following the procedure, the mice were provided with MediGel Sucralose gels infused with carprofen (25 mg/kg +/− 3 mg/kg) in the days following the procedure. Staples were removed 10 days post-implantation. Mice were monitored for signs indicative of a moribund state, including ataxia, hunching, slowed movement, head tilt, and weight loss. The average time to disease onset in mice was 21.5 days.
2.3. Isolation of Tumor-Infiltrating Leukocytes
Once the mice exhibited a moribund state, tumors were harvested by gross dissection. Tumors were weighed and dissociated using the Neural Tissue Dissociation Kit (P) (Miltenyi, Tokyo, Japan) and transferred to a GentleMACS C-tube (Miltenyi) for further dissociation on the GentleMacs Octo Dissociator, with constant stirring at 37 °C for 30 min. Digestion was halted with the addition of 5 mL of cold MACS buffer (1X PBS, 0.5% BSA, 2 mM EDTA) and then strained through a 40 μm filter (Corning, Somerville, MA, USA). Myelin removal was performed by resuspending cells in a 30% Percoll gradient and spinning down at 700× g for 10 min at room temperature (RT). Red blood cell removal was performed using ACK lysis buffer (Quality Biological, Gaithersburg, MD, USA) for 5 min at RT. Either CD45 TIL microbead enrichment (Miltenyi) or CD11b microbead enrichment (Miltenyi) was performed. Both the positive bead-enriched fraction and negative flow-through were collected. Cells were counted and stained for multi-color spectral flow cytometry (see below). For the sham controls, the right cerebellar hemisphere was isolated 21 days following the surgical procedure, and the same downstream processing was performed as for the experimental samples.
2.4. Isolation of Spleens and Bone Marrow
Spleens and bone marrow from all tumor-bearing mice were collected. Bone marrow cells were isolated by flushing the femur with cold 1X PBS using a 25-gauge (BD Hypodermic, Franklin Lakes, NJ, USA) needle and centrifuging the cells at 300× g for 5 min. Spleens were collected and mashed through a 40-micron filter (Corning) and centrifuged at 300× g for 5 min. Red blood cell removal for spleens and bone marrow was performed using ACK lysis (Quality Biological, Gaithersburg, MD, USA) for 5 min at RT. Cells were washed with MACS buffer, counted, and stained for flow cytometry.
2.5. Multi-Color Spectral Flow Cytometry (CD45 Bead Enrichment)
CD45 TIL bead-enriched single-cell suspensions, spleen and bone marrow, were labeled with Zombie NIR Fixable Viability dye (Biolegend, San Diego, CA, USA, cat. no. 423105). Cells were then Fc-blocked with CD16/CD32 (Biolegend, cat. no. 101301). Cells were stained in a 1:1 solution of Brilliant Stain Buffer (BD Bioscience, Franklin Lakes, NJ, USA, cat. no. 563794) and 1X PBS containing the following fluorochrome-conjugated extracellular antibodies: CD45-AlexaFluor532 (ThermoFisher, Waltham, MA, USA, cat. no. 58-0451-80), CD11b-BUV395 (BD Bioscience, cat. no. 565976), CD11c-Brilliant Violet 570 (Biolegend, cat. no. 117331), CX3CR1-AlexaFluor700 (Biolegend, cat. no. 149035), Tmem119-PerCPeFluor710 (ThermoFisher, cat. no. 46-6119-80), CD206-Brilliant Violet 421 (Biolegend, cat. no. 141717), CD3-Brilliant Violet 650 (Biolegend, cat. no. 100229), CD4-Brilliant Violet 605 (Biolegend, cat. no. 100547), CD8-BUV805 (ThermoFisher, cat. no. 368-0081-80), CD19-Brilliant Violet 510 (Biolegend, cat. no. 115545), NK1.1-Pacific Blue (Biolegend, cat. no. 108721), VISTA-APC (Biolegend, cat. no. 150205), and B7H3-PECy7 (Biolegend, cat. no. 135613). Intracellular fixation was performed using the FOXP3/Transcription Factor staining kit (ThermoFisher). Cells were permeabilized and stained with IFNy-PECF594 (BD Bioscience, cat. no. 562333) and FOXP3-PE (ThermoFisher, cat. no. 14-5773-80) diluted in permeabilization buffer. Cells were resuspended in MACS buffer, and data was collected on the Aurora Cytek maintained by the AECOM Flow Core Facility. Immune populations were gated in FlowJo v.10.10.0 (
Figure S1). All gating was based on fluorescence minus one (FMO) controls.
2.6. Multi-Color Spectral Flow Cytometry (CD11b Bead Enrichment)
To more closely look at myeloid cells from tumor-bearing or sham control mice, CD11b bead-enriched single-cell suspensions were labeled with Zombie NIR Fixable Viability dye (Biolegend, cat. no. 423105). Cells were then Fc-blocked with CD16/CD32 (BD Biosciences). Cells were stained in a 1:1 solution of Brilliant Stain Buffer (BD Bioscience, cat. no. 563794) and 1X PBS containing the following extracellular fluorochrome-conjugated antibodies: CD45-AlexaFluor532 (ThermoFisher, cat. no. 58-0451-80), CD11b-BUV395 (BD Bioscience, cat. no. 565976), CD11c-Brilliant Violet 570 (Biolegend, cat. no. 117331), CD3-Pacific Blue (Biolegend, cat. no. 100213), F4/80-PerCPVio700 (Miltenyi cat. no. 130-118-466), Arg1-AlexaFluor700 (ThermoFisher, cat. no. 56-3697-80), PDL1-Brilliant Violet 605 (Biolegend, cat. no. 124321), VISTA-APC (Biolegend, cat. no. 143709), CD40-PECy7 (Biolegend, cat. no. 124621), CD163- PE/Dazzle594 (Biolegend, cat. no. 155315), CD68-Brilliant Violet 711 (Biolegend, cat. no. 137029), Tim3-PE (Biolegend, cat. no. 134003), CD80-Brilliant Violet 650 (Biolegend, cat. no. 104713), and CTLA4-Brilliant Violet 421 (Biolegend, cat. no. 106311).
2.7. Flow Cytometry
For evaluation of VSIG3 and VSIG8 expression on human and mouse cell lines, both D283-MED and mCB DNp53 MYC were collected and stained with Live/Dead Fixable Aqua (ThermoFisher, cat. no. L34965). Cells were washed and then stained with either rabbit anti-VSIG3 (R&D, Santa Clara, CA, USA, cat. no. MAB11226-SP), rabbit anti-VSIG8 (ThermoFisher, cat. no. PA5-51157), or rabbit IgG isotype control (R&D, cat. no. AB-105-C) for 15 min at RT in the dark. Cells were washed and then incubated with goat anti-rabbit-APC IgG (H+L) cross-absorbed secondary antibody (ThermoFisher, cat. no. A-10931) for 15 min at RT in the dark. Cells were fixed with 2% paraformaldehyde and analyzed on the BD LSRII maintained by the AECOM Flow Core Facility. Gating was performed in FlowJo v10.10.0 and based on fluorescence minus one (FMO) controls.
2.8. Western Blot Analysis
Protein was isolated from D283-MED, D425-MED, and mCB DNp53 MYC cells using an extraction buffer (RIPA buffer, 0.01 M NaF, 100 mM sodium vanadate, 1X Sigma protease inhibitor cocktail, 0.1 M PMSF), followed by protein quantification with the DC Protein Assay Kit (BioRad, Hercules, CA, USA) and measurement of absorbance on the SpectraMax M5e at 750 nm. Each sample received 20 µg of respective protein and was run on TGX precast gels on the Mini Protean TetraCell system. Protein was transferred to a PVDF membrane using the Biorad TransBlot Turbo transfer system. The membrane was blocked in 5% milk and incubated with primary antibodies overnight at 4 °C: Rabbit anti-VSIG3 (R&D, cat. no. MAB11226-SP), Rabbit anti-VSIG8 (ThermoFisher, cat. no. PA5-51157), or Sheep anti-VISTA (R&D, cat. no. AF-7005-SP). Next day, the membrane was washed with TBST and incubated with Goat anti-Rabbit IgG (H+L), Peroxidase (Vector Laboratories, Burlingame, CA, USA, cat. no. PI-1000-1), or Donkey anti-Sheep IgG HRP-conjugated (R&D, cat. no. HAF016) for 1 h at RT, followed by three consecutive TBST washes. Chemiluminescence imaging was performed on the Biorad ChemiDoc Touch imaging system.
2.9. Immunohistochemistry
Tumors were harvested and fixed in FormaFixx (ThermoFisher) at 4 °C before submission to the Histology Core for paraffin embedding and sectioning. Sectioning was performed every 5 microns. Briefly, serial sections were deparaffinized and incubated for 10 min in 3% hydrogen peroxide. Antigen retrieval was performed in 1X DAKO (Agilent, Santa Clara, CA, USA) for 30 min using an electric steamer (Oster, Boca Raton, NW, USA), and then the samples were allowed to cool at RT for 30 min. Fc blocking was performed for 30 min at RT, followed by blocking with 2.5% Normal Horse Serum (Vector Laboratories) for 30 min at RT. Slides were incubated overnight at 4 °C with primary antibody: rabbit anti-mouse CD11b (Cell Signaling Technology, Danvers, MA, USA, cat. no. 93169S), rabbit anti-mouse VISTA (Cell Signaling Technology, cat. no. 54979T), anti-mouse CD3 (Cell Signaling Technology, cat. no. 78588T), rabbit anti-mouse FoxP3 (Cell Signaling Technology, cat. no. 12653), or rabbit mAb IgG isotype control (Cell Signaling Technology, cat. no. 3900S). The following day, slides were washed with TBST and incubated with ImPRESS HRP horse anti-rabbit secondary antibodies (Vector Laboratories). Slides were developed with DAB peroxidase substrate (Vector Laboratories) for up to 90 seconds. Slides were counterstained with hematoxylin (Vector Laboratories), dehydrated, and mounted with Surgipath Micromount (Leica, Wetzlar, Germany). Slides were allowed to dry, and then imaging was performed on the EVOSS microscope. White balancing was performed using Adobe Photoshop. Scale bars were incorporated in Fiji by aligning pixels to microns through calibration using a cell-counter slide.
2.10. Tumor Cell and T-Cell Co-Cultures
Naïve splenic CD4 T-cells were isolated from a C57BL/6J wildtype mouse using the EasySepTM Mouse CD4+ Isolation Kit (StemCell Technologies, Vancouver, Canada, cat. no. 19852). Following isolation, 80,000 CD4 T-cells were plated in a 96-well V-plate and stained with CellTrace-Violet (ThermoFisher) for 30 min at RT. T-cells were resuspended in 100 µL of RPMI medium supplemented with 10% FBS, 1% Pen/Strep, 1% HEPES, 1% sodium pyruvate, 1% NEAA, 55 uM B-mercaptoethanol, and 0.45% D-glucose, in addition to 1 ng of recombinant mouse IL2 (BioLegend, San Diego, CA, USA, cat. no. 575402) and 1:1 CD3/CD28 Dynabeads (Gibco, cat. no. 11-452-D). T-cells were then co-cultured with 100 µL of either RPMI medium, EF medium, conditioned medium (isolated by filtering cultured supernatant through a Nalgene 0.2 µm syringe filter), or increasing numbers of mCB MYC DNp53 tumor cells (T-cell/mCB DNp53 MYC ratios: 80:1, 40:1, and 20:1). Negative controls included CD4 T-cells with no CD3/CD28 bead activation. Co-cultures were transferred to a 96-well U-plate. After 5 days of incubation, cells were stained with L/D-Zombie NIR (Biolegend, cat. no. 423105), CD4-Brilliant Violet 605 (Biolegend, cat. no. 100547), and VISTA-APC (Biolegend, cat. no. 143709) and analyzed on the Cytek Aurora. Gating was performed using FlowJo, where CellTrace Violet dilution was measured as a function of T-cell proliferation. To see if blocking VISTA could rescue T-cell proliferation, CD4 T-cells were co-cultured with mCB MYC DNp53 tumor cells at a ratio of 20:1 for 5 days, as described above. Addition of either 0, 0.2, 2, or 20 µg/mL of InVivoMab anti-mouse VISTA blocking Ab (BioXCell, Lebanon, NH, USA, cat. no. BE0310) or 20 µg/mL of an InVivoMab polyclonal Armenian Hamster IgG Isotype control (BioXCell, cat. no. BE0091) was performed in technical triplicates over 6 biological replicates. After 5 days, cells were stained with L/D-Zombie NIR (Biolegend, cat. no. 423105), CD4-Brilliant Violet 605 (Biolegend, cat. no. 100547), and VISTA-APC (Biolegend, cat. no. 143709) and analyzed on the Cytek Aurora maintained by the AECOM Flow Core Facility. Gating was performed using FlowJo, where CellTrace Violet dilution was measured as a function of T-cell proliferation.
2.11. Caris Molecular Profiling Analysis
Biomarker data acquired from the Caris database contained records of 81 patients whose medulloblastoma tumors underwent RNA (whole-transcriptome) and DNA (next-generation) sequencing at Caris Life Sciences (Phoenix, AZ, USA). Of these patients, 53 were aged 30 or under, forming the primary focus of the analysis. One patient was excluded from the analysis due to frontal lobe primary tumor location. This study was conducted in accordance with the guidelines of the Declaration of Helsinki, the Belmont Report, and US Common Rule. In compliance with policy 45 CFR 46.101(b), this study was performed utilizing retrospective, deidentified clinical data, thereby qualifying for IRB exemption, obviating the need for patient consent. To evaluate mRNA expression (WTS), the Qiagen RNA FFPE tissue extraction kit was used on tumor specimens (with a minimum of 10% tumor content for enrichment and extraction of RNA), and the RNA quality and quantity were determined via Agilent TapeStation. Biotinylated RNA baits were hybridized to the synthesized and purified cDNA targets and the bait–target complexes were amplified in a post-capture PCR reaction. The Illumina NovaSeq 6500 was used to sequence the whole transcriptomes from patients to an average of 60 M reads (Illumina, San Diego, CA, USA). Raw data were demultiplexed by the Illumina Dragen BioIT accelerator, trimmed, and counted, and PCR duplicates were removed and aligned to the human reference genome hg19 by STAR aligner. For transcription counting, transcripts per million (TPMs) were generated using the Salmon expression pipeline.
2.12. Nanostring GeoMx® Digital Spatial Profiling
A single formalin-fixed paraffin-embedded slide containing multiple embryonal human tumor cores in the form of a tissue microarray was stained with 4 immunofluorescent markers, including DAPI, synaptophysin, CD45, and Iba1, and 77 additional proteins from NanoString GeoMx® Digital Spatial Profiling curated panels, including Immune Cell Profiling, IO Drug Target, Immune Activation Status, PIK/AKT, and Immune Cell Typing panels (NanoString, Seattle, WA, USA). Twelve samples representing distinct tumors were selected based on tissue integrity: four SHH-activated medulloblastoma samples, five non-WNT/non-SHH medulloblastoma samples, and three medulloblastoma samples of indeterminate subgroups. Analysis and selection were performed in real time based on the appearance of a paired H&E slide. Rare cell masking software identified Iba-1-positive geometric regions within the tumor cores and was applied to selected samples. Photocleavable linkers were activated to collect protein specifically from IBA-1-positive and -negative regions based on masking.
2.13. Statistical Analysis
Results are presented as mean values +/− standard error of the mean (SEM). Statistical tests for in vitro and in vivo experiments were conducted in GraphPad Prism v10. In all cases, a
p-value of less than 0.05 was considered statistically significant. For heatmap generation of in vivo murine data and in situ human data, markers were considered significantly different between compared groups with a log2 fold change less than −0.5 or greater than 0.5 and a
p-value less than 0.05. For Caris Life Sciences transcriptomic data, continuous variables were compared using non-parametric tests, including Mann–Whitney U tests. Adjustments for multiple comparisons were applied using the Benjamini–Hochberg method to avoid type I errors. An adjusted
p-value (q-value) of <0.05 was considered a significant difference. Specifically, for NanoString GeoMx
® Digital Spatial Profiling data analysis, protein expression was transformed with the natural log. Differentially expressed proteins were analyzed with the limma package (v3.48.3) [
31].
4. Discussion
To the best of our knowledge, this is the first study to thoroughly evaluate the expression of VISTA in the MB tumor microenvironment. We have shown that VISTA is highly expressed across infiltrating immune cells within our syngeneic murine model, including CD45
int/CD11b
int microglia-like TAMs, CD45
hi/CD11b
hi macrophage-like TAMs, and CD4+FoxP3+ Tregs [
14]. While all myeloid populations highly express VISTA, we observed decreased expression of VISTA on microglia-like TAMs and an increase in VISTA expression on macrophage-like TAMs, when compared to sham controls, as demonstrated in
Figure 2c. This opposing pattern of VISTA regulation has been observed in other CNS pathologies and may indicate that while microglia downregulate VISTA to allow for T-cell infiltration, infiltrating BMDMs can oppose this response [
33]. Additionally, microglia may enter an activated macrophage-like state in which they contribute to VISTA-mediated immune suppression along with BMDMs. This aligns with the observed percent decrease in the CD45
int/CD11b
int compartment with a concurrent increase in the CD45
hi/CD11b
hi compartment in tumor-bearing mice as compared to sham control mice, as shown in
Figure 2b. Furthermore, we were unable to identify expression of Tmem119 among microglia-like TAMs, aligning with a recent study which used a different mouse MB model, and consistent with the idea that microglia may downregulate Tmem119 and adopt a more macrophage-like phenotype upon entering the tumor microenvironment [
32]. High co-expression of VISTA with MHCII was also observed on both myeloid TAM populations, which suggests to us that tumor-infiltrating myeloid cells in MB have heightened antigen presentation capabilities consistent with an activated macrophage phenotype but do so in the context of a novel inhibitory immune checkpoint molecule, VISTA. Importantly, this high co-expression level was not evident in myeloid cells within the bone marrow, underscoring the tumor-specific nature of heightened VISTA and MHCII. These findings are supported by the human mRNA and protein expression profiling where both VISTA and HLA-DR were highly correlated with IBA-1+ infiltrating immune cells, as shown in
Figure 6.
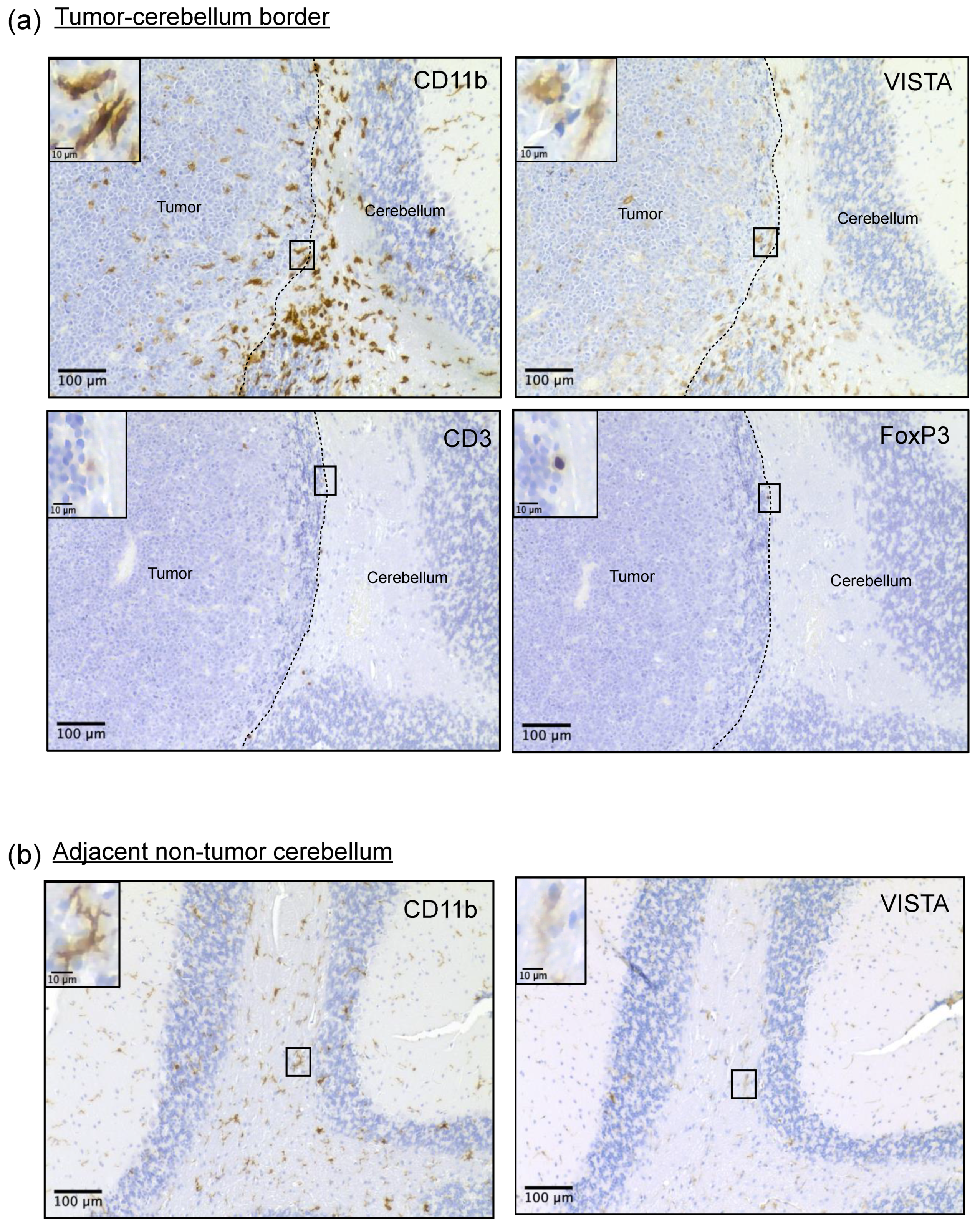
Immunohistochemistry on resected murine brain tumors was used to assess spatial localization of cellular populations within the tumors. We found that VISTA-expressing myeloid cells were present throughout murine MB tumors, but the highest VISTA expressors congregated at the tumor border. The presence of these myeloid cells aggregating at the tumor border has been observed in other brain tumor models with low T-cell presence, including MB [
32,
39]. This spatial distribution along the tumor border has been postulated to form a physical barrier that reduces the activation and infiltration of effector T-cells while promoting a Treg response [
40]. In support of this and similar to human MB, across all murine IHC samples, only a small number of CD3+ lymphocytes were detected within the tumor or at the tumor border. Instead, the majority of CD3 staining, though still sparse, was localized to perivascular areas of the tumor. This may indicate that T-cells are trapped in these areas within the tumor, as has been observed in other cold tumors [
41]. Several of these CD3+ T-cells stained positive for FOXP3, supporting our flow cytometry data, which detected the limited presence of FOXP3+ T
regs. These VISTA
hi-expressing myeloid cells displayed a more ameboid morphology compared to the ramified appearance and weaker VISTA staining of myeloid cells in the adjacent non-tumor cerebellum and remote cortical regions, as demonstrated in
Figure 3b. This supports the idea that these high-expressing VISTA cells may be the opposing force responsible for preventing immune infiltration into the tumor. Unfortunately, we were unable to confirm these findings in our human samples. The use of minimally invasive neurosurgical techniques has improved neurocognitive outcomes by carefully removing tumors from critical brain regions, but this impedes evaluation of the peritumoral border in most diagnostic samples [
42]. Nevertheless, these novel observations in our mouse model support our earlier findings that human MB exhibits poor T-cell infiltration and help explain its poor response to T-cell-targeting immune checkpoint blockade [
11]. Taken together, we believe these findings support the notion that T-cell infiltration and antigen response mediated by CD45
int/CD11b
int microglia-like cells are impeded by the presence of CD45
hi/CD11b
hi macrophage-like TAMs through a VISTA-dependent mechanism in our mouse model. These findings hold translational implications, as they highlight the importance of targeting myeloid cells within the tumor microenvironment of MB and further uncovering the role VISTA may play in these cells by promoting T-cell suppression.
Negative effects on T-cells were also reflected by the remarkable ability of mCB DNp53 MYC tumor cells to override the exogenous solid activation signals provided by CD3/CD28 combined with IL-2 in vitro. These experiments demonstrated for the first time that MB tumor cells can function as direct CD4 T-cell suppressors. This suppression of T-cell proliferation was observed exclusively in the presence of tumor cells rather than soluble factors from conditioned media. This implies a direct interaction between tumor cells and CD4+ T-cells operating independently of cytokine-mediated pathways. It also underscores the likelihood that MB employs multiple mechanisms of immune suppression. Therefore, even if immune modulatory therapies allow T-cells to penetrate the tumor, there will likely be additional hurdles to the persistence of T-cells in this microenvironment due to the significant capacity of tumor cells to hinder T-cell proliferation. These findings have profound implications for therapies such as CAR T-cells, which need to be able to proliferate and expand upon entry into the tumor microenvironment. They also have implications for immune checkpoint therapies, which may not be able to overcome the strong immunosuppressive signals delivered by tumor cells. This was demonstrated in vitro by the inability of VISTA-blocking antibodies to significantly rescue T-cells from tumor-mediated suppression of proliferation. Therefore, while VISTA may be an important mediator of immune suppression in MB, combination therapies will likely be needed to fully overcome the powerful immunosuppressive effects mediated by this tumor.
Identifying the specific mechanism of VISTA-mediated T-cell inhibition in MB was beyond the scope of this study. However, we identified multiple VISTA binding partners in human and murine cell lines as well as in our human cohorts where VISTA signaling likely occurs. Both described tumor-expressed binding partners, VSIG3 and VSIG8, were expressed in vitro. Transcriptomic analysis of human MB tumors revealed that VISTA expression positively correlated with VSIG8 but not VSIG3, suggesting that VSIG8 may be a more translationally relevant target. Moreover, the low expression of VISTA on our tumor cells indicated that VISTA expression is predominantly confined to immune cells within this tumor. This finding aligns with previous studies showing that VISTA is typically exclusive to either tumor cells or immune cells within the tumor microenvironment [
43]. Multiple binding partners on tumor, myeloid, and T-cells have been described for VISTA in various contexts. Two of these, PSGL-1 and LRIG1, also correlated with VISTA expression in human transcriptomic data, as shown in
Figure 5. It will be important to understand which specific binding partner(s) are most relevant in MB for more effective targeting of the VISTA signaling axis. Identifying and investigating the VSIG family in MB is a novel aspect of this work and deserves further consideration in future studies. Whether these binding partners actively engage VISTA in this tumor microenvironment remains unclear.
We recognize several limitations of the current study. Given the paucity of infiltrating immune cells in MB, one major constraint was the limited number of cells isolated from tumors. Despite efforts to increase immune cell recovery through bead enrichment, only a few tens of thousands of total immune cells could be isolated from a single tumor. This challenge extended to the Caris human sequencing database due to the large abundance of tumor cells and the low number of infiltrating immune cells, exacerbated by a small cohort (
n = 52). Additionally, distinguishing microglia from bone marrow-derived macrophages within our flow cytometry data proved challenging, as both upregulate CD45 and CD11b. Conventional markers for microglia, such as Tmem119, were absent in our model. The mixed activating and inhibitory immune signature exhibited by TAMs in both human and mouse MB underscored the difficulty of delineating these states in microglia and macrophages. Despite the overwhelming similarities we observed between our human cohorts and our mouse model, there were some notable differences between mice and humans, including Arg1, CD163, and Tim-3. These markers are not usually associated with tumor cells and were only upregulated in a fraction of the samples. Whether this represents tumor heterogeneity within our mouse model or is an artifact of the bead isolation procedure remains unclear, and further studies focusing on these markers are needed. Together, these issues highlight the limitations of using surface markers in the setting of tumors. Thus, single-cell analysis may offer a more sensitive approach to the immune phenotyping of these complex and dynamic immune cells. Of note, our flow cytometry gating of myeloid subpopulations excluded CD11c cells—a widely established pan-dendritic marker. Given that CD11c is a marker that was upregulated in our human IBA-1+ TAMs as well, it may be that CD11c+ TAMs exist in MB tumors and have a function that has yet to be investigated [
39].



 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}